
Increased Expression of the Mitochondrial Glucocorticoid Receptor Enhances Tumor Aggressiveness in a Mouse Xenograft Model



 ,
, 

Abstract
1. Introduction
2. Results
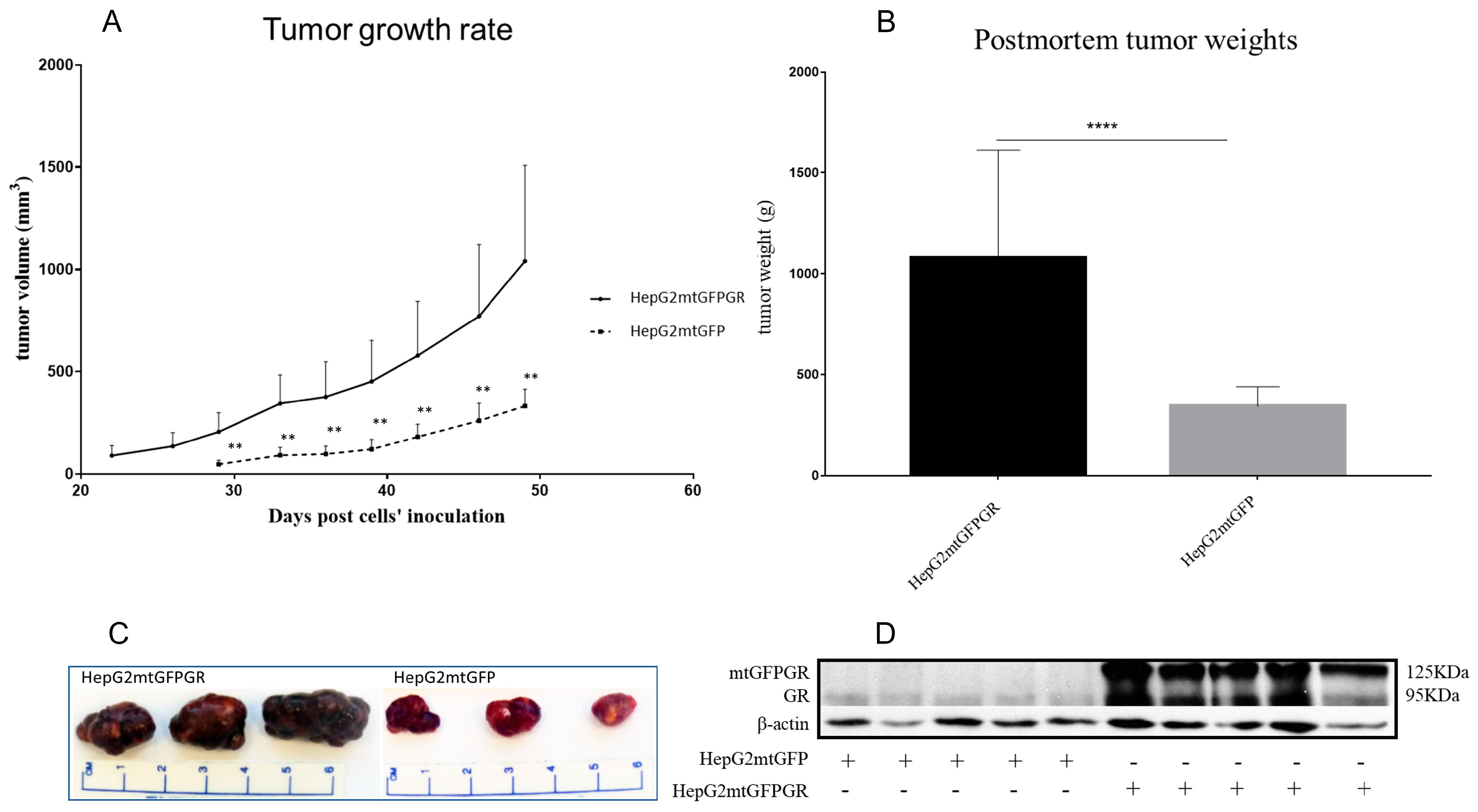
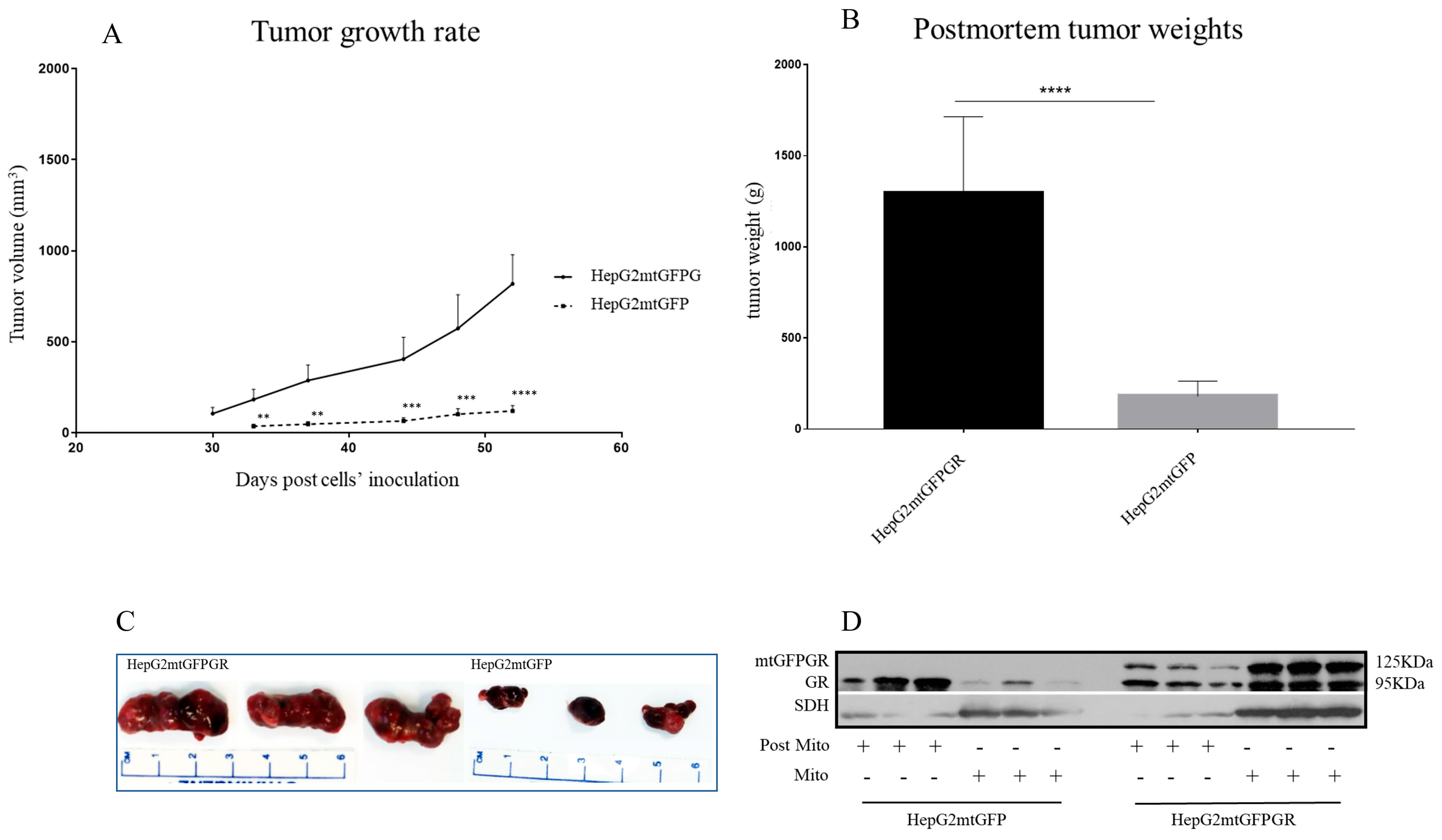
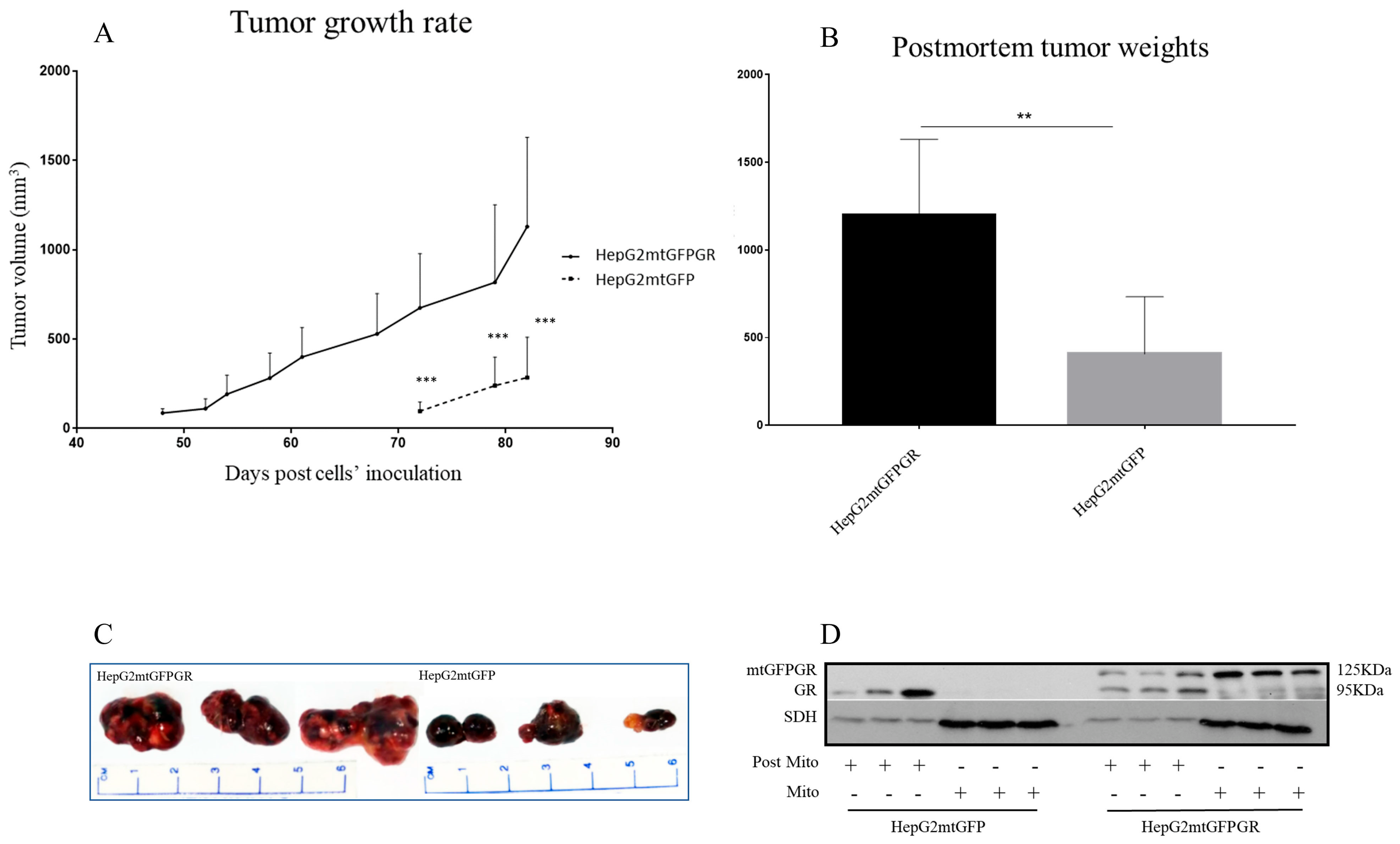
2.1. mtGR Enhances the In Vivo Aggressiveness of HepG2 Cancer Cells
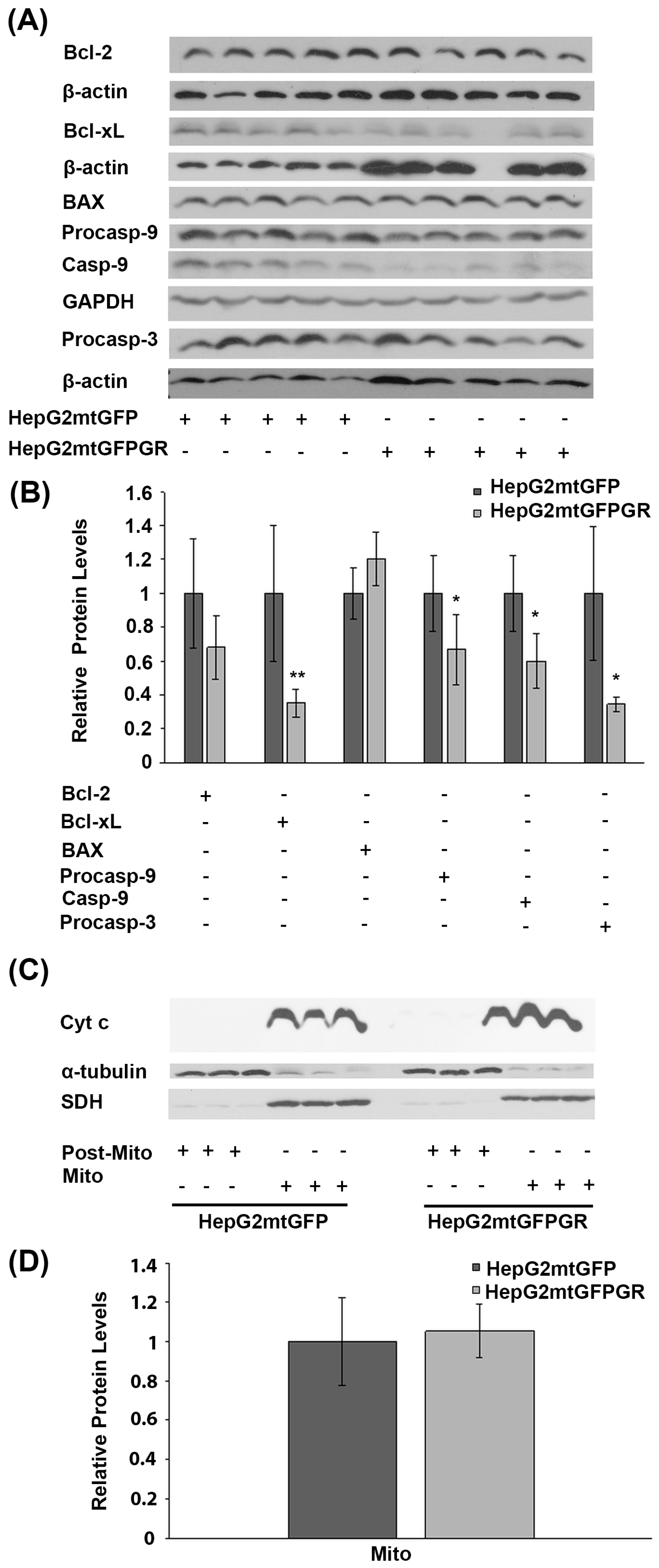
2.2. mtGR-Induced Tumorigenesis Is not Associated with mtGR-Associated Regulation of Apoptosis
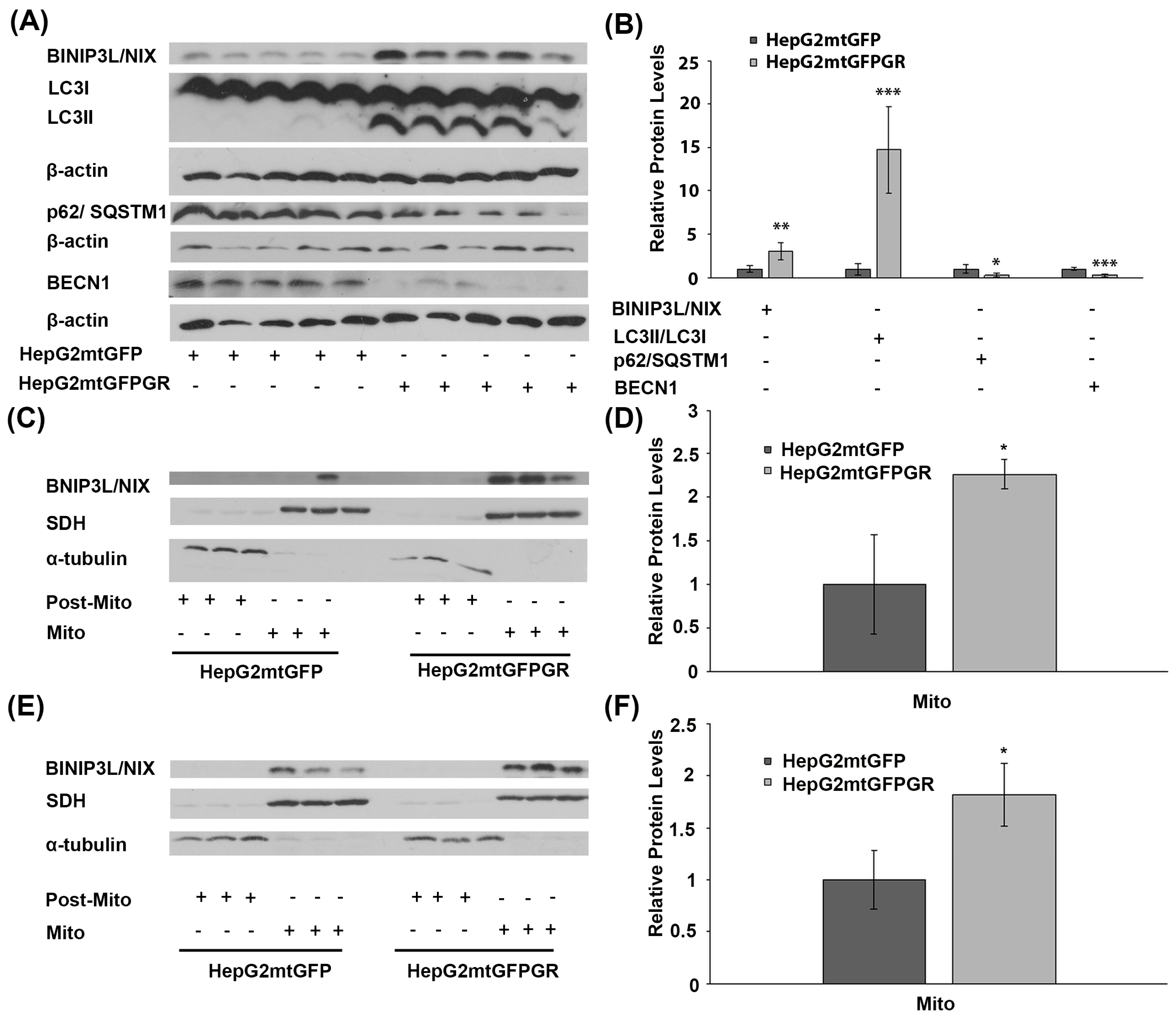
2.3. mtGR-Associated Increase in Tumor Aggressiveness Is Related to Autophagy Induction
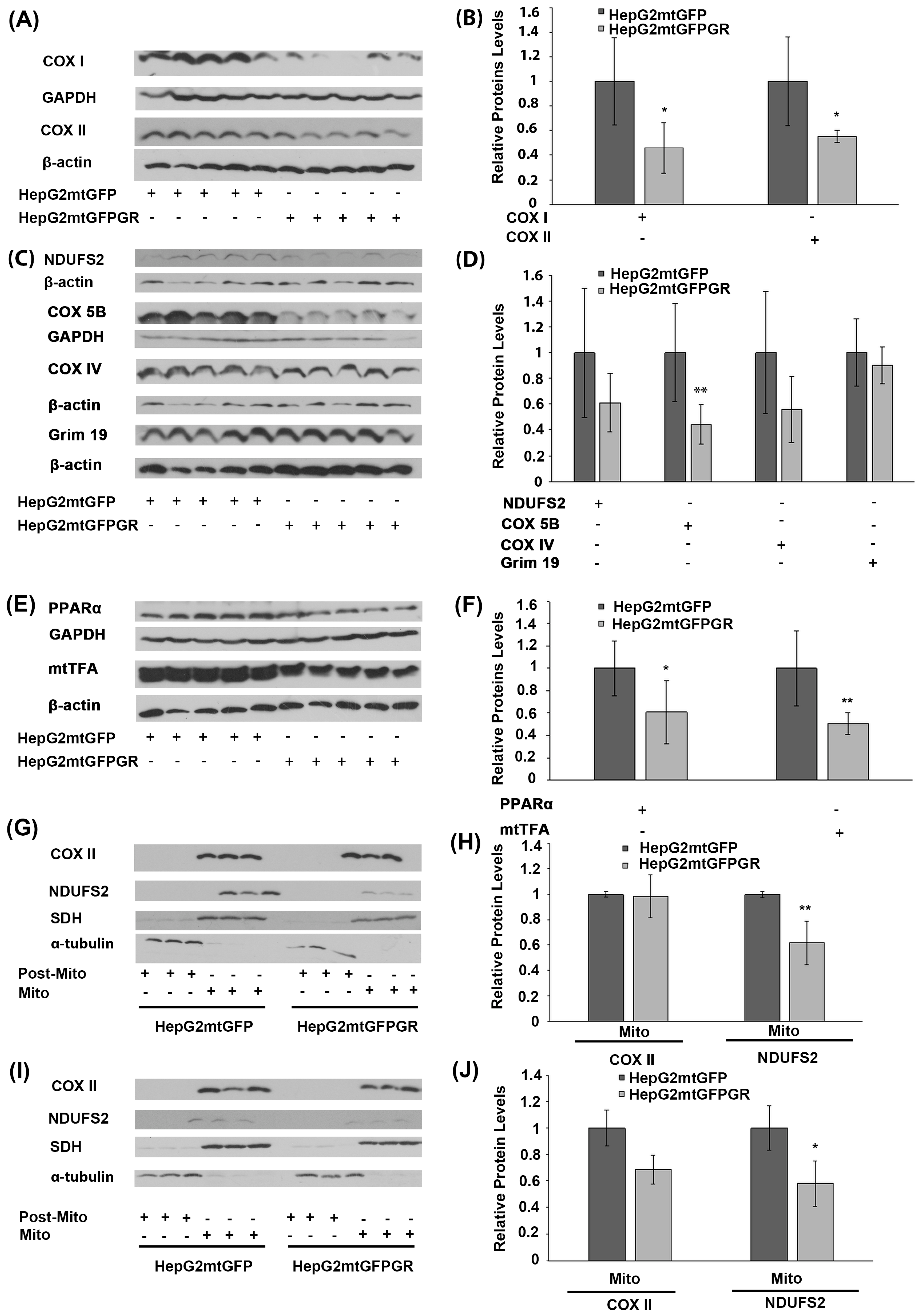
2.4. Regulation of OXPHOS Biosynthesis by mtGR upon Tumor Progression
2.5. Role of mtGR in the Regulation of Krebs Cycle during Tumor Progression
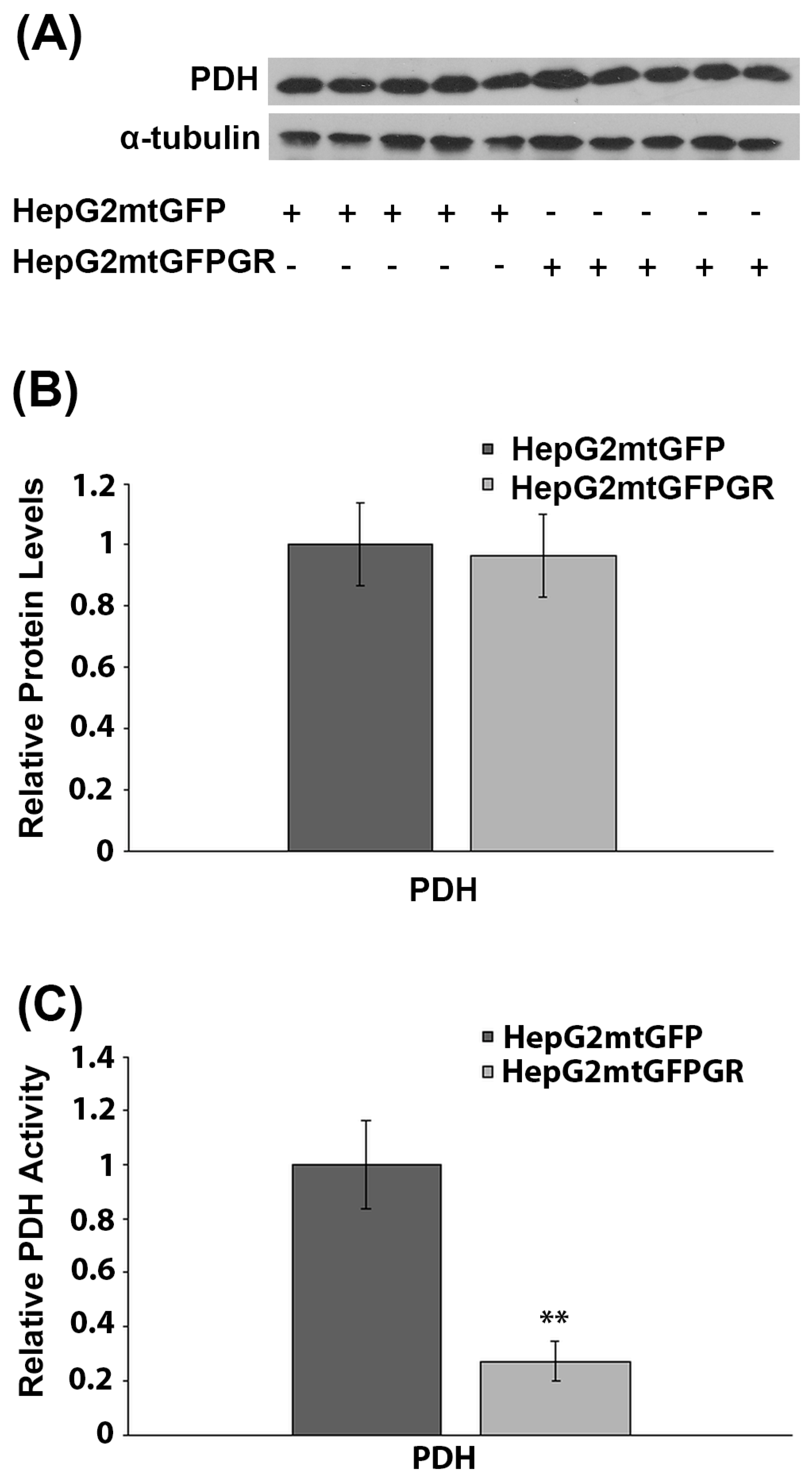
2.6. Effect of mtGR on PDH Activity during Tumor Progression
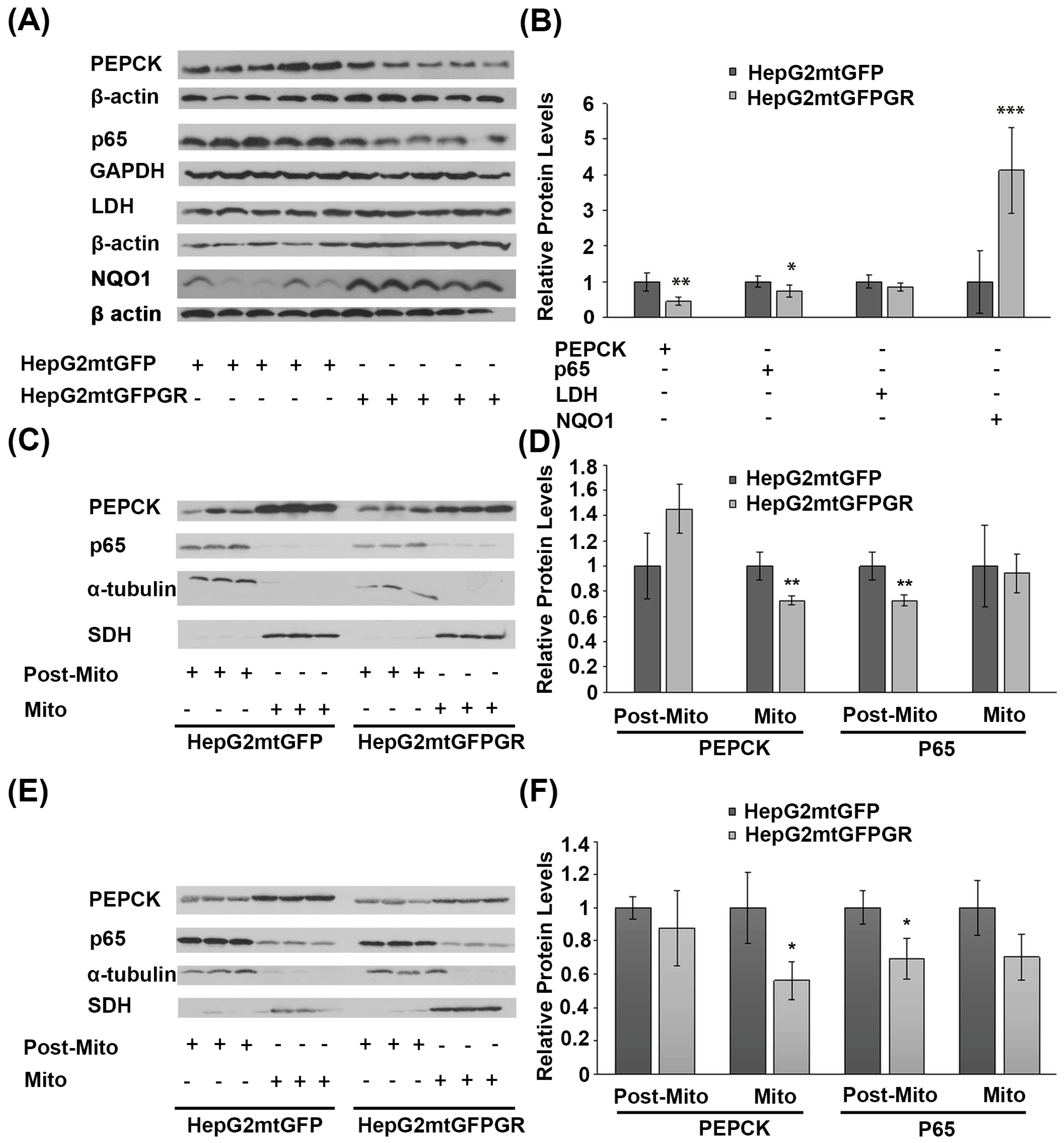
2.7. Effect of mtGR on Glucose Metabolism and Inflammation
3. Discussion
4. Material and Methods
4.1. Chemicals
4.2. Antibodies
4.3. Cell Culture-Mitochondrial Isolation
4.4. Animals
4.5. Xenografts
4.6. Electrophoresis and Western Blot
4.7. Citrate Synthase Enzymatic Assay
4.8. Pyruvate Dehydrogenase Enzymatic Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, R.M. The nuclear receptor superfamily: A rosetta stone for physiology. Mol. Endocrinol. 2005, 19, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Luhder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef] [PubMed]
- Panettieri, R.A.; Schaafsma, D.; Amrani, Y.; Koziol-White, C.; Ostrom, R.; Tliba, O. Non-genomic Effects of Glucocorticoids: An Updated View. Trends Pharmacol. Sci. 2019, 40, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.; Sekeris, C.E. Nuclear receptors and other nuclear transcription factors in mitochondria: Regulatory molecules in a new environment. Biochim. Biophys. Acta 2008, 1783, 1–11. [Google Scholar] [CrossRef]
- Psarra, A.M.; Sekeris, C.E. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: Role of the mitochondrial glucocorticoid receptor. Biochim. Biophys. Acta 2011, 1813, 1814–1821. [Google Scholar] [CrossRef]
- Karra, A.G.; Sioutopoulou, A.; Gorgogietas, V.; Samiotaki, M.; Panayotou, G.; Psarra, A.G. Proteomic analysis of the mitochondrial glucocorticoid receptor interacting proteins reveals pyruvate dehydrogenase and mitochondrial 60 kDa heat shock protein as potent binding partners. J. Proteom. 2022, 257, 104509. [Google Scholar] [CrossRef]
- Kokkinopoulou, I.; Moutsatsou, P. Mitochondrial Glucocorticoid Receptors and Their Actions. Int. J. Mol. Sci. 2021, 22, 6054. [Google Scholar] [CrossRef]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Johnson, J.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Dong, Y.N.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2020, 702, 108698. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Sotgia, F.; Lisanti, M.P. Mitochondrial biogenesis drives tumor cell proliferation. Am. J. Pathol. 2011, 178, 1949–1952. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Gruver-Yates, A.L.; Cidlowski, J.A. Tissue-specific actions of glucocorticoids on apoptosis: A double-edged sword. Cells 2013, 2, 202–223. [Google Scholar] [CrossRef]
- Sionov, R.V.; Cohen, O.; Kfir, S.; Zilberman, Y.; Yefenof, E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J. Exp. Med. 2006, 203, 189–201. [Google Scholar] [CrossRef]
- Du, J.; Wang, Y.; Hunter, R.; Wei, Y.; Blumenthal, R.; Falke, C.; Khairova, R.; Zhou, R.; Yuan, P.; Machado-Vieira, R.; et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 3543–3548. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzym. 2009, 452, 181–197. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, W.; Lu, Y.; Zheng, Y.; Pan, L.; Wu, X.; Yuan, Y.; Shen, Z.; Ma, S.; Zhang, X.; et al. BNIP3L/NIX-mediated mitophagy: Molecular mechanisms and implications for human disease. Cell Death Dis. 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.; Sekeris, C.E. Glucocorticoid receptors and other nuclear transcription factors in mitochondria and possible functions. Biochim. Biophys. Acta 2009, 1787, 431–436. [Google Scholar] [CrossRef]
- Ma, Y.C.; Tian, P.F.; Chen, Z.P.; Yue, D.S.; Liu, C.C.; Li, C.G.; Chen, C.; Zhang, H.; Liu, H.L.; Zhang, Z.F.; et al. Urinary malate dehydrogenase 2 is a new biomarker for early detection of non-small-cell lung cancer. Cancer Sci. 2021, 112, 2349–2360. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Z.M.; Yu, J.L.; Lu, K.; Xu, S.J.; Lu, Y.; Liu, T.; Xia, B.J.; Huang, Z.; Zhao, X.Y.; et al. Malic enzyme 2 promotes the progression of hepatocellular carcinoma via increasing triglyceride production. Cancer Med. 2021, 10, 6795–6806. [Google Scholar] [CrossRef]
- Sutendra, G.; Michelakis, E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013, 3, 38. [Google Scholar] [CrossRef]
- Sainero-Alcolado, L.; Liano-Pons, J.; Ruiz-Perez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef]
- Michaud, D.S.; Houseman, E.A.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. Understanding the Role of the Immune System in the Development of Cancer: New Opportunities for Population-Based Research. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1811–1819. [Google Scholar] [CrossRef]
- Mut-Salud, N.; Alvarez, P.J.; Garrido, J.M.; Carrasco, E.; Aranega, A.; Rodriguez-Serrano, F. Antioxidant Intake and Antitumor Therapy: Toward Nutritional Recommendations for Optimal Results. Oxidative Med. Cell. Longev. 2016, 2016, 6719534. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Zhang, C.; Li, N.; Liu, Y.Y.; Yuan, T.; Yang, S.; Wang, X.P. Cox15 is a novel oncogene that required for lung cancer cell proliferation. Biochem. Biophys. Res. Commun. 2021, 578, 70–76. [Google Scholar] [CrossRef]
- Lu, N.; Wang, J.; Zhu, B.; Zhang, M.; Qi, F.; Wang, X.; Gu, J. Whole-exome sequencing to identify novel mutations of nevoid basal cell carcinoma syndrome in a Chinese population. Cancer Biomark. 2017, 21, 161–168. [Google Scholar] [CrossRef]
- Demonacos, C.; Djordjevic-Markovic, R.; Tsawdaroglou, N.; Sekeris, C.E. The mitochondrion as a primary site of action of glucocorticoids: The interaction of the glucocorticoid receptor with mitochondrial DNA sequences showing partial similarity to the nuclear glucocorticoid responsive elements. J. Steroid Biochem. Mol. Biol. 1995, 55, 43–55. [Google Scholar] [CrossRef]
- Hunter, R.G.; Seligsohn, M.; Rubin, T.G.; Griffiths, B.B.; Ozdemir, Y.; Pfaff, D.W.; Datson, N.A.; McEwen, B.S. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 9099–9104. [Google Scholar] [CrossRef]
- Morgan, D.J.; Poolman, T.M.; Williamson, A.J.; Wang, Z.; Clark, N.R.; Ma’ayan, A.; Whetton, A.D.; Brass, A.; Matthews, L.C.; Ray, D.W. Glucocorticoid receptor isoforms direct distinct mitochondrial programs to regulate ATP production. Sci. Rep. 2016, 6, 26419. [Google Scholar] [CrossRef]
- Elrebehy, M.A.; Al-Saeed, S.; Gamal, S.; El-Sayed, A.; Ahmed, A.A.; Waheed, O.; Ismail, A.; El-Mahdy, H.A.; Sallam, A.M.; Doghish, A.S. miRNAs as cornerstones in colorectal cancer pathogenesis and resistance to therapy: A spotlight on signaling pathways interplay—A review. Int. J. Biol. Macromol. 2022, 214, 583–600. [Google Scholar] [CrossRef]
- Kovalski, J.R.; Kuzuoglu-Ozturk, D.; Ruggero, D. Protein synthesis control in cancer: Selectivity and therapeutic targeting. EMBO J. 2022, 41, e109823. [Google Scholar] [CrossRef]
- Lin, C.C.; Cheng, T.L.; Tsai, W.H.; Tsai, H.J.; Hu, K.H.; Chang, H.C.; Yeh, C.W.; Chen, Y.C.; Liao, C.C.; Chang, W.T. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci. Rep. 2012, 2, 785. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.M.; McDonnell, A.M.; Lake, R.A.; Nowak, A.K. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology 2016, 5, e1066062. [Google Scholar] [CrossRef]
- Hudson, W.A.; Li, Q.; Le, C.; Kersey, J.H. Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia 1998, 12, 2029–2033. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Psarra, A.M.; Solakidi, S.; Trougakos, I.P.; Margaritis, L.H.; Spyrou, G.; Sekeris, C.E. Glucocorticoid receptor isoforms in human hepatocarcinoma HepG2 and SaOS-2 osteosarcoma cells: Presence of glucocorticoid receptor alpha in mitochondria and of glucocorticoid receptor beta in nucleoli. Int. J. Biochem. Cell Biol. 2005, 37, 2544–2558. [Google Scholar] [CrossRef]
- Srere, P. Citrate Synthase Enzyme; Elsevier: Amsterdam, The Netherlands, 1969; Volume 13, pp. 3–11. [Google Scholar]
- Hinman, L.M.; Blass, J.P. An NADH-linked spectrophotometric assay for pyruvate dehydrogenase complex in crude tissue homogenates. J. Biol. Chem. 1981, 256, 6583–6586. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Source | Catalog Number | Working Dilution |
|---|---|---|---|
| α-tubulin | Santa Cruz | SC-9104 | 1/1000 |
| β-actin | Sigma | A5316 | 1/4000 |
| Bax | Cell Signaling | 2772 | 1/1000 |
| BcL-xS/L(D-3) | Santa Cruz | SC-271121 | 1/250 |
| Bcl-2 | Cell Signaling | 2876 | 1/1000 |
| Caspase 3 | Cell Signaling | 9662 | 1/1000 |
| Caspasae 9 | Cell Signaling | 9508 | 1/1000 |
| GR-H300 | Santa Cruz | SC-8992 | 1/1000 |
| GR (G-5) | Santa Cruz | SC-393232 | 1/1000 |
| PEPCK | Santa Cruz | H300 | 1/1000 |
| PDH | Santa Cruz | SC-65242 | 1/1000 |
| SDH | Invitrogen | A11142 | 1/1000 |
| COX II | Invitrogen | A-6404 | 1/1000 |
| Citrate synthase | Santa Cruz | SC-390693 | 1/1000 |
| Grim 19 (F10) | Santa Cruz | SC-365978 | 1/250 |
| NQO1 (H9) | Santa Cruz | SC-376023 | 1/250 |
| COX 5b (C-5) | Santa Cruz | SC-374416 | 1/250 |
| BECN1 (E-8) | Santa Cruz | SC-48341 | 1/250 |
| GAPDH (G-9) | Santa Cruz | SC-365062 | 1/100 |
| COX 15 (G10) | Santa Cruz | SC-390987 | 1/250 |
| PPARα (H2) | Santa Cruz | SC-398394 | 1/500 |
| MDH2 (1G12) | Santa Cruz | SC-293474 | 1/250 |
| mtTFA (C-9) | Santa Cruz | SC-376672 | 1/250 |
| Anti-LC3B | Abcam | Ab48394 | 1/1000 |
| BNIP3L/NIX (D4R4B) | Cell Signaling | 12396 | 1/1000 |
| P65 | Santa Cruz | SC-109 | 1/1000 |
| LDH | Santa Cruz | Sc-133123 | 1/250 |
| P62/SQSTM1 | MBC | PM045 | 1/1000 |
| NDUFS2 | Thermo scientific | PA5-19342 | 1/500 |
| COXIV | Abcam | Ab33985 | 1/1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karra, A.G.; Tsialtas, I.; Kalousi, F.D.; Georgantopoulos, A.; Sereti, E.; Dimas, K.; Psarra, A.-M.G. Increased Expression of the Mitochondrial Glucocorticoid Receptor Enhances Tumor Aggressiveness in a Mouse Xenograft Model. Int. J. Mol. Sci. 2023, 24, 3740. https://doi.org/10.3390/ijms24043740
Karra AG, Tsialtas I, Kalousi FD, Georgantopoulos A, Sereti E, Dimas K, Psarra A-MG. Increased Expression of the Mitochondrial Glucocorticoid Receptor Enhances Tumor Aggressiveness in a Mouse Xenograft Model. International Journal of Molecular Sciences. 2023; 24(4):3740. https://doi.org/10.3390/ijms24043740
Chicago/Turabian StyleKarra, Aikaterini G., Ioannis Tsialtas, Foteini D. Kalousi, Achilleas Georgantopoulos, Evangelia Sereti, Konstantinos Dimas, and Anna-Maria G. Psarra. 2023. "Increased Expression of the Mitochondrial Glucocorticoid Receptor Enhances Tumor Aggressiveness in a Mouse Xenograft Model" International Journal of Molecular Sciences 24, no. 4: 3740. https://doi.org/10.3390/ijms24043740
APA StyleKarra, A. G., Tsialtas, I., Kalousi, F. D., Georgantopoulos, A., Sereti, E., Dimas, K., & Psarra, A.-M. G. (2023). Increased Expression of the Mitochondrial Glucocorticoid Receptor Enhances Tumor Aggressiveness in a Mouse Xenograft Model. International Journal of Molecular Sciences, 24(4), 3740. https://doi.org/10.3390/ijms24043740