
Integrative Transcriptome, miRNAs, Degradome, and Phytohormone Analysis of Brassica rapa L. in Response to Plasmodiophora brassicae

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
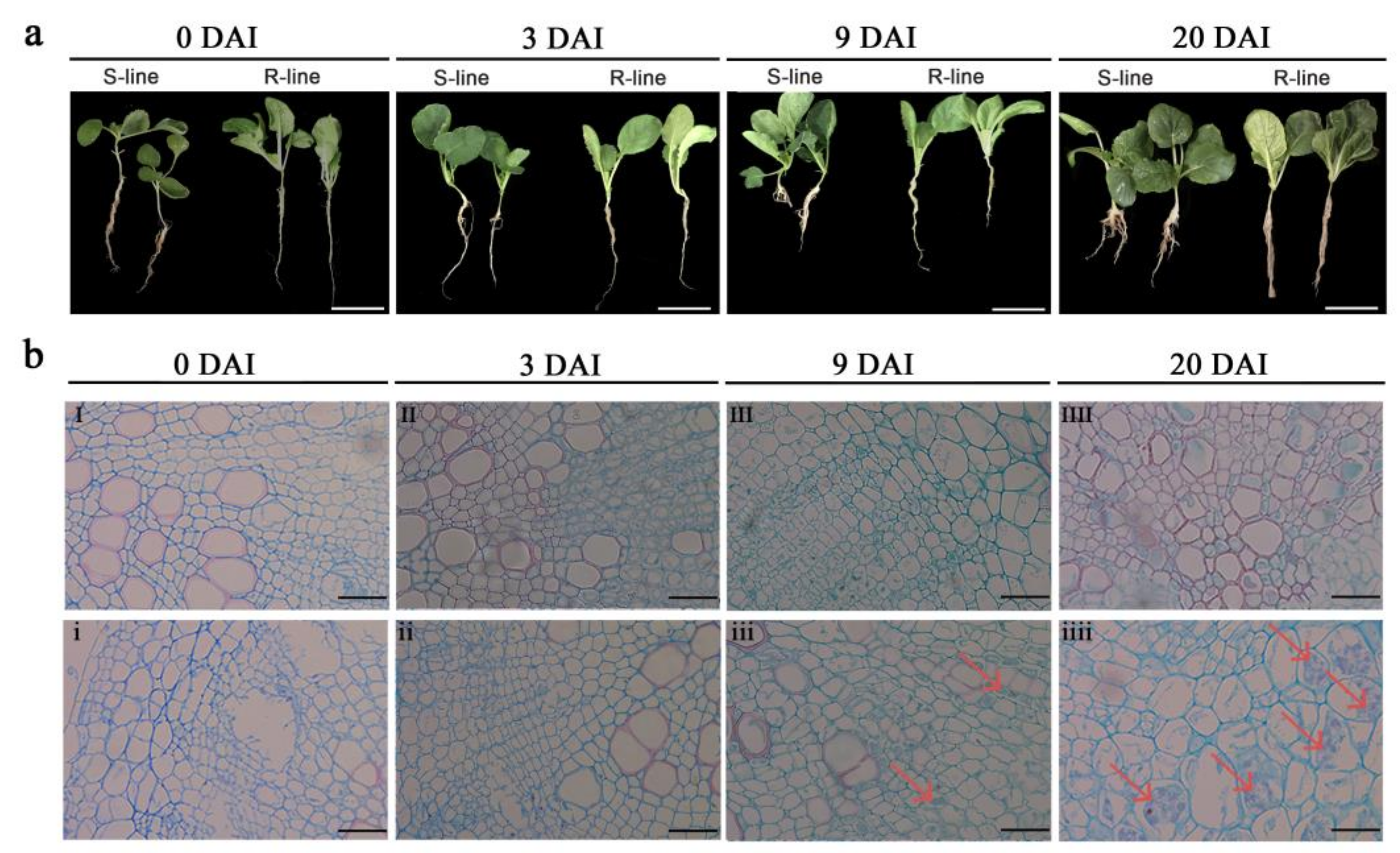
2.1. Phenotypic and Cytological Observation of R- and S-Lines
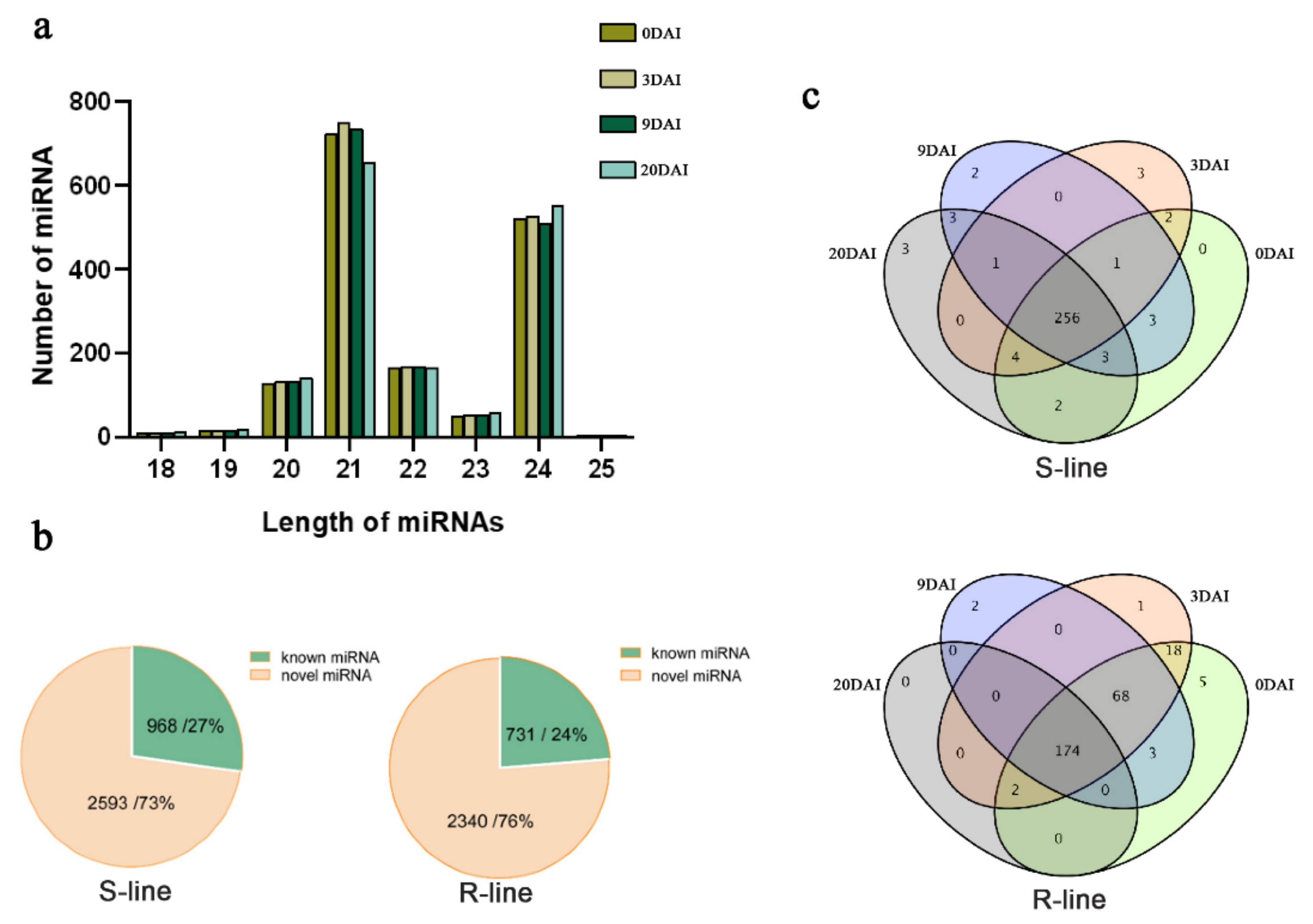
2.2. Small RNA Sequencing Profile
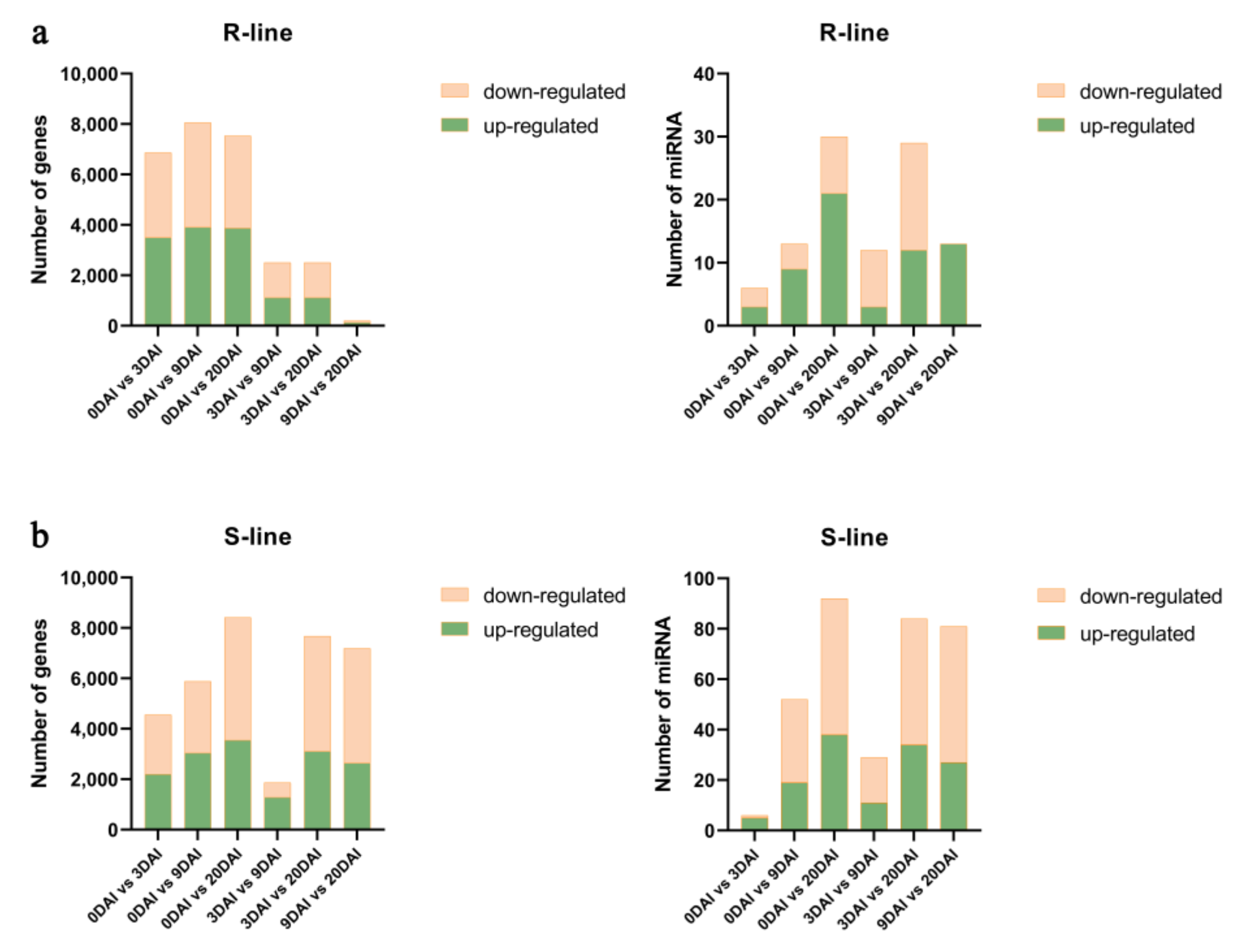
2.3. Co-Analysis of the Differential Expression of the Transcriptome and miRNAs between R- and S-Lines under Different DAIs
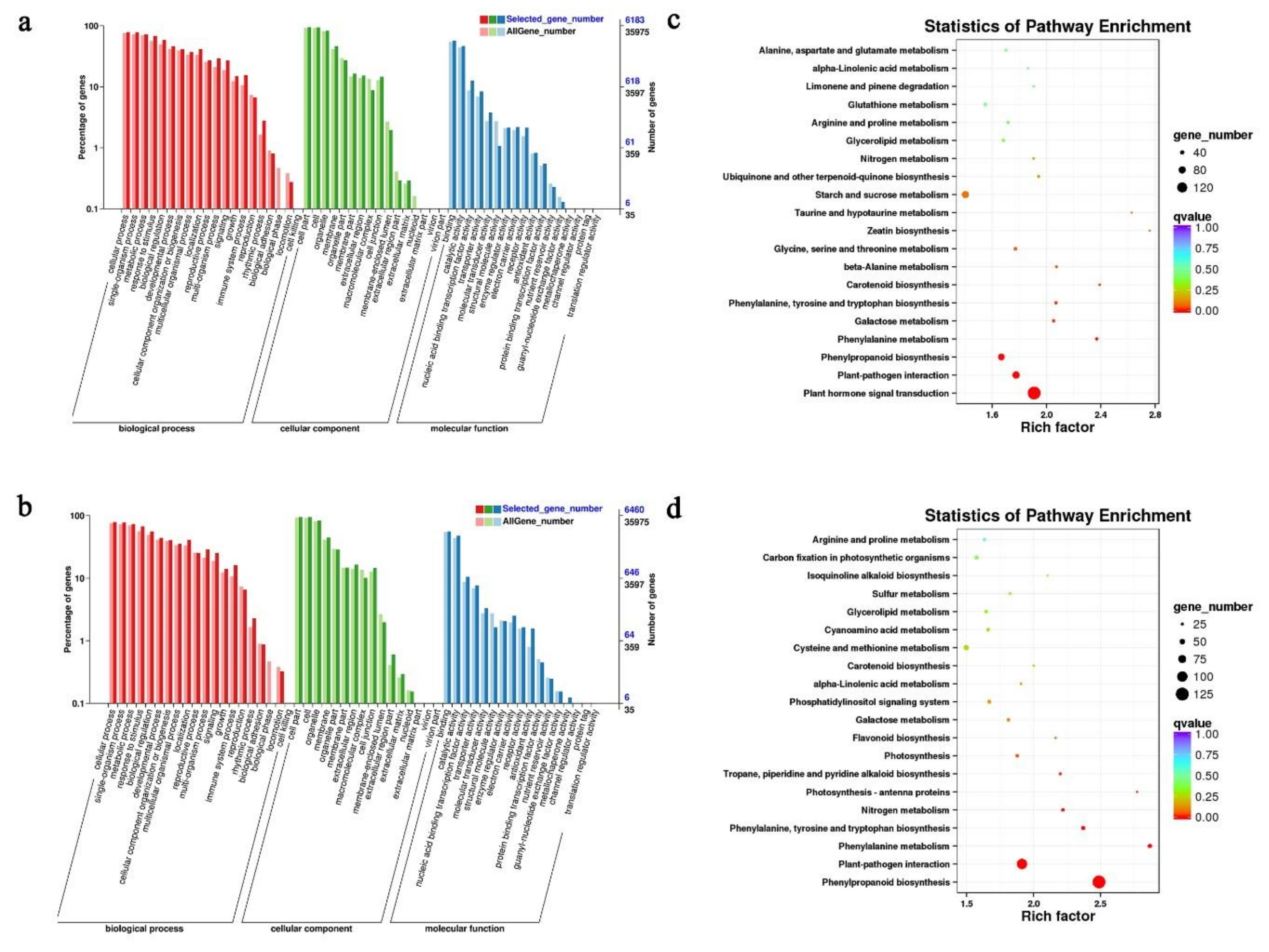
2.4. GO and KEGG Pathway Analyses of DEGs
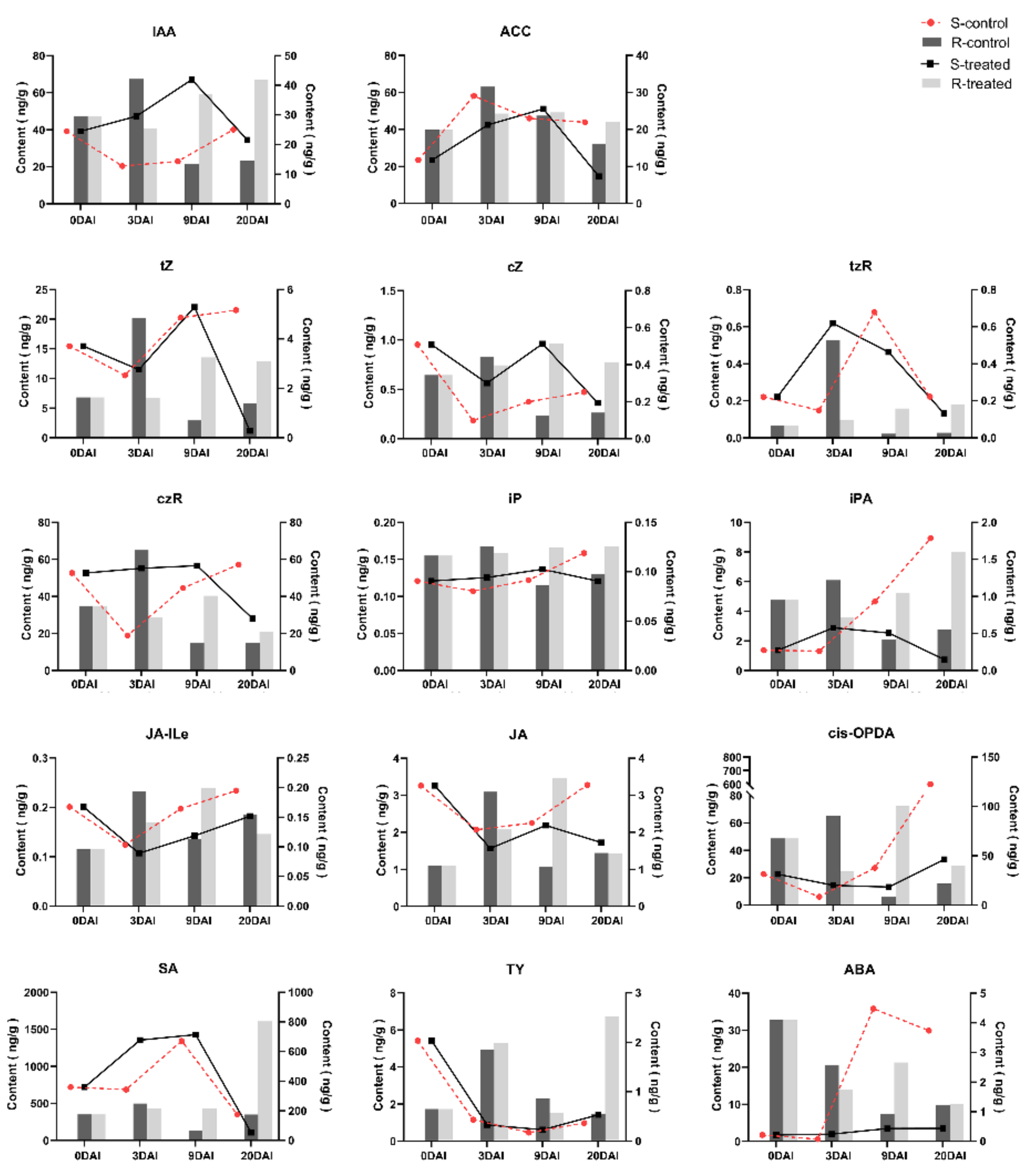
2.5. Phytohormone Analysis in R- and S-Lines at Four DAIs
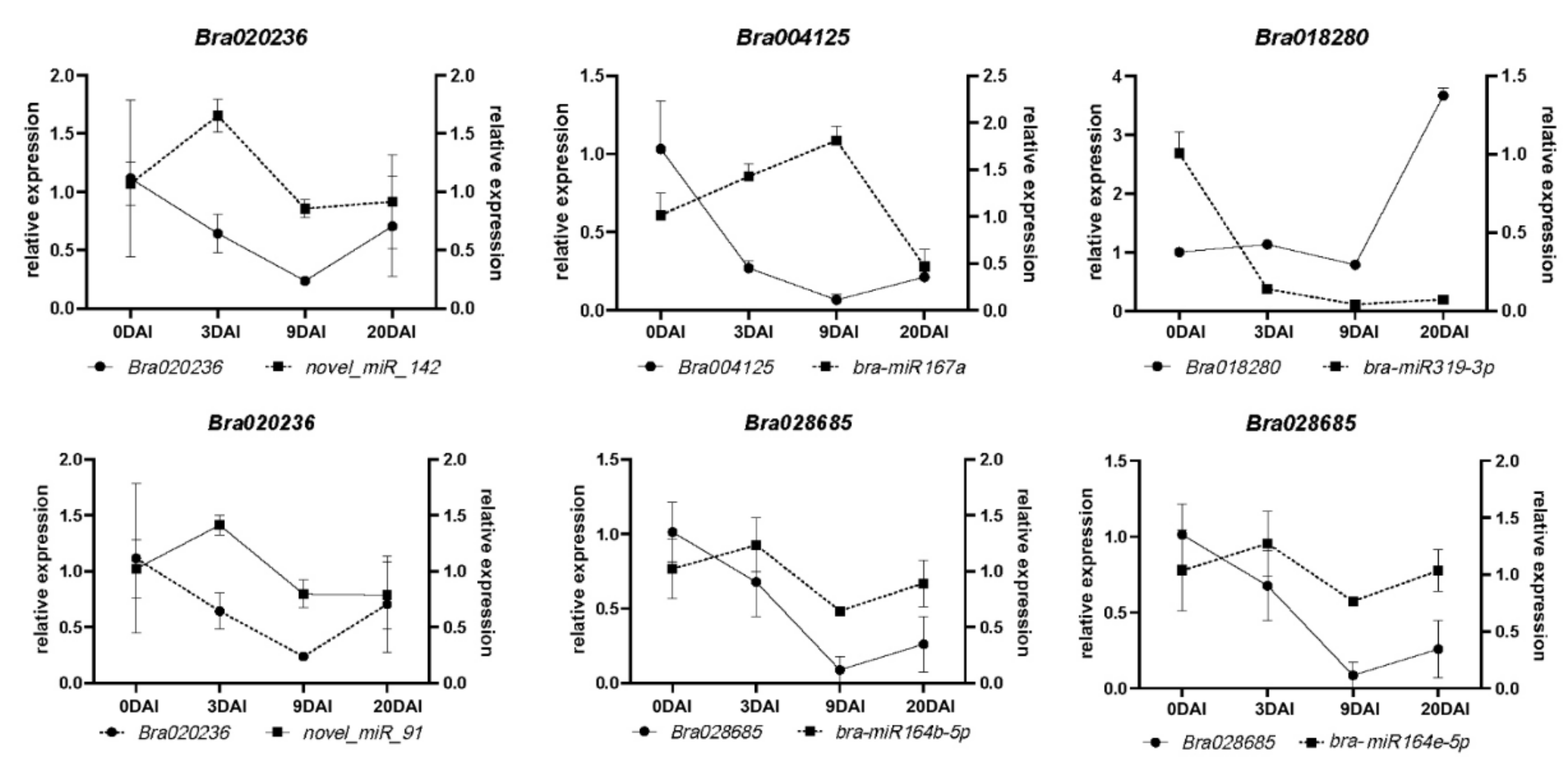
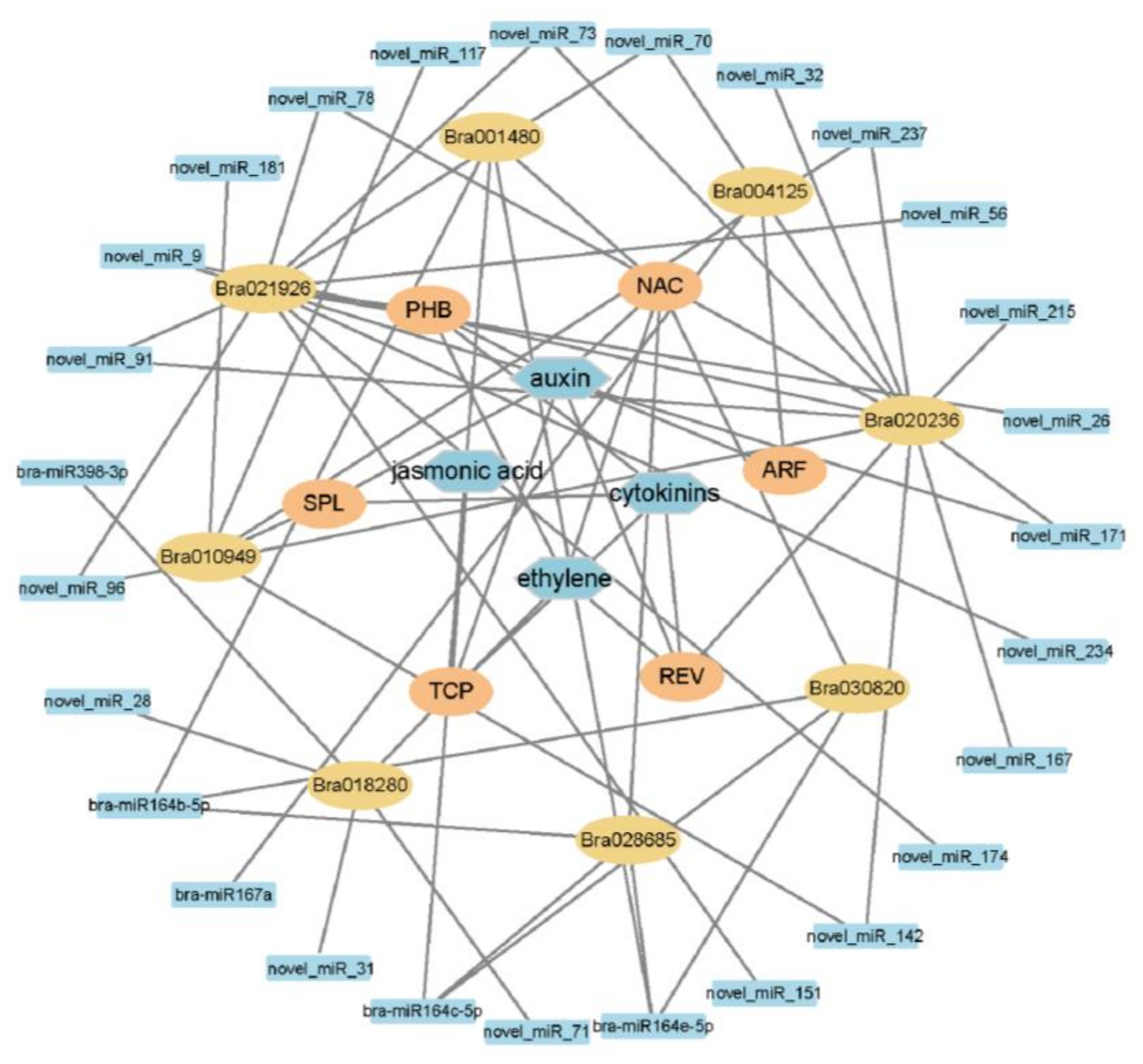
2.6. miRNAs and Target Genes of the S-Line under P. brassicae Infection
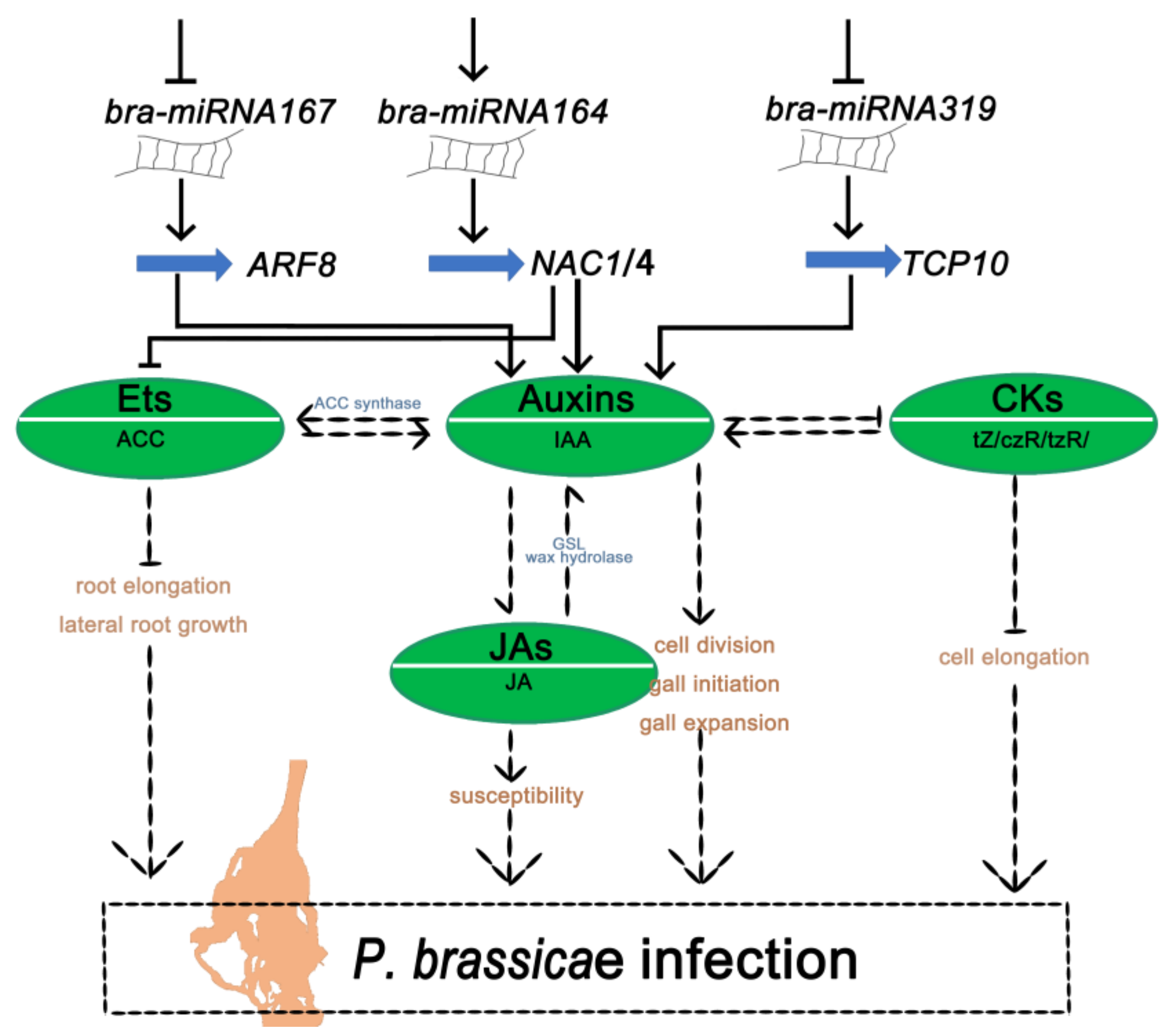
3. Discussion
4. Materials and Methods
4.1. Plant Materials and P. brassicae Treatment
4.2. RNA Extraction and Construction of the cDNA Library
4.3. Transcriptome Sequencing and Assembly Analysis
4.4. Identification of Known and Novel miRNAs and Predication of Their Target Genes
4.5. Degradome Sequencing
4.6. Quantification of Phytohormones
4.7. qRT-PCR Validation of miRNAs and mRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Javed, M.A.; Schwelm, A.; Zamani-Noor, N.; Salih, R.; Silvestre Vano, M.; Wu, J.; Gonzalez Garcia, M.; Heick, T.M.; Luo, C.; Prakash, P.; et al. The clubroot pathogen Plasmodiophora brassicae: A profile update. Mol. Plant Pathol. 2022, 24. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Wei, X.; Yang, Y.; Yuan, Y.; Wei, F.; Zhao, Y.; Yang, S.; Yao, Q.; Wang, Z.; Tian, B.; et al. Root RNA-seq analysis reveals a distinct transcriptome landscape between clubroot-susceptible and clubroot-resistant Chinese cabbage lines after Plasmodiophora brassicae infection. Plant Soil 2017, 421, 93–105. [Google Scholar] [CrossRef]
- Wei, X.; Xu, W.; Yuan, Y.; Yao, Q.; Zhao, Y.; Wang, Z.; Jiang, W.; Zhang, X. Genome-wide investigation of microRNAs and their targets in Brassica rapa ssp. pekinensis root with Plasmodiophora brassicae infection. Hortic. Plant J. 2016, 2, 209–216. [Google Scholar] [CrossRef]
- Kageyama, K.; Asano, T. Life Cycle of Plasmodiophora brassicae. J. Plant Growth Regul. 2009, 28, 203–211. [Google Scholar] [CrossRef]
- Zhang, S.W.; Li, C.H.; Cao, J.; Zhang, Y.C.; Zhang, S.Q.; Xia, Y.F.; Sun, D.Y.; Sun, Y. Altered architecture and enhanced drought tolerance in rice via the down-regulation of indole-3-acetic acid by TLD1/OsGH3.13 activation. Plant Physiol. 2009, 151, 1889–1901. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.; Li, F.; Zhang, S.; Li, G.; Ma, X.; Liu, X.; Sun, R. Research progress on clubroot disease resistance breeding of Brassica rapa. Acta Hortic. Sin. 2020, 47, 1648–1662. [Google Scholar]
- Zhu, M.; He, Y.; Li, Y.; Ren, T.; Liu, H.; Huang, J.; Jiang, D.; Hsiang, T.; Zheng, L. Two new biocontrol agents against clubroot caused by Plasmodiophora brassicae. Front. Microbiol. 2019, 10, 3099. [Google Scholar] [CrossRef]
- Yuan, Y.; Qin, L.; Su, H.; Yang, S.; Zhang, X. Transcriptome and coexpression network analyses reveal hub genes in Chinese cabbage (Brassica rapa L. ssp. pekinensis) during different stages of Plasmodiophora brassicae infection. Front. Plant Sci. 2021, 12, 650252. [Google Scholar] [CrossRef]
- Khraiwesh, B.; Zhu, J.K.; Zhu, J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 2012, 1819, 137–148. [Google Scholar] [CrossRef]
- Zhou, J.M.; Zhang, Y. Plant Immunity: Danger Perception and Signaling. Cell 2020, 181, 978–989. [Google Scholar] [CrossRef]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, Y.G.; Shi, Y.; Wu, L.; Xu, Y.J.; Huang, F.; Guo, X.Y.; Zhang, Y.; Fan, J.; Zhao, J.Q.; et al. Multiple rice microRNAs are involved in immunity against the blast fungus Magnaporthe oryzae. Plant Physiol. 2014, 164, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, H.; Gao, S.; Wang, W.-C.; Katiyar-Agarwal, S.; Huang, H.-D.; Raikhel, N.; Jin, H. Arabidopsis Argonaute 2 regulates innate immunity via miRNA393∗-mediated silencing of a Golgi-Localized SNARE Gene, MEMB12. Mol. Cell 2011, 42, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhang, Y.; Zhao, Y.; Xie, Z.; Hossain, M.R.; Yang, S.; Shi, G.; Lv, Y.; Wang, Z.; Tian, B.; et al. Root transcriptome and metabolome profiling reveal key phytohormone-related genes and pathways involved clubroot resistance in Brassica rapa L. Front. Plant Sci. 2021, 12, 759623. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Genetic Improvement of Clubroot Resistance of Huayouza 62 Restorer Line and Analysis of Molecular Mechanism of miRNA Response. Doctoral Dissertation, Huazhong Agricultural University, Wuhan, China, 2021. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, X.; Yu, H.; Wu, F. Root exudates of potato onion are involved in the suppression of clubroot in a Chinese cabbage-potato onion-Chinese cabbage crop rotation. Eur. J. Plant Pathol. 2017, 150, 765–777. [Google Scholar] [CrossRef]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef]
- Li, T.; Gonzalez, N.; Inze, D.; Dubois, M. Emerging connections between small RNAs and phytohormones. Trends Plant Sci. 2020, 25, 912–929. [Google Scholar] [CrossRef]
- Qixin, G.; Xinxi, H.; Huanyan, W.; Ke, C.; Chang, S.; Zheng, H. Research progress in mechanism of plant disease resistance induced by Salicylic acid. Chin. Potato J. 2014, 28, 238–242. [Google Scholar]
- Ludwig-Muller, J.; Julke, S.; Geiss, K.; Richter, F.; Mithofer, A.; Sola, I.; Rusak, G.; Keenan, S.; Bulman, S. A novel methyltransferase from the intracellular pathogen Plasmodiophora brassicae methylates salicylic acid. Mol. Plant Pathol. 2015, 16, 349–364. [Google Scholar] [CrossRef]
- Djavaheri, M.; Ma, L.; Klessig, D.F.; Mithofer, A.; Gropp, G.; Borhan, H. Mimicking the host regulation of Salicylic acid: A virulence strategy by the clubroot pathogen Plasmodiophora brassicae. Mol. Plant Microbe Interact 2019, 32, 296–305. [Google Scholar] [CrossRef]
- Ludwig-Müller, J.; Schuller, A. What Can We Learn from Clubroots: Alterations in Host Roots and Hormone Homeostasis Caused by Plasmodiophora brassicae; Springer: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Siemens, J.; Keller, I.; Sarx, J.; Kunz, S.; Schuller, A.; Nagel, W.; Schmülling, T.; Parniske, M.; Ludwig-Müller, J. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol. Plant-Microbe Interact. 2006, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Staswick, P.E.; Tiryaki, I. The oxylipin signal jasmonic acid is activated by an enzyme that conjugates it to isoleucine in Arabidopsis. Plant Cell 2004, 16, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; Davies, W.J. Drought, ozone, ABA and ethylene: New insights from cell to plant to community. Plant Cell Environ. 2010, 33, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Rock, C.D. Jacalin lectin At5g28520 is regulated by ABA and miR846. Plant Signal. Behav. 2013, 8, e24563. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhou, J.-J.; Zhang, J.-Z. Aux/IAA gene family in plants: Molecular structure, regulation, and function. International Journal of Molecular Sciences. Int. J. Mol. Sci. 2018, 19, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, H.; Ren, L.; Chen, W.; Liu, L.; Liu, F.; Zeng, L.; Yan, R.; Chen, K.; Fang, X. Jasmonic acid-mediated aliphatic glucosinolate metabolism is involved in clubroot disease development in Brassica napus L. Front. Plant Sci. 2018, 9, 750. [Google Scholar] [CrossRef]
- Ludwig-Müller, J.; Prinsen, E.; Rolfe, S.A.; Scholes, J.D. Metabolism and plant hormone action during clubroot disease. J. Plant Growth Regul. 2009, 28, 229–244. [Google Scholar] [CrossRef]
- Zhang, X.L.; Wu, Q.; Tao, Y.; Zhu, X.F.; Takahashi, N.; Umeda, M.; Shen, R.F.; Ma, J.F. ANAC044 is associated with P reutilization in P deficient Arabidopsis thaliana root cell wall in an ethylene dependent manner. Environ. Exp. Bot. 2021, 185, 104386. [Google Scholar] [CrossRef]
- Tian, Q.; Uhlir, N.J.; Reed, J.W. Arabidopsis SHY2/IAA3 inhibits auxin-regulated gene expression. Plant Cell 2002, 14, 301–319. [Google Scholar] [CrossRef]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N.H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, T.; Zhao, M.; Zhang, W. Ethylene-responsive miRNAs in roots of Medicago truncatula identified by high-throughput sequencing at whole genome level. Plant Sci. 2012, 184, 14–19. [Google Scholar] [CrossRef]
- Müller, C.J.; Valdés, A.E.; Wang, G.; Ramachandran, P.; Beste, L.; Uddenberg, D.; Carlsbecker, A. PHABULOSA mediates an auxin signaling loop to regulate vascular patterning in Arabidopsis. Plant Physiol. 2016, 170, 956–970. [Google Scholar] [CrossRef]
- Baulies, J.L.; Bresso, E.G.; Goldy, C.; Palatnik, J.F.; Schommer, C. Potent inhibition of TCP transcription factors by miR319 ensures proper root growth in Arabidopsis. Plant Mol. Biol. 2022, 108, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Study on the Lateral Root Development Regulated by miR319a in Poplar. Master’s Thesis, Southwest University, Chongqing, China, 2020. [Google Scholar] [CrossRef]
- Gautam, V.; Singh, A.; Verma, S.; Kumar, A.; Kumar, P.; Mahima; Singh, S.; Mishra, V.; Sarkar, A.K. Role of miRNAs in root development of model plant Arabidopsis thaliana. Indian J. Plant Physiol. 2017, 22, 382–392. [Google Scholar] [CrossRef]
- Mathesius, U. Auxin: At the root of nodule development? Funct. Plant Biol. 2008, 35, 651–668. [Google Scholar] [CrossRef]
- Turner, M.; Nizampatnam, N.R.; Baron, M.; Coppin, S.; Damodaran, S.; Adhikari, S.; Arunachalam, S.P.; Yu, O.; Subramanian, S. Ectopic expression of miR160 results in auxin hypersensitivity, cytokinin hyposensitivity, and inhibition of symbiotic nodule development in soybean. Plant Physiol. 2013, 162, 2042–2055. [Google Scholar] [CrossRef]
- Malinowski, R.; Novák, O.; Borhan, M.H.; Spíchal, L.; Strnad, M.; Rolfe, S.A. The role of cytokinins in clubroot disease. Eur. J. Plant Pathol. 2016, 145, 543–557. [Google Scholar] [CrossRef]
- Ludwig-Muller, J. Auxin homeostasis, signaling, and interaction with other growth hormones during the clubroot disease of Brassicaceae. Plant Signal. Behav. 2014, 9, e28593. [Google Scholar] [CrossRef]
- Ruzicka, K.; Ljung, K.; Vanneste, S.; Podhorska, R.; Beeckman, T.; Friml, J.; Benkova, E. Ethylene regulates root growth through effects on auxin biosynthesis and transport-dependent auxin distribution. Plant Cell 2007, 19, 2197–2212. [Google Scholar] [CrossRef]
- Stepanova, A.N.; Hoyt, J.M.; Hamilton, A.A.; Alonso, J.M. A Link between ethylene and auxin uncovered by the characterization of two root-specific ethylene-insensitive mutants in Arabidopsis. Plant Cell 2005, 17, 2230–2242. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Devos, S.; Laukens, K.; Deckers, P.; Dominique, V.; Beeckman, T.; Inzé, D.; Onckelen, H.V.; Witters, E.; Prinsen, E. A hormone and proteome approach to picturing the initial metabolic events during Plasmodiophora brassicae infection on Arabidopsis. Mol. Plant Microbe Interact. 2006, 19, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhao, Y.; Wei, X.; Yao, Q.; Jiang, W.; Wang, Z.; Yang, L.I.; Qian, X.U.; Yang, S.; Zhang, X. Pathotype identification of Plasmodiophora brassicae Woron.collected from Chinese cabbage in Henan province. J. Henan Agric. Sci. 2017, 46, 71–76. [Google Scholar]
- Zhang, B. Focus on the Production of Plant Paraffin Sections. J. Educ. Inst. Jilin Prov. 2013, 29, 153–154. [Google Scholar]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb prot5439. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000 Res. 2013, 2, 188. [Google Scholar] [CrossRef]
- Friedlnder, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Nikolaus, R. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Burge, S.W.; Jennifer, D.; Ruth, E.; John, T.; Lars, B.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Alex, B. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013, 41, D226–D232. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.; Liao, R.; Zhang, X.; Zhao, Y.; Xie, Z.; Yang, S.; Su, H.; Wang, Z.; Zhang, L.; Tian, B.; et al. Integrative Transcriptome, miRNAs, Degradome, and Phytohormone Analysis of Brassica rapa L. in Response to Plasmodiophora brassicae. Int. J. Mol. Sci. 2023, 24, 2414. https://doi.org/10.3390/ijms24032414
Wei X, Liao R, Zhang X, Zhao Y, Xie Z, Yang S, Su H, Wang Z, Zhang L, Tian B, et al. Integrative Transcriptome, miRNAs, Degradome, and Phytohormone Analysis of Brassica rapa L. in Response to Plasmodiophora brassicae. International Journal of Molecular Sciences. 2023; 24(3):2414. https://doi.org/10.3390/ijms24032414
Chicago/Turabian StyleWei, Xiaochun, Rujiao Liao, Xiaowei Zhang, Yanyan Zhao, Zhengqing Xie, Shuangjuan Yang, Henan Su, Zhiyong Wang, Luyue Zhang, Baoming Tian, and et al. 2023. "Integrative Transcriptome, miRNAs, Degradome, and Phytohormone Analysis of Brassica rapa L. in Response to Plasmodiophora brassicae" International Journal of Molecular Sciences 24, no. 3: 2414. https://doi.org/10.3390/ijms24032414
APA StyleWei, X., Liao, R., Zhang, X., Zhao, Y., Xie, Z., Yang, S., Su, H., Wang, Z., Zhang, L., Tian, B., Wei, F., & Yuan, Y. (2023). Integrative Transcriptome, miRNAs, Degradome, and Phytohormone Analysis of Brassica rapa L. in Response to Plasmodiophora brassicae. International Journal of Molecular Sciences, 24(3), 2414. https://doi.org/10.3390/ijms24032414