
Design, Synthesis, and Antitumor Activity Evaluation of Proteolysis-Targeting Chimeras as Degraders of Extracellular Signal-Regulated Kinases 1/2


Abstract
:
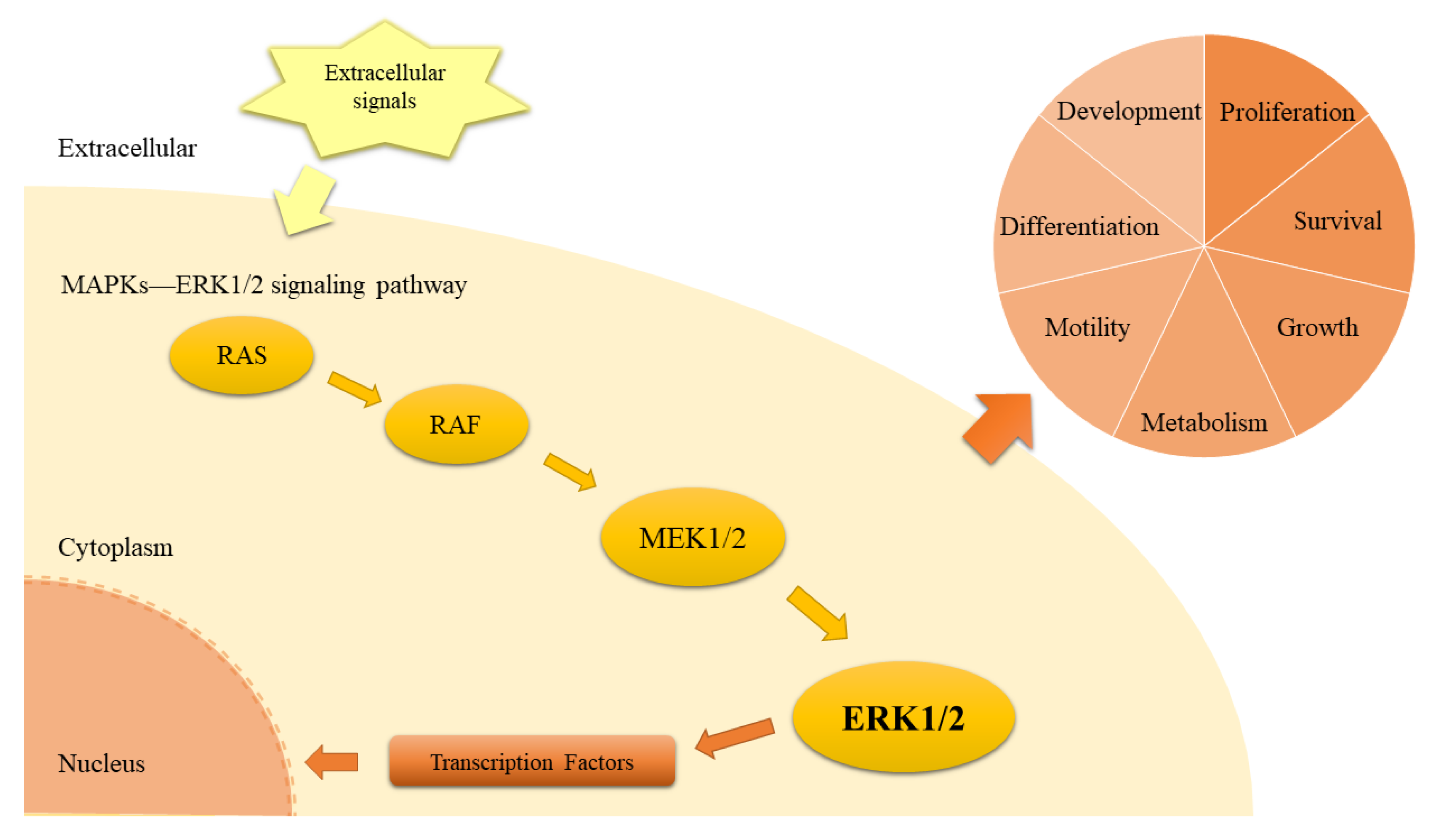
1. Introduction
2. Results and Discussion
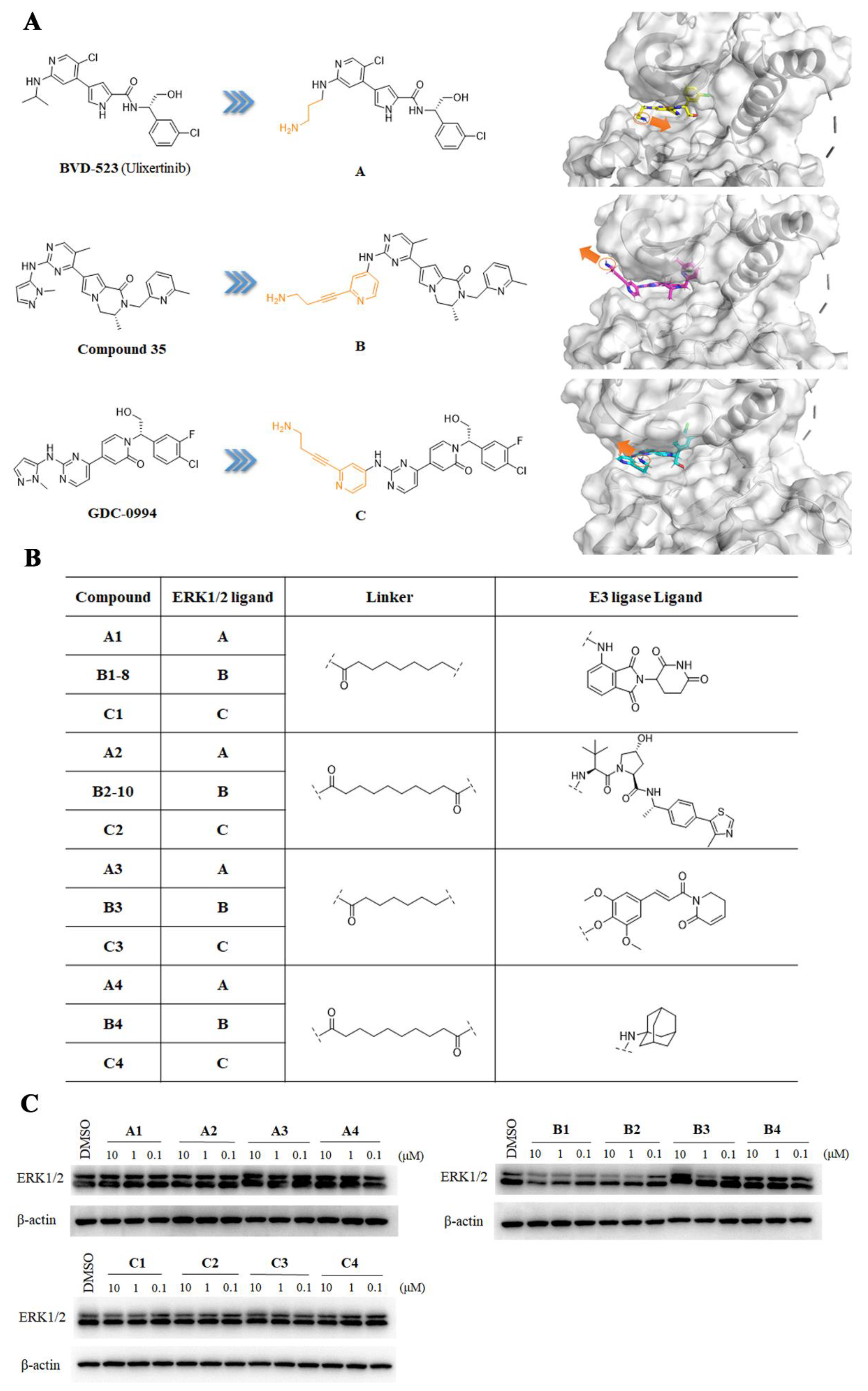
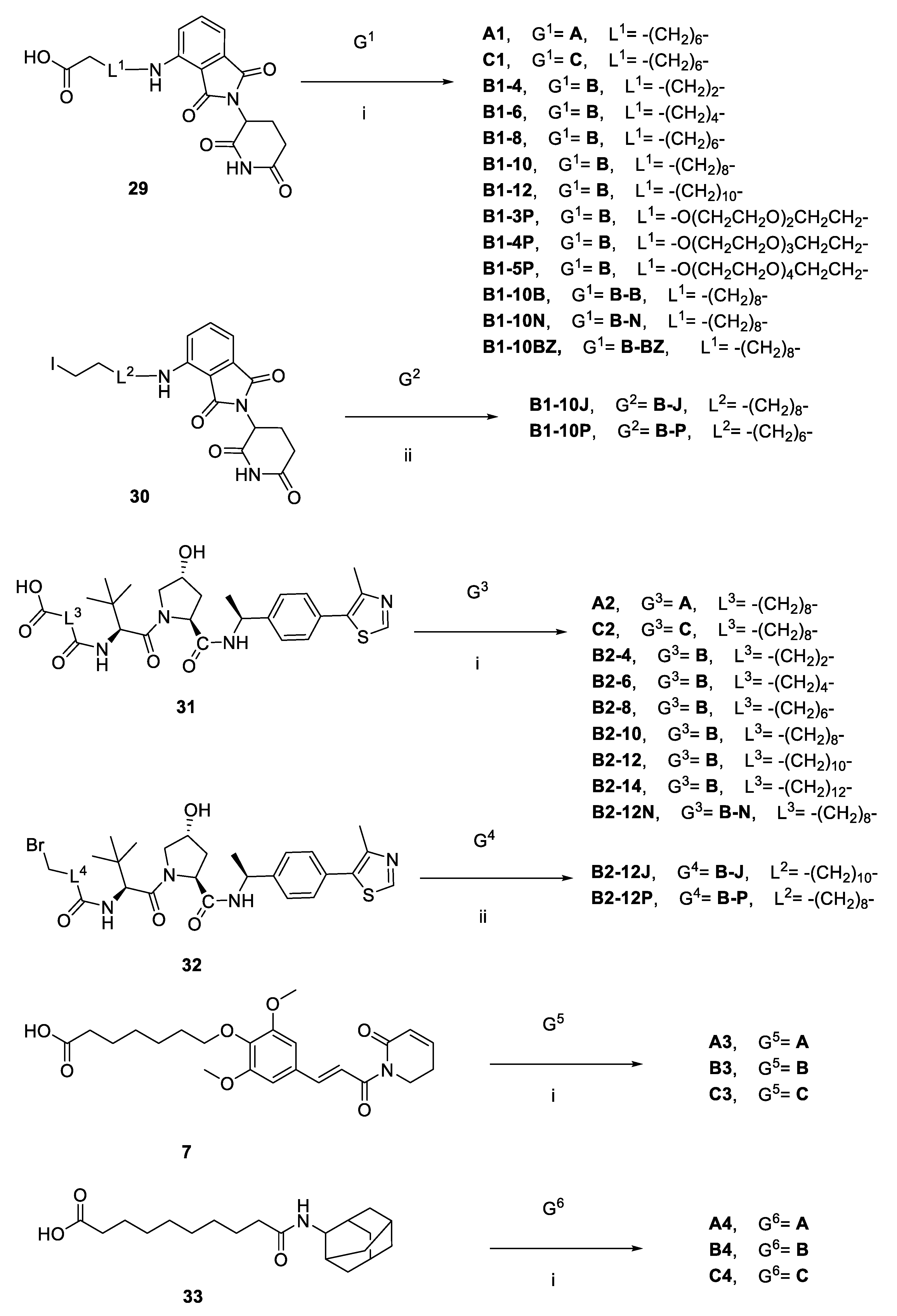
2.1. Design and Discovery of ERK1/2 Degraders
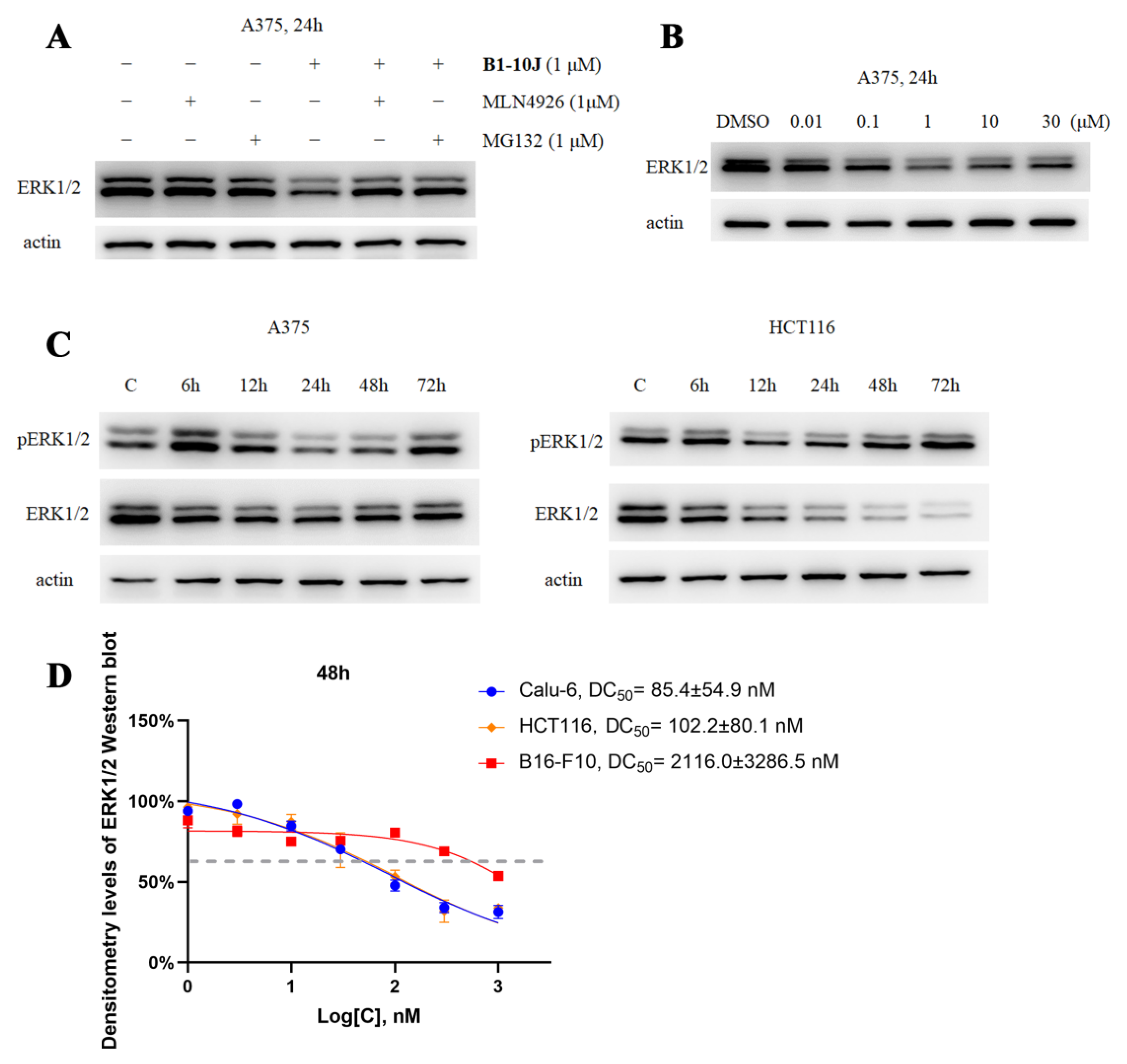
2.2. Characterization of the Degradative Activity of B1-10J
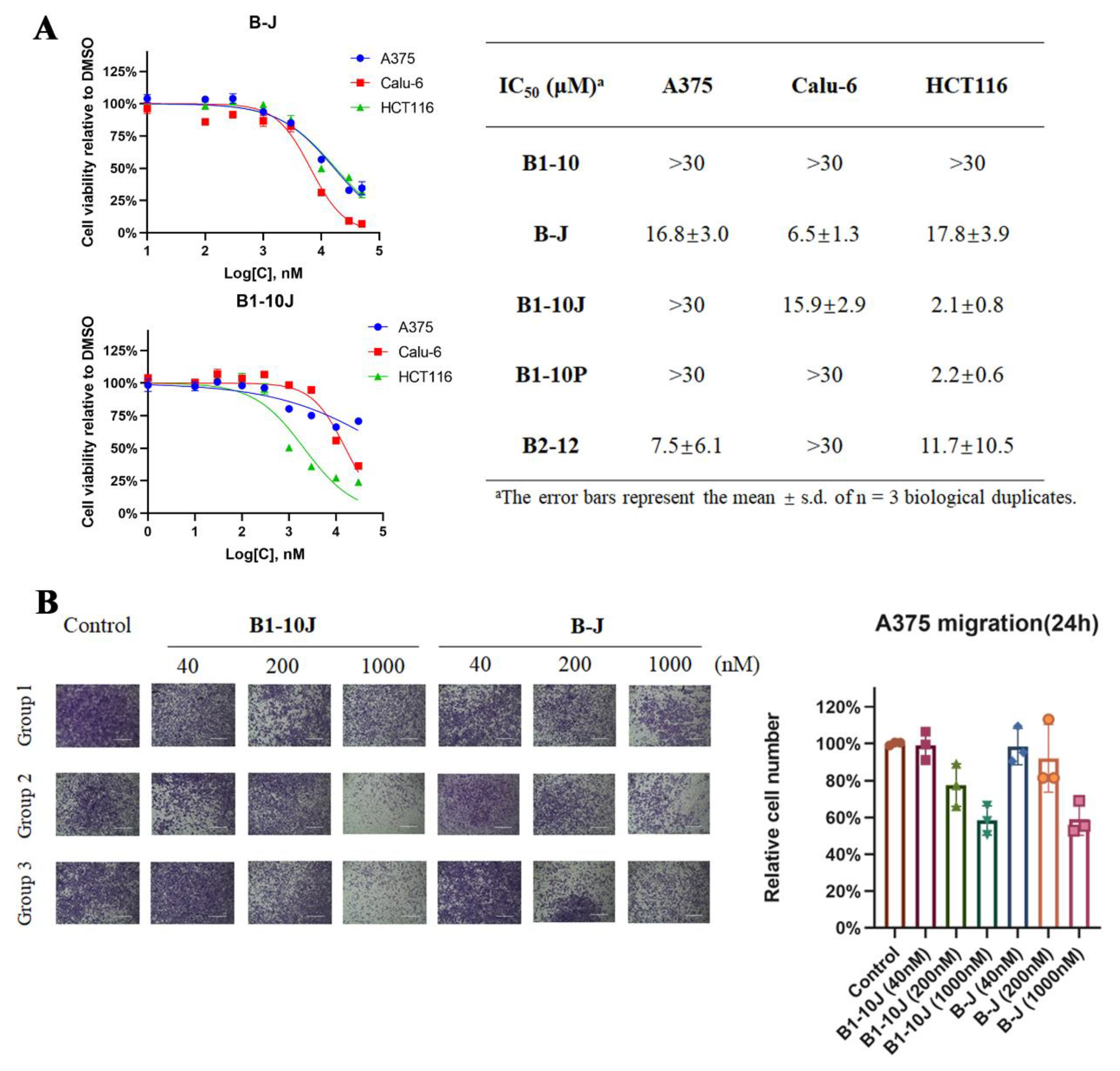
2.3. Effects of B1-10J on the Growth and Migration of Tumor Cells
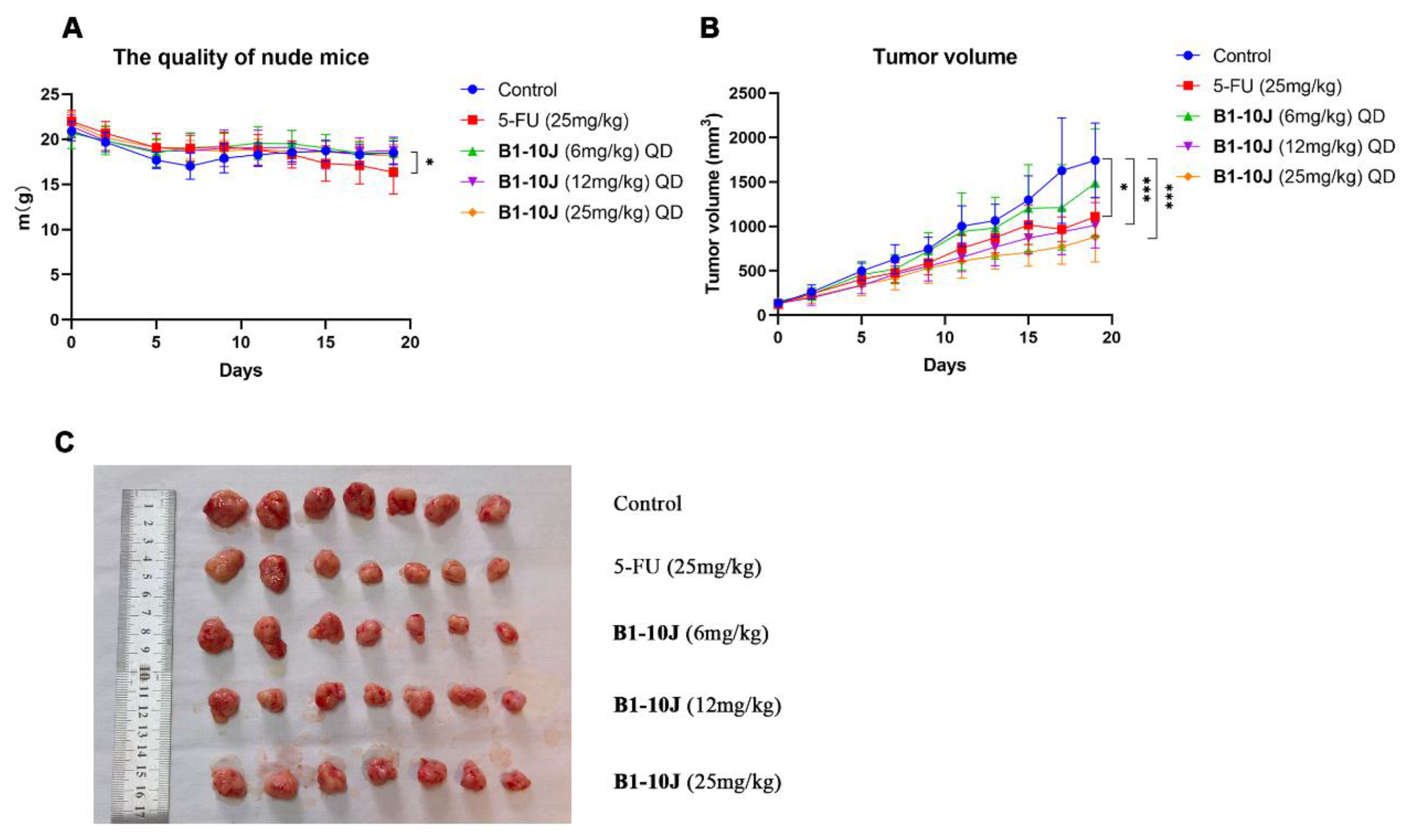
2.4. In Vivo Testing of B1-10J
3. Materials and Methods
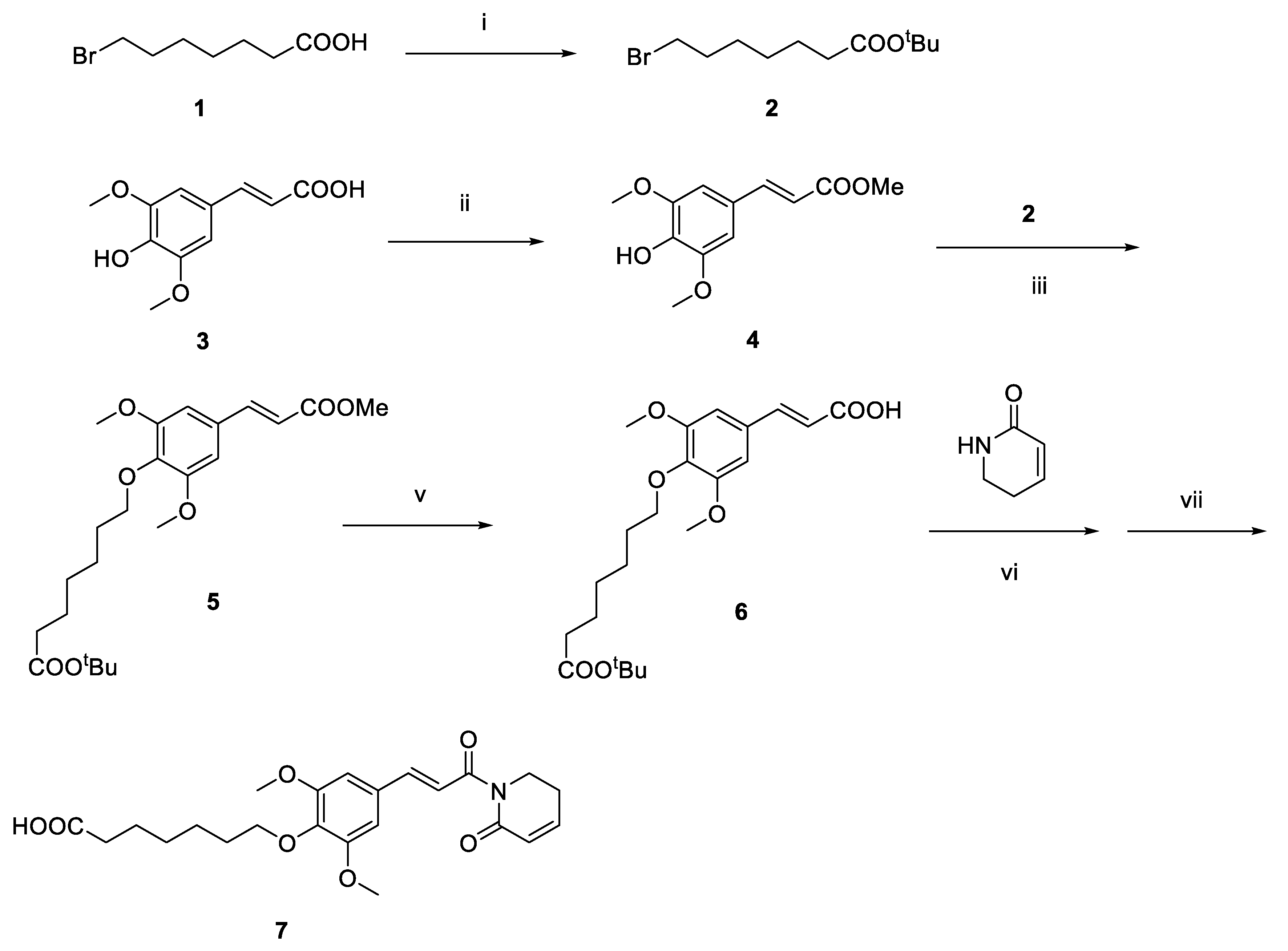
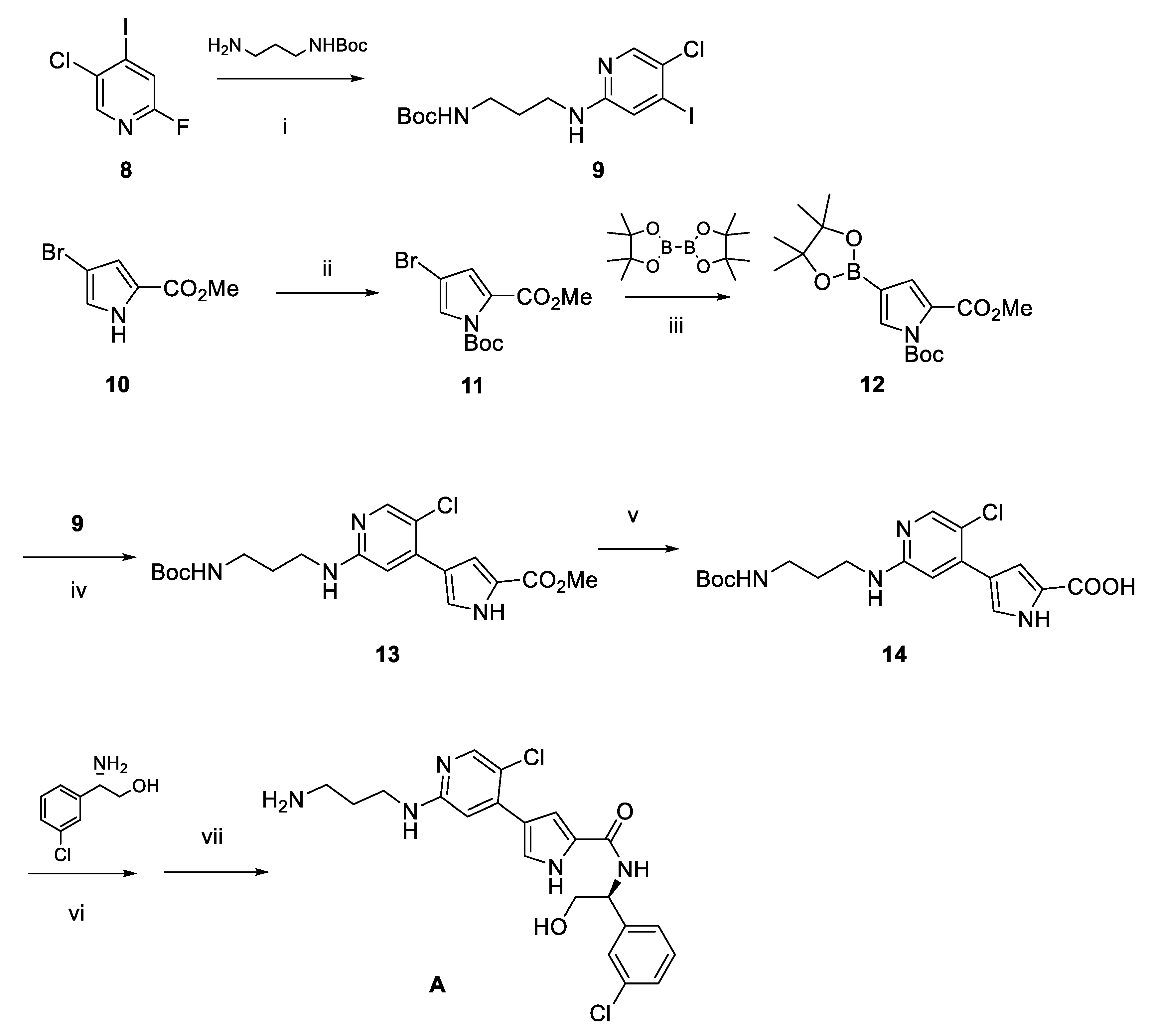
3.1. Chemistry
3.2. Molecular Docking
3.3. Cell Culture
3.4. Western Blot and Protein Degradation Assay
3.5. Cell Proliferation Assay
3.6. Transwell Migration Assay
3.7. Xenograft Mouse Studies
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Jivan, A.; Shao, C.; Duan, L.; Goad, D.; Zaganjor, E.; Osborne, J.; McGlynn, K.; Stippec, S.; Earnest, S.; et al. The roles of MAPKs in disease. Cell Res. 2008, 18, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Little, A.S.; Smith, P.D.; Cook, S.J. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2013, 32, 1207–1215. [Google Scholar] [CrossRef]
- Fu, L.; Chen, S.; He, G.; Chen, Y.; Liu, B. Targeting Extracellular Signal-Regulated Protein Kinase 1/2 (ERK1/2) in Cancer: An Update on Pharmacological Small-Molecule Inhibitors. J. Med. Chem. 2022, 65, 13561–13573. [Google Scholar] [CrossRef]
- Pan, X.; Pei, J.; Wang, A.; Shuai, W.; Feng, L.; Bu, F.; Zhu, Y.; Zhang, L.; Wang, G.; Ouyang, L. Development of small molecule extracellular signal-regulated kinases (ERKs) inhibitors for cancer therapy. Acta Pharm. Sin. B 2022, 12, 2171–2192. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef]
- Blake, J.F.; Burkard, M.; Chan, J.; Chen, H.; Chou, K.J.; Diaz, D.; Dudley, D.A.; Gaudino, J.J.; Gould, S.E.; Grina, J.; et al. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J. Med. Chem. 2016, 59, 5650–5660. [Google Scholar] [CrossRef]
- Ward, R.A.; Bethel, P.; Cook, C.; Davies, E.; Debreczeni, J.E.; Fairley, G.; Feron, L.; Flemington, V.; Graham, M.A.; Greenwood, R.; et al. Structure-Guided Discovery of Potent and Selective Inhibitors of ERK1/2 from a Modestly Active and Promiscuous Chemical Start Point. J. Med. Chem. 2017, 60, 3438–3450. [Google Scholar] [CrossRef]
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer 2022, 21, 99. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- He, S.; Dong, G.; Cheng, J.; Wu, Y.; Sheng, C. Strategies for designing proteolysis targeting chimaeras (PROTACs). Med. Res. Rev. 2022, 42, 1280–1342. [Google Scholar] [CrossRef]
- Liao, Y.; Niu, X.; Chen, B.; Edwards, H.; Xu, L.; Xie, C.; Lin, H.; Polin, L.; Taub, J.W.; Ge, Y.; et al. Synthesis and Antileukemic Activities of Piperlongumine and HDAC Inhibitor Hybrids against Acute Myeloid Leukemia Cells. J. Med. Chem. 2016, 59, 7974–7990. [Google Scholar] [CrossRef]
- Wang, M.; Lin, R.; Li, J.; Suo, Y.; Gao, J.; Liu, L.; Zhou, L.; Ni, Y.; Yang, Z.; Zheng, J.; et al. Discovery of LL-K8-22: A Selective, Durable, and Small-Molecule Degrader of the CDK8-Cyclin C Complex. J. Med. Chem. 2023, 66, 4932–4951. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct Target Ther. 2022, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M.; Tang, Q.; Martinez-Botella, G.; Bemis, G.W.; Cao, J.; Chen, G.; Ewing, N.P.; Ford, P.J.; Germann, U.A.; Green, J.; et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J. Med. Chem. 2009, 52, 6362–6368. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.A.; Anderton, M.J.; Bethel, P.; Breed, J.; Cook, C.; Davies, E.J.; Dobson, A.; Dong, Z.; Fairley, G.; Farrington, P.; et al. Discovery of a Potent and Selective Oral Inhibitor of ERK1/2 (AZD0364) That Is Efficacious in Both Monotherapy and Combination Therapy in Models of Nonsmall Cell Lung Cancer (NSCLC). J. Med. Chem. 2019, 62, 11004–11018. [Google Scholar] [CrossRef]
- Lebraud, H.; Surova, O.; Courtin, A.; O’Reilly, M.; Valenzano, C.R.; Nordlund, P.; Heightman, T.D. Quantitation of ERK1/2 inhibitor cellular target occupancies with a reversible slow off-rate probe. Chem. Sci. 2018, 9, 8608–8618. [Google Scholar] [CrossRef]
- Tsubuki, S.; Saito, Y.; Tomioka, M.; Ito, H.; Kawashima, S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 1996, 119, 572–576. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Heightman, T.D.; Berdini, V.; Braithwaite, H.; Buck, I.M.; Cassidy, M.; Castro, J.; Courtin, A.; Day, J.E.H.; East, C.; Fazal, L.; et al. Fragment-Based Discovery of a Potent, Orally Bioavailable Inhibitor That Modulates the Phosphorylation and Catalytic Activity of ERK1/2. J. Med. Chem. 2018, 61, 4978–4992. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.M.; Al Doghaither, H.A.; Al-Ghafari, A.B.; Pushparaj, P.N. 5-Fluorouracil and capecitabine therapies for the treatment of colorectal cancer (Review). Oncol. Rep. 2023, 50, 175. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
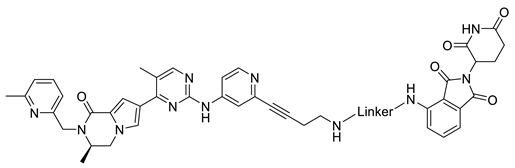 | |
| Compound | Linker |
| B1-4 | 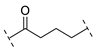 |
| B1-6 | 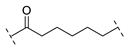 |
| B1-10 |  |
| B1-12 | 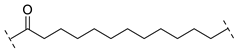 |
| B1-3P | 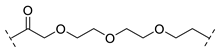 |
| B1-4P | 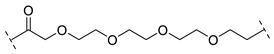 |
| B1-5P | 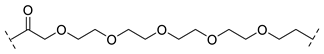 |
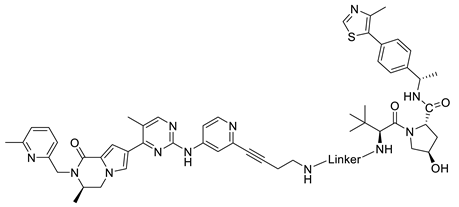 | |
| Compound | Linker |
| B2-4 | 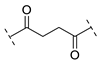 |
| B2-6 | 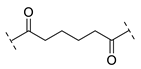 |
| B2-8 | 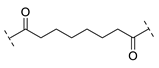 |
| B2-12 | 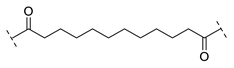 |
| B2-14 |  |
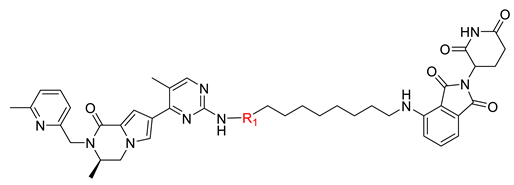 | ||
| Compound | R1 | DC50 (nM) a |
| B1-10J | 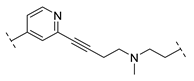 | 498.4 ± 369.3 |
| B1-10P | 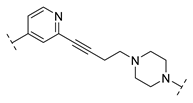 | 616.0 ± 389.4 |
| B1-10N |  | - b |
| B1-10B | 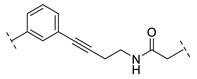 | - |
| B1-10BZ | 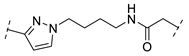 | - |
 | ||
| Compound | R2 | DC50 (nM) a |
| B2-12J | 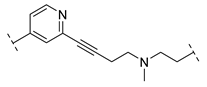 | - |
| B2-12P |  | 3800 ± 1824 |
| B2-12N | 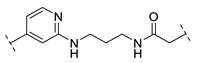 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, P.; He, Y.; Geng, T.; Li, Z.; Li, Z.; Meng, X. Design, Synthesis, and Antitumor Activity Evaluation of Proteolysis-Targeting Chimeras as Degraders of Extracellular Signal-Regulated Kinases 1/2. Int. J. Mol. Sci. 2023, 24, 16290. https://doi.org/10.3390/ijms242216290
Pan P, He Y, Geng T, Li Z, Li Z, Meng X. Design, Synthesis, and Antitumor Activity Evaluation of Proteolysis-Targeting Chimeras as Degraders of Extracellular Signal-Regulated Kinases 1/2. International Journal of Molecular Sciences. 2023; 24(22):16290. https://doi.org/10.3390/ijms242216290
Chicago/Turabian StylePan, Pengming, Yichao He, Tongtong Geng, Zhongtang Li, Zhongjun Li, and Xiangbao Meng. 2023. "Design, Synthesis, and Antitumor Activity Evaluation of Proteolysis-Targeting Chimeras as Degraders of Extracellular Signal-Regulated Kinases 1/2" International Journal of Molecular Sciences 24, no. 22: 16290. https://doi.org/10.3390/ijms242216290
APA StylePan, P., He, Y., Geng, T., Li, Z., Li, Z., & Meng, X. (2023). Design, Synthesis, and Antitumor Activity Evaluation of Proteolysis-Targeting Chimeras as Degraders of Extracellular Signal-Regulated Kinases 1/2. International Journal of Molecular Sciences, 24(22), 16290. https://doi.org/10.3390/ijms242216290