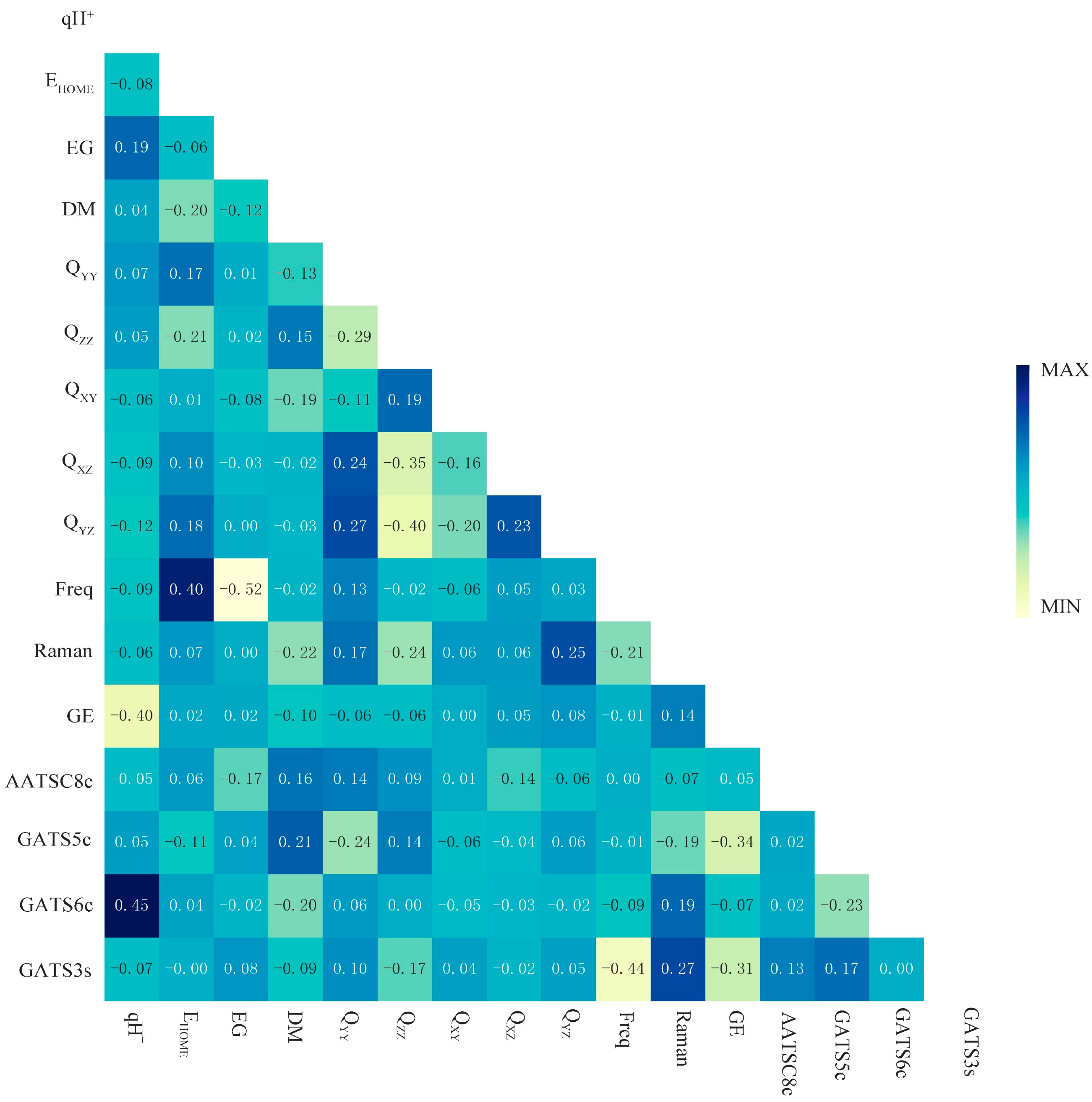
2.4.1. Sensitivity Analysis of the Synthesizability of SM Derivatives
To further analyze the influence of the 16 key descriptors (i.e., qH
+, E
HOMO, energy gap (EG), dipole moment (DM), Q
YY, Q
ZZ, Q
XY, Q
XZ, Q
YZ, positive frequency (Freq), Raman, GE, AATSC8c, GATS5c, GATS6c, and GATS3s) screened by the Pearson correlation coefficient method in
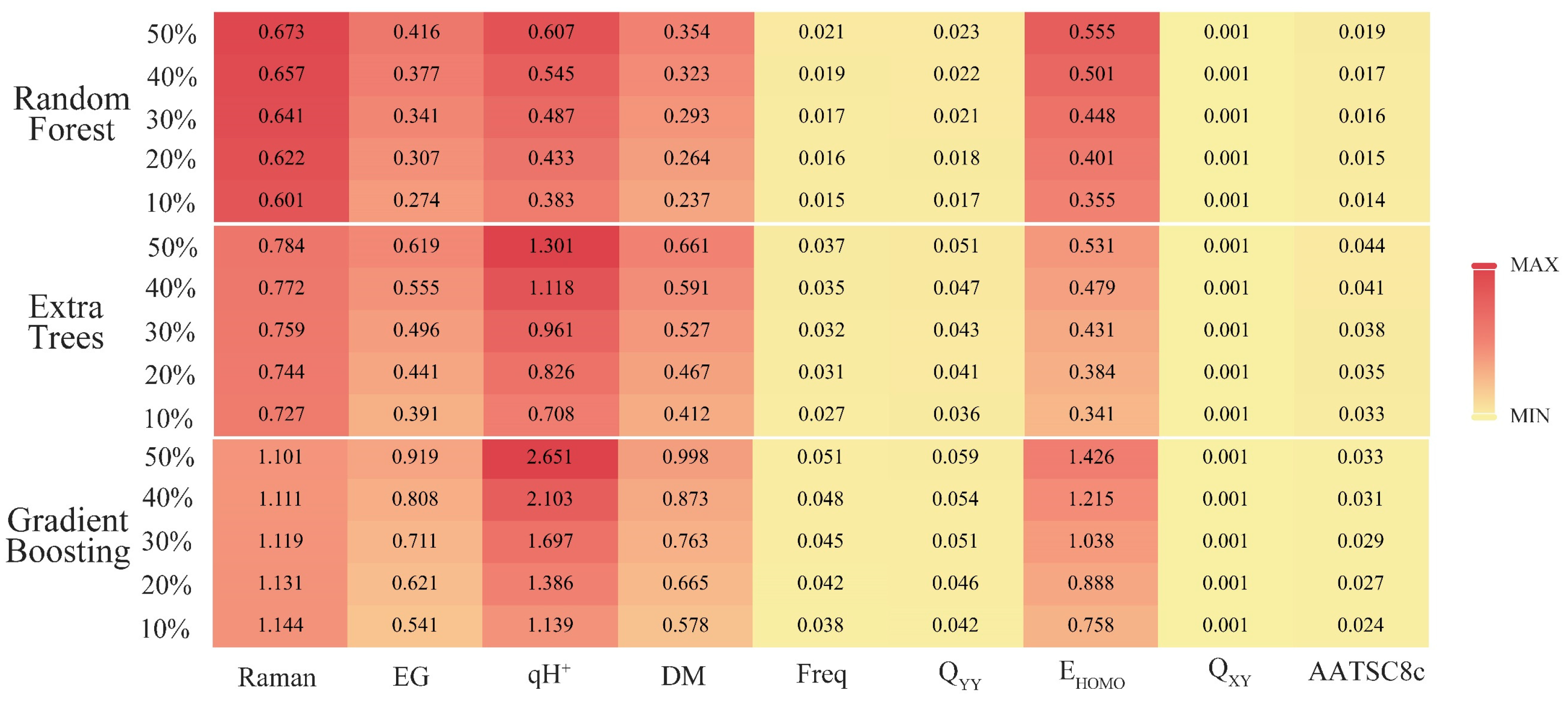
Section 3.1, the python software was used to output the correlation ranking between the 16 key descriptors and synthesizability. Then the sensitivity analysis of the top 50% most important key descriptors was analyzed by SPSS software to explore the important factors affecting the synthesis of SM derivatives. With the help of the “sklearn.feature_selection” package in the ML tool library scikit-learn, the relationship between the synthesizability of SM derivatives and 16 key descriptors in the bagging-RF, bagging-ERT, and bagging-GBC algorithms [
36] was analyzed. The SelectKBest function was used to output the correlation ranking of 16 key descriptors in the three algorithm models (
Table 6).
Raman, energy gap, qH
+, dipole moment, positive frequency, Q
YY, and E
HOMO are the top 50% of the key descriptors in three models; Q
XY is the top 50% of the key descriptors in two models; and AATSC8c ranks the top 50% of the descriptors of the key feature correlation of the bagging-GBC model. Therefore, this paper selected nine key eigenvalues (i.e., Raman, energy gap, qH
+, dipole moment, positive frequency, Q
YY, E
HOMO, Q
XY, and AATSC8c) for the sensitivity analysis. The above nine key descriptor values were used as independent variables, and the synthesizability of SM derivatives was used as a dependent variable for constructing three linear regression models by SPSS software. The correlation coefficients R of the bagging-RF, bagging-ERT, and bagging-GBC algorithms were 0.793, 0.825, and 0.805, respectively, all of which met the statistical requirements, and the Sig. of these models were all 0.000, passing the significance test [
37]. The linear relationship between the synthesizability of SM derivatives and key descriptors was shown in Formula (1) (bagging-RF), Formula (2) (bagging-ERT), and Formula (3) (bagging-GBC). The coefficients of Raman, positive frequency, and E
HOMO are positive, indicating a positive correlation between these three descriptors and the synthesizability of SM derivatives. The coefficients of the energy gap, qH
+, dipole moment, Q
YY, Q
XY, and AATSC8c are negative, indicating a negative correlation between these three descriptors and the synthesizability of SM derivatives.
The absolute value of the sensitivity coefficient under different variation degrees of key descriptors was calculated by Formulas (1)–(3) (
Figure 3). As shown in
Figure 3, when the degree of key eigenvalues increased, the sensitivity coefficients of all key eigenvalues showed an upward trend, except for the Raman descriptor in the bagging-ERT model. Among the descriptors, the characteristics of Raman, energy gap, qH
+, dipole moment, and E
HOMO were more prominent, and their sensitivity coefficients were all greater than 0.2. However, the characteristics of positive frequency, Q
YY, Q
XY, and AATSC8c descriptors were not obvious, and the sensitivity coefficients were all less than 0.05. Therefore, it can be inferred that the key descriptors (i.e., Raman, energy gap, qH
+, dipole moment, and E
HOMO) have a more significant impact on the synthesizability of SM derivatives and are in a higher position in the ranking order of the key descriptors’ correlations (
Table 6). In contrast, the positive frequencies, Q
YY, Q
XY, and AATSC8c had relatively little effect on the synthesizable properties of SM derivatives.
Raman, energy gap, qH
+, dipole moment, and E
HOMO significantly impact the synthesizability of SM derivatives when the change degree of key descriptors is less than 50%. In order to further analyze the changing trend of the synthesizability of SM derivatives when the parameter eigenvalues vary greatly, this paper analyzed the growth rate of the sensitivity coefficients of the characteristic parameters. Thus, the eigenvalues that most significantly affect the synthesizability of SM derivatives were screened, and the growth rates of the sensitivity coefficients of the three models were calculated (
Table 7). Results showed that the average growth rates of sensitivity coefficients for the energy gap, qH
+, dipole moment, and E
HOMO were all higher than 10%. However, the average growth rate of Raman’s sensitivity coefficient was less than 3% and even showed a negative growth trend in the bagging-GBC algorithm. After comparison, in the case of significant changes in key eigenvalues, the potential impact of Raman on the molecular synthesis of SM derivatives is much smaller than that of the energy gap, qH
+, dipole moment, and E
HOMO. Therefore, energy gap, qH
+, dipole moment, and E
HOMO were the most significant key descriptors affecting the synthesizability of SMs and their derivatives. E
HOMO refers to the energy of the highest occupied orbital of a molecule, which is one of the important quantum chemical properties of molecules [
38]. The energy gap is the difference in energy between the highest and the lowest occupied orbitals of a molecule. Studies have shown that molecular E
HOMO and energy gap values are closely related to molecular stability [
39]. The descriptor qH
+ refers to the maximum charge number of molecular hydrogen ions, and intramolecular hydrogen bonds can enhance the stability of molecules [
39]. The dipole moment is the product of the distance between the positive and negative charge centers in a molecule and the charge at the charge center, which is closely related to the effective charge carried by the molecule [
40]. Li et al. [
24] found that those mentioned above as key characteristic values with significant influence belong to the electrons of the molecule parameter. The molecular structure is associated with key eigenvalues such as E
HOMO, energy gap, and other properties [
41]. Laikov [
42] developed a new molecular electronic structure model using the electronic parameters of the molecule and found that molecular structures were inseparable from their electronic parameters. In quantum machine learning, molecular structures and electronic parameters such as E
HOMO, energy gap, and dipole moment play an extremely important role in studying molecular physico-chemical properties [
43]. In summary, the eigenvalues of the molecular electronic descriptors of SM derivatives played an essential role in the training process of ML models. They had a significant impact on the synthetic probability of SM derivatives.
2.4.2. The Mechanism Analysis for the Synthesizability of SMs Derivatives Based on 3D-QSAR Model
In
Section 2.4.1, energy gap, qH
+, dipole moment, and E
HOMO were screened out as the key descriptors that significantly affect the synthesis of theoretically designed SM derivatives. In order to verify the above results, the 11 commercially synthesized SMs (positive samples), the top seven SM derivatives with high synthesizability, and the last seven SM derivatives with low synthesizability predicted by the three models were selected. The energy gap, qH
+, dipole moment, and E
HOMO values of the 14 SM derivatives and 11 commercially synthesized SMs are given in
Table 8. Compared with the 11 synthesized SMs, the energy gap, qH
+, dipole moment, and E
HOMO eigenvalues of the seven synthesizable SM derivatives changed by 3.72%, −0.66%, −5.70%, and −7.57%, respectively. The energy gap, qH
+, dipole moment, and E
HOMO eigenvalues of the seven none synthesizable SM derivatives changed by 3.72%, −0.66%, −5.70%, and −7.57%, respectively, compared with the 11 commercially synthesized SMs. The results further showed that the eigenvalues of qH
+, dipole moment, and E
HOMO have relatively significant effects on the synthesizable properties of SMs. The higher the value of qH
+, the larger the value of the dipole moment, and the lower the value of E
HOMO, the lower the synthesizability of SM derivatives, which is consistent with the conclusion of the sensitivity analysis of the constructed machine learning models.
The 3D-QSAR model can effectively analyze the relationship between molecular structural features and physico-chemical activities [
23]. Therefore, the synthesizability of unlabeled samples (SM derivatives) was used as the input to construct an environment-friendly 3D-QSAR prediction model (CoMSIA) for predicting the synthesis of SM derivatives. The data set (n = 35) was composed of a training set (27 SMs) and a test set (9 SMs) for 3D-QSAR model construction and validation, and the template molecule (SM 12) existed in both the training set and test set. The cross-validation coefficient q
2 is 0.753 > 0.5, indicating that the constructed 3D-QSAR model had a good prediction ability [
23]. Relatively high values of non-cross-validation (R
2 = 0.971 > 0.9 and close to 1.000) and the external test coefficient (r
2pred = 0.940 > 0.6) further proved the good predictive ability and robustness of the generated models. The standard error of estimate (SEE) of the model was 0.047 < 0.95, which confirmed the good fit ability and predictive ability of the constructed 3D-QSAR model. In addition, in order to verify the rationality of setting the positive samples and unlabeled samples of the machine learning prediction model, this paper also used the constructed 3D-QSAR model to predict the synthesizability of the 11 positive samples (i.e., Phantolide, Celestolide, Tonalid, Galaxolide, Versalide, Musk xylene, Muscone, Musk methy, Musk ambrette, Moskene, and Musk ketone). However, since the 3D-QSAR model can only predict molecules with a common skeleton (
Table 9), it cannot predict Muscone without a common benzene ring. Based on the 3D-QSAR model prediction, it was found that 90% of the positive samples had a synthesizability greater than 0.5, indicating that the constructed 3D-QSAR model had high prediction accuracy. At the same time, it showed that the setting of positive and unlabeled samples of SMs and SM derivatives used for machine learning model training is reasonable.
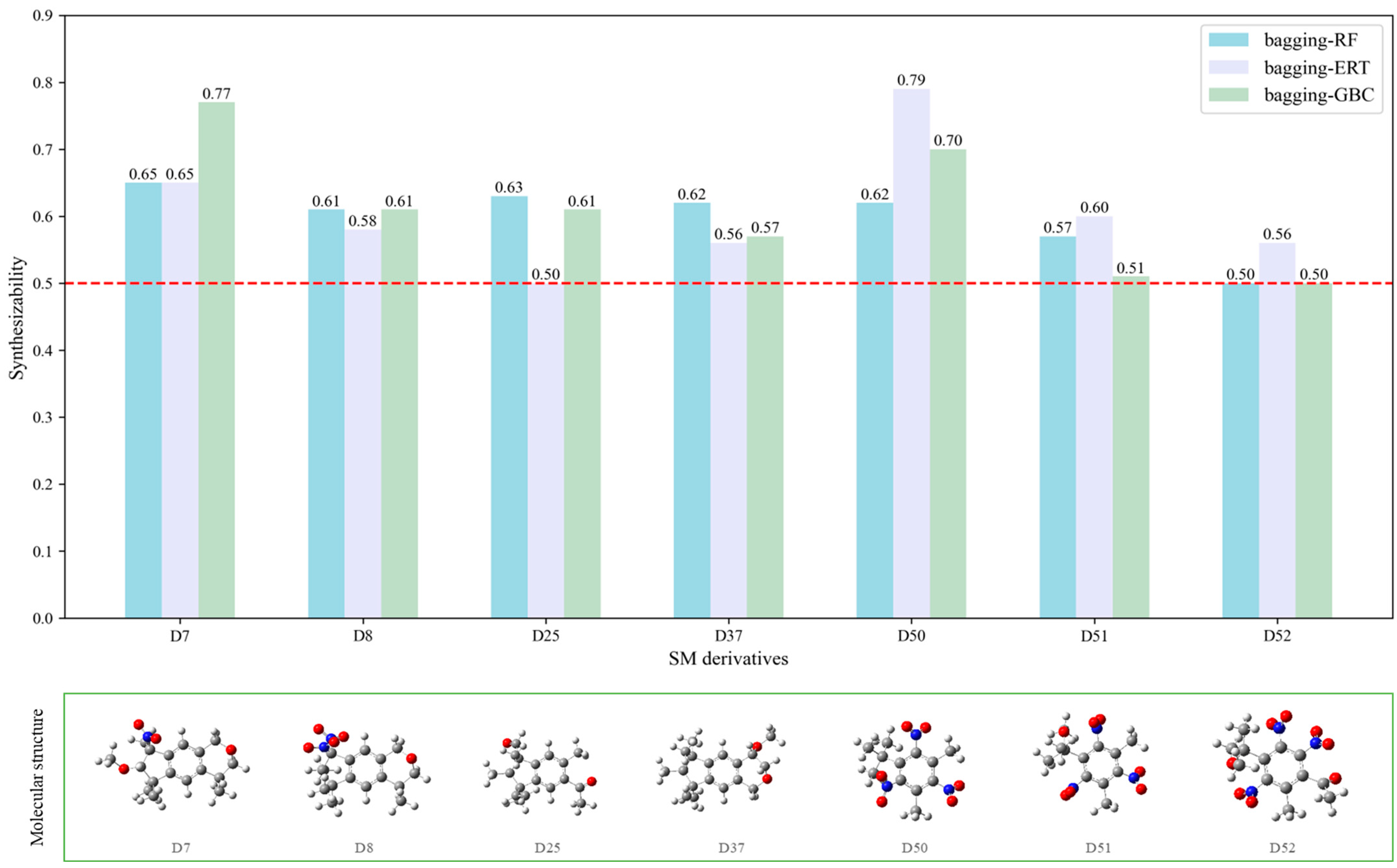
In the contour maps of the CoMSIA model for the molecular synthesizability of environmentally friendly SM derivatives, the contributions of hydrophobic, electrostatic, hydrogen bond acceptor, hydrogen bond donor, and steric fields were 22.6%, 19.8%, 33.2%, 20.4%, and 4.0%, respectively. The results indicated that the hydrophobic, electrostatic, hydrogen bond acceptor, and hydrogen bond donor fields significantly impacted the synthesizability of SM derivatives. In this study, unlabeled sample D50 was taken as a template molecule for analysis (
Figure 4). Seven derivatives of MK (i.e., D50, D51, D57, D61, D62, D67, and D76) were selected as examples (
Table 10) to analyze the synthesizability mechanism based on the contour maps. Studies showed that increasing the positive electric groups in the blue area in the contour maps was beneficial to chemical activity [
22,
44]. As shown in
Table 10 and
Figure 4a, b, compared with the synthesizability of D50 and D51, the non-synthesizable SM derivatives (i.e., D67, D61, D62, D57, and D76) had increased electron-withdrawing groups (-NO
2) at position 3, which led to the decreased synthesizability of D67, D61, D62, D57, and D76. Previous studies have shown that adding strong electron-withdrawing groups can reduce the E
HOMO value of the molecule [
45]. Then the energy gap value of the molecule will change significantly, which will increase the charge of some hydrogen atoms in the molecule. According to Long and Niu [
46], the higher the qH
+ value of a molecule, the easier it is to accept electrons and generate electrophilic reactions. The lower the energy gap value of the molecule, the easier it is for nucleophilic and electrophilic reactions to occur, resulting in poor stability (or synthesizability) of the molecule [
47,
48]. Compared with the synthesizable D50 and D51, the E
HOMO values of non-synthesizable SM derivatives (i.e., D57, D61, D62, and D76) were all smaller than D50 and D51. Furthermore, D57, D61, D62, D67, and D76 substitute molecules had lower energy gap values and higher qH
+ values than D50 and D51. The higher the value of qH
+, the lower the value of E
HOMO, and the smaller the energy gap value, the lower the synthesis ability of SMs substitute molecules. It showed that the higher the value of qH
+, the lower the value of E
HOMO, and the smaller the energy gap value, the lower the synthesizability of SM derivatives.
Yellow patches appeared surrounding positions 2 and 4 of D50 in the contour map (
Figure 4c), indicating that substituting hydrophobic groups in this region are conducive to synthesizing SMs molecules [
23]. Compared with the synthesizability of D50, hydrophilic groups (e.g., methoxyl and carboxyl groups) of D67, D61, and D62 were found at position 4. This should be why the synthesizable properties of the D67, D61, and D62 molecules were predicted as “non-synthesizable”. It has been found that the higher the dipole moment value of the molecule, the stronger the polarity of the molecule [
49]. Compared with D50, the dipole moment values of the remaining non-synthesizable D67, D61, and D62 were 4.313, 5.726, and 5.915, respectively, which were significantly higher than the dipole moment values of D50 (1.192). Thus, the possible replacement of hydrophilic groups may enhance the polarity of SMs (increased dipole moment value). The change will decrease the synthesis of SMs molecules, which is consistent with the previous conclusions. Thus, the 3D-QSAR model of the molecular synthesizability of environmentally friendly SM derivatives constructed in this paper has a good predictive ability. The results obtained from sensitivity analysis were consistent with those obtained from contour maps: the higher the value of qH
+, the larger the value of the dipole moment, the lower the value of E
HOMO, and the smaller the value of the energy gap, the lower the synthesizability of SM molecules.
2.4.3. Mechanism Verification Analysis for the Synthesizability of SM Derivatives Based on Intramolecular Hydrogen Bond Theory
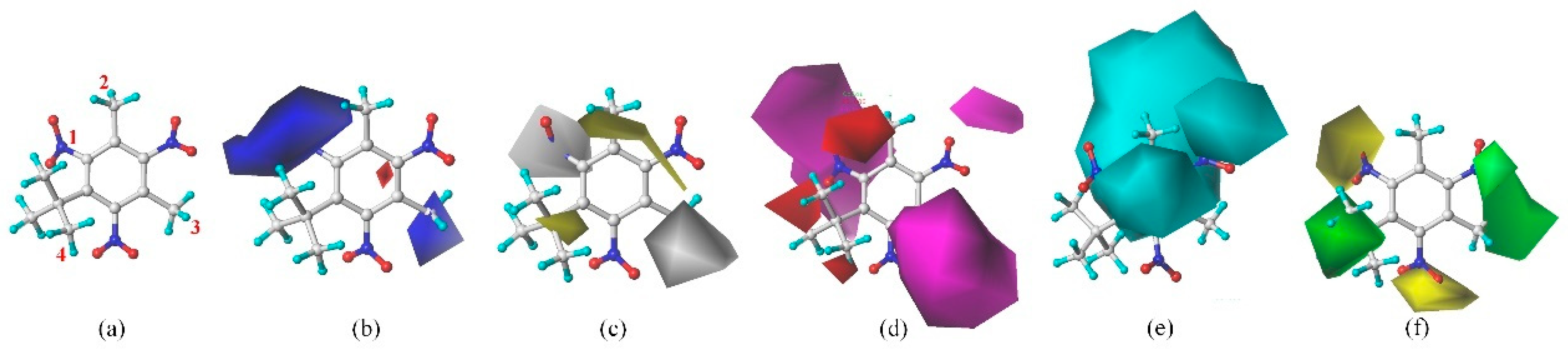
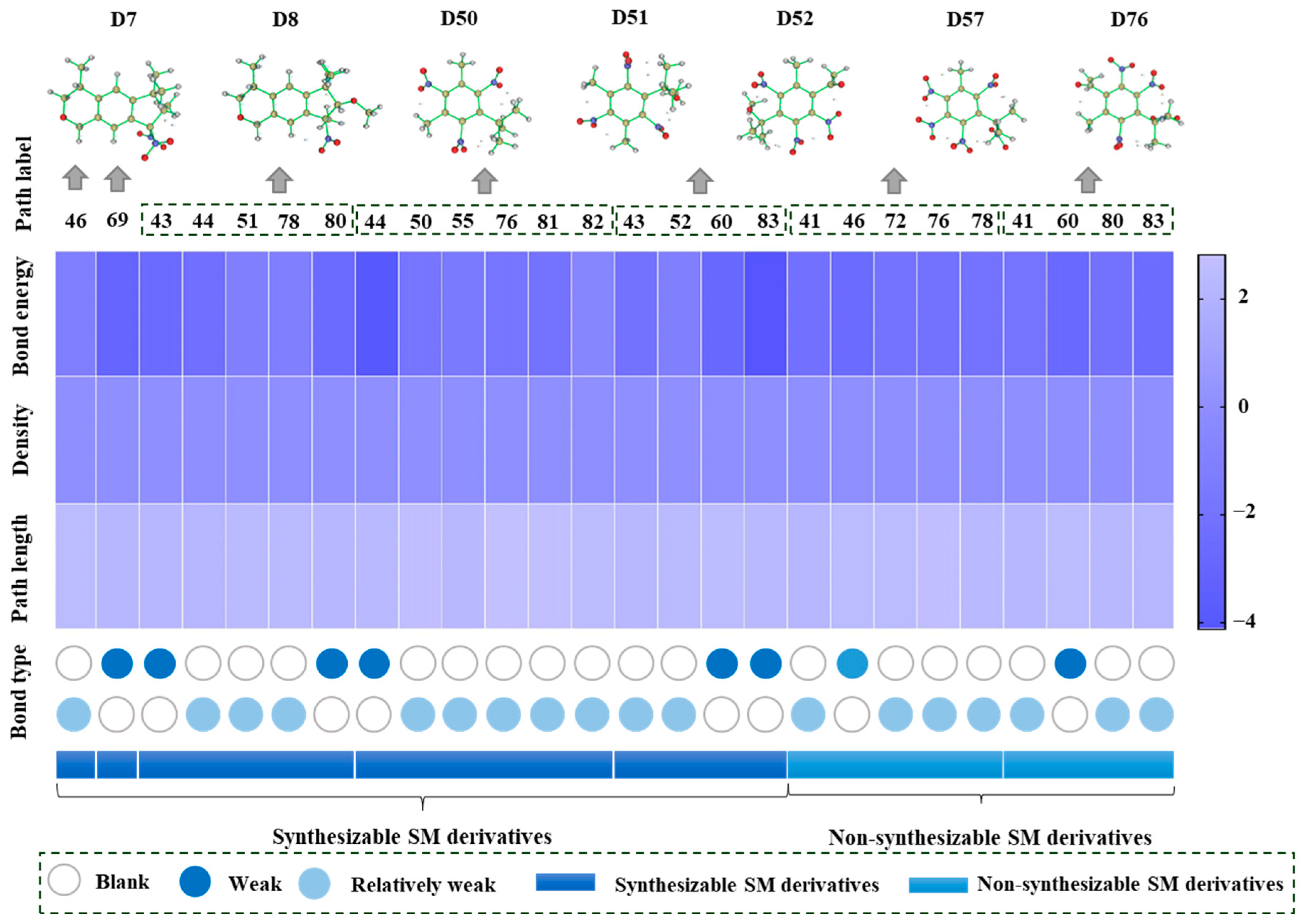
In order to further analyze the synthesizable mechanism of SM derivatives, they were screened based on bagging-RF, bagging-ERT, and bagging-GBC models. The intramolecular hydrogen bond differences of the top seven SM substitute molecules (D7 > D50 > D8 > D25 > D37 > D51 > D52) and the last seven non-synthesizable SM derivatives (D67 < D62 < D39 < D57 < D61 < D76 < D49) were compared to analyze the intrinsic factors affecting the internal reasons of synthesizability. HHCB and MK derivatives from the above 14 SM derivatives were selected as representatives. D7, D8 (synthesizable), and D39, D49 (non-synthesizable) were the derivatives of HHCB. D50, D51, and D52 (synthesizable) and D57 and D76 (non-synthesizable) were the derivatives of MK. For the synthesizable derivatives, D7 contained one intramolecular hydrogen bond C3−H23···O13 (H23···O13 bond length is 2.46 Å), D8 contained one intramolecular hydrogen bond C16–H29···O15 (H29···O15 bond length is 2.22 Å), D50 had five intramolecular hydrogen bonds C10–H28···O17 (H28···O17 bond length is 2.22 Å), C1−H24···O20 (H24···O20 bond length is 2.21 Å), C12−H36···O17 (H36···O17 bond length is 2.42 Å), C12−H35···O14 (H35···O14 bond length is 2.42 Å), C11–H33···O14 (H33···O14 O14 bond length is 2.22 Å), D51 had six intramolecular hydrogen bonds C10–H30···O19 (H30···O19 bond length is 2.36 Å), C13–H35···O22 (H35···O22 bond length is 2.70 Å), C10–H30···O18 (H30···O18 bond length is 2.30 Å), C1–H25···O21 (H25···O21 bond length is 2.82 Å), C9–H27 ···O16 (H27···O16 bond length is 2.73 Å), C8–H31···O16 (H31···O16 bond length is 2.54 Å), and D52 had four intramolecular hydrogen bonds C10−H31·· O20 (H31···O20 bond length is 2.31 Å), C9−H30···O20 (H30···O20 bond length is 2.43 Å), C14–H38···O17 (H38···O17 bond length is 2.49 Å), C12–H34···O23 (the bond length of H34···O23 is 2.34 Å). Among the non-synthesizable SM derivatives, D39 and D49 had no intramolecular hydrogen bonds, while D57 had five intramolecular hydrogen bonds C10–H33··O18 (the bond length of H33···O18 is 2.18 Å), C10–H33···O19 (H33···O19 bond length is 2.48 Å), C1–H28···O21 (H28···O21 bond length is 2.45 Å), C9−H29···O15 (H29···O15 bond length 2.67 Å), C9–H29···O16 (H29···O16 bond length is 2.34 Å), and D76 contained four intramolecular hydrogen bonds C10–H33···O21 (H33···O21 bond length is 2.22 Å), C12–H36···O24 (H36···O24 bond length is 2.51 Å), C9–H30···O19 (H30···O19 bond length is 2.27 Å), C1–H28···O13 (H28···O13 bond length is 2.19 Å) (
Table 11). Comparing the number of intramolecular hydrogen bonds, it was found that the formation rate of intramolecular hydrogen bonds in the synthesizable SM derivatives was 100%. The predicted rate of intramolecular hydrogen bond formation in non-synthesizable SM derivatives was only 50% (
Figure 5). Since the basic forms of intramolecular hydrogen bonds in the SM derivatives were all C–H···O, it was speculated that the hydrogen bond between the methyl group and the nitro group restricts the rotation of the C–C bond between the benzene ring and the nitro group in the molecular center of the SM derivatives. Thus, D7, D8, D50, D51, D52, D57, and D76 have stable, planar molecular conformations that allow for synthesis. However, the lack of intramolecular hydrogen bonds in D39 and D49 resulted in unclear molecular conformation and unstable structures. Thus, D39 and D49 had low synthesizability. The above inference was consistent with a previous study [
50]. The expected molecular conformation and arrangement can be obtained by rationally designing the intramolecular hydrogen bond between the amide and alkoxy groups. The bispyridyl aromatic dicarboxamide derivatives and their complexes can thus be designed [
50].
Although the formation rate of intramolecular hydrogen bonds in non-synthesizable SM derivatives was only 50%, D57 and D76 still had intramolecular hydrogen bonds due to structural reasons. Therefore, we further analyzed the influences of intramolecular hydrogen bonds on the synthesizability of MK derivatives (i.e., D50, D51, D52, D57, and D76). Intramolecular hydrogen bond strength is difficult to calculate accurately due to the complex and strict operation process. If the system needs to be cut off and the cutoff point should be saturated, the structure needs to be adjusted to avoid severe steric hindrance. Emamian et al. [
51] first proposed that the bond critical point (BCP) electron density can be defined by AIM theory to estimate the intramolecular hydrogen bond strength (E_HB). Emamian et al. [
51] also redefined the standard definition of intramolecular hydrogen bond strength, where E_HB > −2.5 kcal/mol is weak strength and E_HB < −2.5 kcal/mol is weak strength. Among these, E_HB > −2.5 kcal/mol is “very weak intensity”, and −14 < E_HB < −2.5 kcal/mol is “weak to medium intensity”. The BCP electron densities of D7, D8, D50, D51, D52, D57, and D76 were calculated by density functional theory (DFT). The strength of the intramolecular hydrogen bonds of these derivatives was then estimated (
Table 11,
Figure 5). Among the synthesizable SM derivatives, D50 formed three extremely weak intramolecular hydrogen bonds with E_HB of −2.336, −1.102, and −1.101 kcal/mol, respectively, and two weak intramolecular hydrogen bonds with E_HB of −2.704 and −2.707 kcal/mol, respectively. D51 formed five extremely weak intramolecular hydrogen bonds (with the E_HB of −1.960, −1.602, −1.919, −2.100, and −0.677 kcal/mol) and one weak intramolecular hydrogen bond (with E_HB of −4.039 kcal/mol). D52 formed two extremely weak intramolecular hydrogen bonds (E_HB were −2.182, −1.091 kcal/mol) and two weak intramolecular hydrogen bonds (E_HB were −2.885, −4.127 kcal/mol). Among the non-synthesizable SM derivatives, D57 formed four “very weak intensity” intramolecular hydrogen bonds (E_HB were −2.280, −2.105, −2.167, and −2.128 kcal/mol) and one weak intramolecular hydrogen bond (E_HB was −2.576 kcal/mol). D76 formed three “very weak intensity” intramolecular hydrogen bonds (E_HB were −2.049, −2.200, and −2.485 kcal/mol) and one weak intramolecular hydrogen bond (E_HB was −2.781 kcal/mol). Statistical analysis found that the formation rate of weak intramolecular hydrogen bonds in predicted synthesizable SM derivatives was 33.33%, and the weak intramolecular hydrogen bond formation rate in non-synthesized SM derivatives was 22.22%. It showed that “very weak intensity” intramolecular hydrogen bonds are more conducive to synthesizing SM derivatives than weak intramolecular hydrogen bonds. Studies have confirmed that when the intramolecular hydrogen bond is weak, it is mainly dominated by electrostatic interaction forces [
51]. This result is consistent with the conclusion in
Section 2.4.2 that the electrostatic field can affect the synthesizability of SMs and their derivatives. In addition, based on the sensitivity analysis results of the synthesizability-machine learning model, it can be seen that the energy gap, qH
+, dipole moment, and E
HOMO are key descriptors affecting the synthesizability of SM derivatives. Among them, the size of E
HOMO and the energy gap are closely related to molecular stability, and qH
+ affects the interaction between donor and acceptor in intramolecular hydrogen bonds [
39]. Therefore, the intramolecular hydrogen bond strength analysis, the sensitivity analysis of the three synthesizability-machine learning models, and the contour map analysis by the 3D-QSAR model were mutually verified. The higher the intramolecular hydrogen bond strength, electrostatic interaction, qH
+ value, and energy gap value, the lower the E
HOMO value, the more stable the SMs substitute molecule, and the higher the synthesis probability will be.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











































