

Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment


 ,
,  ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Pathophysiological Hallmarks of AD: Therapeutic Implications
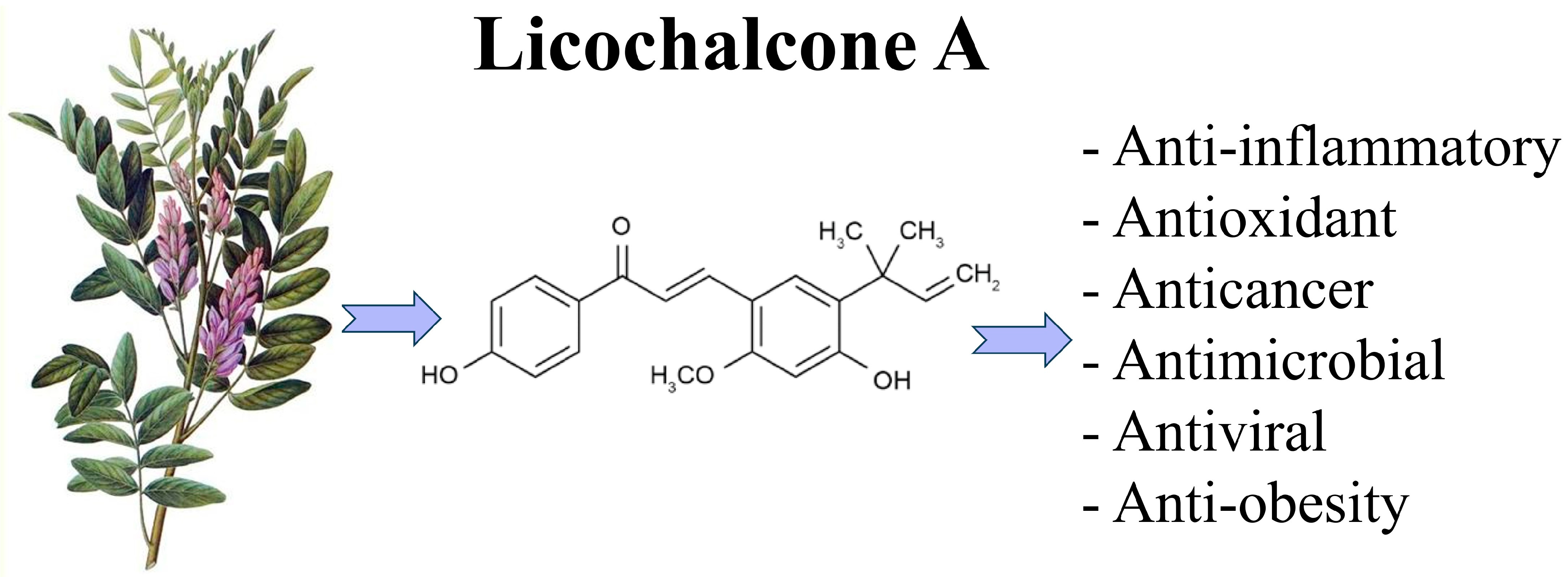
3. Licochalcone A: A Natural Compound with a Multitarget Side
3.1. Licochalcone A against Diabesity-Associated Cognitive Loss
3.2. Licochalcone A as a PTP1B Inhibitor
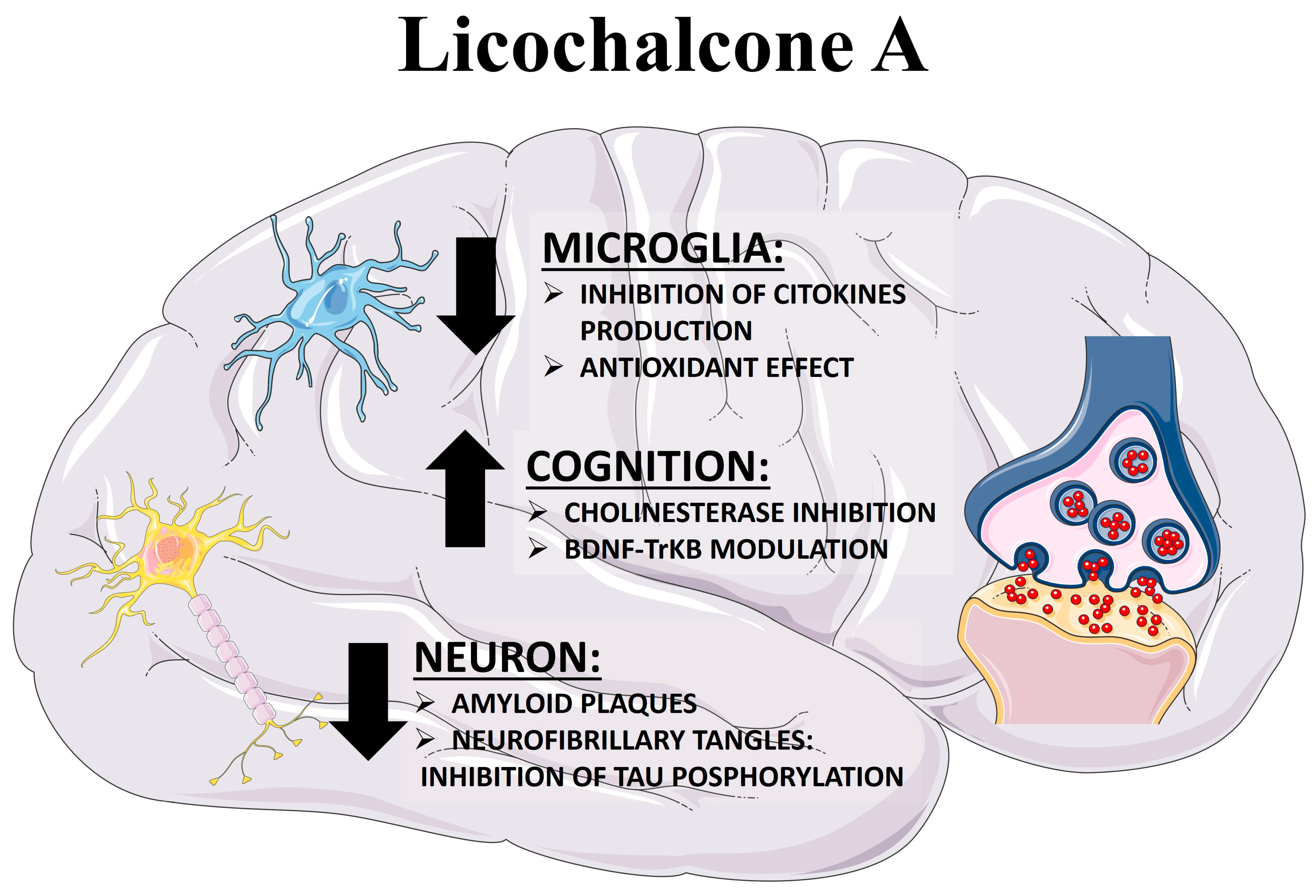
3.3. Licochalcone A as an Anti-Inflammatory Compound
3.4. Licochalcone A as an Antioxidant Compound
3.5. Licochalcone A as Amyloid Inhibitor
3.6. Licochalcone A as an Acetylcholinesterase Inhibitor and Memory Enhancer
4. Current Clinical Application of Licochalcone A
5. Conclusions
Funding
Conflicts of Interest
References
- World Health Organization. Dementia. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 26 February 2020).
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimer’s Dement. 2023, 19, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. 2022 Jun 5. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gunes, S.; Aizawa, Y.; Sugashi, T.; Sugimoto, M.; Rodrigues, P.P. Biomarkers for Alzheimer’s Disease in the Current State: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 4962. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 2, Erratum in Mol. Neurodegener. 2021, 16, 21. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Central Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- McShane, R.; Westby, M.J.; Roberts, E.; Minakaran, N.; Schneider, L.; E Farrimond, L.; Maayan, N.; Ware, J.; Debarros, J. Memantine for dementia. Cochrane Database Syst. Rev. 2019, 3, CD003154. [Google Scholar] [CrossRef]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443, Erratum in: Drugs 2021, 81, 1701. [Google Scholar] [CrossRef]
- Tampi, R.R.; Forester, B.P.; Agronin, M. Aducanumab: Evidence from clinical trial data and controversies. Drugs Context 2021, 10, 1–9. [Google Scholar] [CrossRef]
- Li, M.-T.; Xie, L.; Jiang, H.-M.; Huang, Q.; Tong, R.-S.; Li, X.; Xie, X.; Liu, H.-M. Role of Licochalcone A in Potential Pharmacological Therapy: A Review. Front. Pharmacol. 2022, 13, 878776. [Google Scholar] [CrossRef] [PubMed]
- Budziak-Wieczorek, I.; Kamiński, D.; Skrzypek, A.; Ciołek, A.; Skrzypek, T.; Janik-Zabrotowicz, E.; Arczewska, M. Naturally Occurring Chalcones with Aggregation-Induced Emission Enhancement Characteristics. Molecules 2023, 28, 3412. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chiu, Y.; Yang, S.; Chen, C.; Huang, C.; Lee-Chen, G.; Lin, W.; Chang, K. Novel synthetic chalcone-coumarin hybrid for Aβ aggregation reduction, antioxidation, and neuroprotection. CNS Neurosci. Ther. 2018, 24, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, Y.; Wei, X.; Fan, T. Neuroprotective effect of licochalcone A against oxygen-glucose deprivation/reperfusion in rat primary cortical neurons by attenuating oxidative stress injury and inflammatory response via the SIRT1/Nrf2 pathway. J. Cell. Biochem. 2018, 119, 3210–3219. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Gene–environment interactions in Alzheimer disease: The emerging role of epigenetics. Nat. Rev. Neurol. 2022, 18, 643–660. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-beta Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid Beta in Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12924. [Google Scholar] [CrossRef]
- Frontzkowski, L.; Ewers, M.; Brendel, M.; Biel, D.; Ossenkoppele, R.; Hager, P.; Steward, A.; Dewenter, A.; Römer, S.; Rubinski, A.; et al. Earlier Alzheimer’s disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat. Commun. 2022, 13, 4899. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of neurodegeneration—Insights from familial Alzheimer’s disease. Semin. Cell Dev. Biol. 2020, 105, 75–85. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Babaei, P. NMDA and AMPA receptors dysregulation in Alzheimer’s disease. Eur. J. Pharmacol. 2021, 908, 174310. [Google Scholar] [CrossRef]
- Badimon, A.; Torrente, D.; Norris, E.H. Vascular Dysfunction in Alzheimer’s Disease: Alterations in the Plasma Contact and Fibrinolytic Systems. Int. J. Mol. Sci. 2023, 24, 7046. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of metals in Alzheimer’s disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Marcello, E.; Epis, R.; Saraceno, C.; Di Luca, M. Synaptic Dysfunction in Alzheimer’s Disease. Synaptic Plast. Dyn. Dev. Dis. 2012, 970, 573–601. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Deane, R.; Gottesdiener, A.J.; Verghese, P.B.; Stewart, F.R.; West, T.; Paoletti, A.C.; Kasper, T.R.; DeMattos, R.B.; Zlokovic, B.V.; et al. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc. Natl. Acad. Sci. USA 2012, 109, 15502–15507. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Altmann, A.; Ng, B.; Landau, S.M.; Jagust, W.J.; Greicius, M.D. Alzheimer’s Disease Neuroimaging Initiative. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 2015, 138 Pt 12, 3734–3746. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Hoy, S.M. Lecanemab: First Approval. Drugs 2023, 83, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356, Erratum in: Science 2002, 297, 2209. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Boros, B.D.; Greathouse, K.M.; Gentry, E.G.; Curtis, K.A.; Bs, E.L.B.; Gearing, M.; Herskowitz, J.H. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann. Neurol. 2017, 82, 602–614. [Google Scholar] [CrossRef]
- Boros, B.D.; Greathouse, K.M.; Gearing, M.; Herskowitz, J.H. Dendritic spine remodeling accompanies Alzheime’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol. Aging 2019, 73, 92–103. [Google Scholar] [CrossRef]
- Walker, C.K.; Herskowitz, J.H. Dendritic Spines: Mediators of Cognitive Resilience in Aging and Alzheimer’s Disease. Neuroscientist 2021, 27, 487–505. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Small, D.H.; Mok, S.S.; Bornstein, J.C. Alzheimer’s disease and Aβ toxicity: From top to bottom. Nat. Rev. Neurosci. 2001, 2, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Meyer-Luehmann, M.; Stern, E.A.; McLean, P.J.; Skoch, J.; Nguyen, P.T.; Bacskai, B.J.; Hyman, B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 2005, 25, 7278–7287. [Google Scholar] [CrossRef]
- Ten Kate, M.; Dicks, E.; Visser, P.J.; van der Flier, W.M.; Teunissen, C.E.; Barkhof, F.; Scheltens, P.; Tijms, B.M.; Alzheimer’s Disease Neuroimaging, I. Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain 2018, 141, 3443–3456. [Google Scholar] [CrossRef]
- Fantini, J.; Chahinian, H.; Yahi, N. Progress toward Alzheimer’s disease treatment: Leveraging the Achilles’ heel of Aβ oligomers? Protein Sci. 2020, 29, 1748–1759. [Google Scholar] [CrossRef]
- Cacabelos, R.; Naidoo, V.; Martínez-Iglesias, O.; Corzo, L.; Cacabelos, N.; Pego, R.; Carril, J.C. Personalized Management and Treatment of Alzheimer’s Disease. Life 2022, 12, 460. [Google Scholar] [CrossRef]
- Yamali, C.; Donmez, S. Recent developments in tacrine-based hybrids as a therapeutic option for Alzheimer’s disease. Mini-Rev. Med. Chem. 2023, 23, 869–880. [Google Scholar] [CrossRef]
- Dkhil, M.A.; Delic, D.; El Enshasy, H.A.; Moneim, A.E.A. Medicinal Plants in Therapy: Antioxidant Activities. Oxidative Med. Cell. Longev. 2016, 2016, 7468524. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Son, D.-H.; Chung, T.-H.; Lee, Y.-J. A Review of the Pharmacological Efficacy and Safety of Licorice Root from Corroborative Clinical Trial Findings. J. Med. Food 2020, 23, 12–20. [Google Scholar] [CrossRef]
- Souza, J.M.; de Carvalho, A.A.; Candido, A.C.B.B.; de Mendonça, R.P.; da Silva, M.F.; Parreira, R.L.T.; Dias, F.G.G.; Ambrósio, S.R.; Arantes, A.T.; Filho, A.A.d.S.; et al. Licochalcone a Exhibits Leishmanicidal Activity in vitro and in Experimental Model of Leishmania (Leishmania) Infantum. Front. Veter-Sci. 2020, 7, 527. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liang, J.; Zhang, J.; Wang, Y.; Chai, X. Natural Chalcones in Chinese Materia Medica: Licorice. Evid. -Based Complement. Altern. Med. 2020, 2020, 3821248. [Google Scholar] [CrossRef]
- Muto, E.; Okada, T.; Yamanaka, T.; Uchino, H.; Inazu, M. Licochalcone E, a β-Amyloid Aggregation Inhibitor, Regulates Microglial M1/M2 Polarization via Inhibition of CTL1-Mediated Choline Uptake. Biomolecules 2023, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xue, J.; Ding, Z.; Li, X.; Wang, X.; Xue, H. Activated Phosphoinositide 3-Kinase/Akt/Mammalian Target of Rapamycin Signal and Suppressed Autophagy Participate in Protection Offered by Licochalcone A Against Amyloid-β Peptide Fragment 25–35–Induced Injury in SH-SY5Y Cells. World Neurosurg. 2022, 157, e390–e400. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Ren, H.; Wang, L.; Chen, W.; Ci, X. Lico A Enhances Nrf2-Mediated Defense Mechanisms against t-BHP-Induced Oxidative Stress and Cell Death via Akt and ERK Activation in RAW 264.7 Cells. Oxid. Med. Cell Longev. 2015, 2015, 709845. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Z.; Meng, R.; Shi, C.; Guo, N. Antioxidative and anticancer properties of Licochalcone A from licorice. J. Ethnopharmacol. 2017, 198, 331–337. [Google Scholar] [CrossRef]
- Busquets, O.; Ettcheto, M.; Verdaguer, E.; Castro-Torres, R.D.; Auladell, C.; Beas-Zarate, C.; Folch, J.; Camins, A. JNK1 inhibition by Licochalcone A leads to neuronal protection against excitotoxic insults derived of kainic acid. Neuropharmacology 2018, 131, 440–452. [Google Scholar] [CrossRef]
- Han, J.Y.; Park, S.H.; Yang, J.H.; Kim, M.G.; Cho, S.S.; Yoon, G.; Cheon, S.H.; Ki, S.H. Licochalcone Suppresses LXRα-Induced Hepatic Lipogenic Gene Expression through AMPK/Sirt1 Pathway Activation. Toxicol. Res. 2014, 30, 19–25. [Google Scholar] [CrossRef]
- Bisht, D.; Rashid, M.; Arya, R.K.K.; Kumar, D.; Chaudhary, S.K.; Rana, V.S.; Sethiya, N.K. Revisiting liquorice (Glycyrrhiza glabra L.) as anti-inflammatory, antivirals and immunomodulators: Potential pharmacological applications with mechanistic insight. Phytomedicine Plus 2022, 2, 100206. [Google Scholar] [CrossRef]
- Furusawa, J.-I.; Funakoshi-Tago, M.; Tago, K.; Mashino, T.; Inoue, H.; Sonoda, Y.; Kasahara, T. Licochalcone A significantly suppresses LPS signaling pathway through the inhibition of NF-κB p65 phosphorylation at serine 276. Cell. Signal. 2009, 21, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yu, C.; Zeng, F.-S.; Fu, X.; Yuan, X.-J.; Wang, Q.; Fan, C.; Sun, B.-L.; Sun, Q.-S. Licochalcone A Attenuates Chronic Neuropathic Pain in Rats by Inhibiting Microglia Activation and Inflammation. Neurochem. Res. 2021, 46, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Cano, A.; Busquets, O.; Manzine, P.R.; Sánchez-López, E.; Castro-Torres, R.D.; Beas-Zarate, C.; Verdaguer, E.; García, M.L.; Olloquequi, J.; et al. A metabolic perspective of late onset Alzheimer’s disease. Pharmacol. Res. 2019, 145, 104255. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef]
- Hölscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert. Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.M.; González-Rodríguez, A. IRS2 and PTP1B: Two opposite modulators of hepatic insulin signalling. Arch. Physiol. Biochem. 2011, 117, 105–115. [Google Scholar] [CrossRef]
- Luo, Z.; Fu, C.; Li, T.; Gao, Q.; Miao, D.; Xu, J.; Zhao, Y. Hypoglycemic Effects of Licochalcone A on the Streptozotocin-Induced Diabetic Mice and Its Mechanism Study. J. Agric. Food Chem. 2021, 69, 2444–2456. [Google Scholar] [CrossRef]
- Luo, Z.; Li, T.; Gao, Q.; Chen, Y.; Su, G.; Zhao, Y. Impact of licochalcone A on the progression of diabetic nephropathy in type 2 diabetes mellitus of C57BL/6 mice. Food Funct. 2021, 12, 10676–10689. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.-J.; Lee, Y.-K.; Ting, N.-C.; Chen, Y.-L.; Shen, S.-C.; Wu, S.-J.; Huang, W.-C. Protective Effects of Licochalcone A Ameliorates Obesity and Non-Alcoholic Fatty Liver Disease Via Promotion of the Sirt-1/AMPK Pathway in Mice Fed a High-Fat Diet. Cells 2019, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K. Inflammation and Insulin Resistance: An Old Story with New Ideas. Korean Diabetes J. 2010, 34, 137–145. [Google Scholar] [CrossRef][Green Version]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Manieri, E.; Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 2015, 55, R11–R22. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. cJun NH2-terminal kinase 1 (JNK1): Roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010, 35, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Sabio, G. Brain JNK and metabolic disease. Diabetologia 2021, 64, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Sze, C.-I.; Su, M.; Pugazhenthi, S.; Jambal, P.; Hsu, L.-J.; Heath, J.; Schultz, L.; Chang, N.-S. Down-regulation of WW domain-containing oxidoreductase induces tau phosphorylation in vitro. J. Biol. Chem. 2004, 279, 30498–30506. [Google Scholar] [CrossRef]
- Yao, K.; Chen, H.; Lee, M.-H.; Li, H.; Ma, W.; Peng, C.; Song, N.R.; Lee, K.W.; Bode, A.M.; Dong, Z.; et al. Licochalcone A, a natural inhibitor of c-Jun N-terminal kinase 1. Cancer Prev. Res. 2014, 7, 139–149. [Google Scholar] [CrossRef]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef]
- Pandey, N.R.; Zhou, X.; Qin, Z.; Zaman, T.; Gomez-Smith, M.; Keyhanian, K.; Anisman, H.; Brunel, J.M.; Stewart, A.F.R.; Chen, H.-H. The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J. Neurosci. 2013, 33, 12647–12655. [Google Scholar] [CrossRef]
- Pandey, N.R.; Zhou, X.; Zaman, T.; Cruz, S.A.; Qin, Z.; Lu, M.; Keyhanian, K.; Brunel, J.M.; Stewart, A.F.; Chen, H.-H. LMO4 is required to maintain hypothalamic insulin signaling. Biochem. Biophys. Res. Commun. 2014, 450, 666–672. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Goodman, E.K.; Mitchell, C.S.; Teo, J.D.; Gladding, J.M.; Abbott, K.N.; Rafiei, N.; Zhang, L.; Herzog, H.; Begg, D.P. The effect of insulin receptor deletion in neuropeptide Y neurons on hippocampal dependent cognitive function in aging mice. J. Integr. Neurosci. 2022, 21, 6. [Google Scholar] [CrossRef]
- Fuentes, F.; Zimmer, D.; Atienza, M.; Schottenfeld, J.; Penkala, I.; Bale, T.; Bence, K.K.; Arregui, C.O. Protein tyrosine phosphatase PTP1B is involved in hippocampal synapse formation and learning. PLoS ONE 2012, 7, e41536. [Google Scholar] [CrossRef]
- Ozek, C.; Kanoski, S.E.; Zhang, Z.-Y.; Grill, H.J.; Bence, K.K. Protein-tyrosine phosphatase 1B (PTP1B) is a novel regulator of central brain-derived neurotrophic factor and tropomyosin receptor kinase B (TrkB) signaling. Health Educ. 2014, 289, 31682–31692. [Google Scholar] [CrossRef]
- Krishnan, N.; Krishnan, K.; Connors, C.R.; Choy, M.S.; Page, R.; Peti, W.; Van Aelst, L.; Shea, S.D.; Tonks, N.K. PTP1B inhibition suggests a therapeutic strategy for Rett syndrome. J. Clin. Investig. 2015, 125, 3163–3177. [Google Scholar] [CrossRef] [PubMed]
- Koss, D.J.; Riedel, G.; Bence, K.; Platt, B. Store-operated Ca2+ entry in hippocampal neurons: Regulation by protein tyrosine phosphatase PTP1B. Cell Calcium 2013, 53, 125–138. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef]
- Zhang, H. Polarity Determinants in Dendritic Spine Development and Plasticity. Neural Plast. 2016, 2016, 3145019. [Google Scholar] [CrossRef]
- Vieira, M.N.N.; e Silva, N.M.L.; Ferreira, S.T.; De Felice, F.G. Protein Tyrosine Phosphatase 1B (PTP1B): A Potential Target for Alzheimer’s Therapy? Front. Aging Neurosci. 2017, 9, 7. [Google Scholar] [CrossRef]
- Lourenco, M.V. Preface: Special issue “Brain Proteostasis in Health and Disease”. J. Neurochem. 2023, 166, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, Z.; Sharmin, F.; Lin, W.; Ricke, K.M.; Zasloff, M.A.; Stewart, A.F.; Chen, H.-H. Tyrosine phosphatase PTP1B impairs presynaptic NMDA receptor-mediated plasticity in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 156, 105402. [Google Scholar] [CrossRef]
- Pei, J.-J.; Sersen, E.; Iqbal, K.; Grundke-Iqbal, I. Expression of protein phosphatases (PP-1, PP-2A, PP-2B and PTP-1B) and protein kinases (MAP kinase and P34cdc2) in the hippocampus of patients with Alzheimer disease and normal aged individuals. Brain Res. 1994, 655, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Jung, M.; Kim, J.-H.; Park, H.; Rahman, H.; Zhang, S.; Zhang, Z.-Y.; Park, D.H.; Kook, H.; Lee, I.-K.; et al. A novel role for protein tyrosine phosphatase 1B as a positive regulator of neuroinflammation. J. Neuroinflammation 2016, 13, 86. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cano, A.; Sanchez-López, E.; Carrasco, M.; Verdaguer, E.; Fortuna, A.; Folch, J.; Bulló, M.; Auladell, C.; Camins, A.; et al. Protein tyrosine phosphatase 1B (PTP1B) as a potential therapeutic target for neurological disorders. BioMedicine 2022, 155, 113709. [Google Scholar] [CrossRef]
- Yoon, G.; Lee, W.; Kim, S.-N.; Cheon, S.H. Inhibitory effect of chalcones and their derivatives from Glycyrrhiza inflata on protein tyrosine phosphatase 1B. Bioorganic Med. Chem. Lett. 2009, 19, 5155–5157. [Google Scholar] [CrossRef]
- Lin, Y.; Kuang, Y.; Li, K.; Wang, S.; Song, W.; Qiao, X.; Sabir, G.; Ye, M. Screening for bioactive natural products from a 67-compound library of Glycyrrhiza inflata. Bioorganic Med. Chem. 2017, 25, 3706–3713. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Liu, J.; Liu, L.; Wang, X.; Jiang, R.; Bai, Q.; Wang, G. Microglia: A Double-Edged Sword in Intracerebral Hemorrhage from Basic Mechanisms to Clinical Research. Front. Immunol. 2021, 12, 675660. [Google Scholar] [CrossRef]
- Perry, V.H.; Nicoll, J.A.R.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross Talk Between Brain Innate Immunity and Serotonin Signaling Underlies Depressive-Like Behavior Induced by Alzheimer’s Amyloid-β Oligomers in Mice. J. Neurosci. 2016, 36, 12106–12116. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Hong, H.; Kim, B.S.; Im, H.-I. Pathophysiological Role of Neuroinflammation in Neurodegenerative Diseases and Psychiatric Disorders. Int. Neurourol. J. 2016, 20 (Suppl. S1), S2–S7. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Sun, Y.; Peng, G. Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease. Clin. Interv. Aging 2022, 17, 665–674. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Gardoni, F.; Viviani, B.; Tozzi, A.; Giampà, C.; Costa, C.; Tantucci, M.; Zianni, E.; Boraso, M.; et al. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol. Dis. 2013, 52, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Erion, J.R.; Wosiski-Kuhn, M.; Dey, A.; Hao, S.; Davis, C.L.; Pollock, N.K.; Stranahan, A.M. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J. Neurosci. 2014, 34, 2618–2631. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, J.Y.; Yoon, J.H.; Lim, H.J.; Kim, T.H.; Jin, C.; Kwak, W.-J.; Han, C.-K.; Ryu, J.-H. Inhibition of nitric oxide synthase expression in activated microglia and peroxynitrite scavenging activity byOpuntia ficus indica var.saboten. Phytother. Res. 2006, 20, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, M.; Jin, L.; Yang, B.; Bai, B.; Mutsinze, R.N.; Zuo, W.; Chattipakorn, N.; Huh, J.Y.; Liang, G.; et al. Licochalcone A protects against LPS-induced inflammation and acute lung injury by directly binding with myeloid differentiation factor 2 (MD2). Br. J. Pharmacol. 2023, 180, 1114–1131. [Google Scholar] [CrossRef]
- Huang, B.; Liu, J.; Ju, C.; Yang, D.; Chen, G.; Xu, S.; Zeng, Y.; Yan, X.; Wang, W.; Liu, D.; et al. Licochalcone A Prevents the Loss of Dopaminergic Neurons by Inhibiting Microglial Activation in Lipopolysaccharide (LPS)-Induced Parkinson’s Disease Models. Int. J. Mol. Sci. 2017, 18, 2043. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, J.; Liu, H.; Liu, H. Licochalcone A improves the cognitive ability of mice by regulating T- and B-cell proliferation. Aging 2021, 13, 8895–8915. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Wes, P.D.; Holtman, I.R.; Boddeke, E.W.; Möller, T.; Eggen, B.J. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016, 64, 197–213. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Shah, S.A.; Amin, F.U.; Khan, M.; Abid, M.N.; Rehman, S.U.; Kim, T.H.; Kim, M.W.; Kim, M.O. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflammation 2016, 13, 286. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, H.; Ishikawa, H.; Mizutani, K.; Tamura, Y.; Kinoshita, T. Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorganic Med. Chem. 1998, 6, 339–347. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L.; Tan, R.; Liang, G.; Fang, S.; Li, W.; Xie, M.; Wen, Y.; Wu, J.; Chen, Y. Design, Synthesis, and Evaluation of Chalcone Derivatives as Multifunctional Agents against Alzheimer’s Disease. Chem. Biodivers. 2021, 18, e2100341. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Ara, I.; Mondal, M.S.A.; Kabir, Y. Phytochemistry, pharmacological activity, and potential health benefits of Glycyrrhiza glabra. Heliyon 2021, 7, e07240. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Buneeva, O.A.; Kopylov, A.T.; Mitkevich, V.A.; Kozin, S.A.; Zgoda, V.G.; Makarov, A.A. Chemical modifications of amyloid-β(1-42) have a significant impact on the repertoire of brain amyloid-β(1-42) binding proteins. Biochimie 2016, 128–129, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Su, K.; Wang, X.; Guan, P.; Hu, X. Study on molecular mechanisms of destabilizing Aβ(1–42) protofibrils by licochalcone A and licochalcone B using molecular dynamics simulations. J. Mol. Graph. Model. 2023, 122, 108500. [Google Scholar] [CrossRef]
- Tsai, J.-P.; Lee, C.-H.; Ying, T.-H.; Lin, C.-L.; Lin, C.-L.; Hsueh, J.-T.; Hsieh, Y.-H. Licochalcone A induces autophagy through PI3K/Akt/mTOR inactivation and autophagy suppression enhances Licochalcone A-induced apoptosis of human cervical cancer cells. Oncotarget 2015, 6, 28851–28866. [Google Scholar] [CrossRef]
- Lin, T.; Chiu, Y.; Lin, C.; Lin, C.; Chao, C.; Chen, Y.; Yang, S.; Lin, W.; Hsieh-Li, H.M.; Wu, Y.; et al. Exploration of multi-target effects of 3-benzoyl-5-hydroxychromen-2-one in Alzheimer’s disease cell and mouse models. Aging Cell 2020, 19, e13169. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Wang, X.; Daley, C.; Gakhar, V.; Lange, H.S.; Vardigan, J.D.; Pearson, M.; Zhou, X.; Warren, L.; Miller, C.O.; Belden, M.; et al. Pharmacological Characterization of the Novel and Selective α7 Nicotinic Acetylcholine Receptor–Positive Allosteric Modulator BNC375. Experiment 2020, 373, 311–324. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Miyazaki, A.; Eerdunbayaer; Shiokawa, T.; Tada, H.; Lian, Y.; Taniguchi, S.; Hatano, T. High-performance liquid chromatographic profile and 1H quantitative nuclear magnetic resonance analyses for quality control of a Xinjiang licorice extract. Biosci. Biotechnol. Biochem. 2020, 84, 2128–2138. [Google Scholar] [CrossRef]
- Weber, T.M.; I Ceilley, R.; Buerger, A.; Kolbe, L.; Trookman, N.S.; Rizer, R.L.; Schoelermann, A. Skin tolerance, efficacy, and quality of life of patients with red facial skin using a skin care regimen containing Licochalcone A. J. Cosmet. Dermatol. 2006, 5, 227–232. [Google Scholar] [CrossRef]
- Kolbe, L.; Immeyer, J.; Batzer, J.; Wensorra, U.; Dieck, K.T.; Mundt, C.; Wolber, R.; Stäb, F.; Schönrock, U.; Ceilley, R.I.; et al. Anti-inflammatory efficacy of Licochalcone A: Correlation of clinical potency and in vitro effects. Arch. Dermatol. Res. 2006, 298, 23–30. [Google Scholar] [CrossRef]
- Chularojanamontri, L.; Tuchinda, P.; Kulthanan, K.; Varothai, S.; Winayanuwattikun, W. A double-blinded, randomized, vehicle-controlled study to access skin tolerability and efficacy of an anti-inflammatory moisturizer in treatment of acne with 0.1% adapalene gel. J. Dermatol. Treat. 2016, 27, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Kulthanan, K.; Trakanwittayarak, S.; Tuchinda, P.; Chularojanamontri, L.; Limphoka, P.; Varothai, S. A Double-Blinded, Randomized, Vehicle-Controlled Study of the Efficacy of Moisturizer Containing Licochalcone A, Decanediol, L-Carnitine, and Salicylic Acid for Prevention of Acne Relapse in Asian Population. BioMed Res. Int. 2020, 2020, 2857812. [Google Scholar] [CrossRef] [PubMed]
- Angelova-Fischer, I.; Rippke, F.; Fischer, T.; Neufang, G.; Zillikens, D. A double-blind, randomized, vehicle-controlled efficacy assessment study of a skin care formulation for improvement of mild to moderately severe acne. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 6–11. [Google Scholar] [CrossRef]
- Lu, W.J.; Wu, G.J.; Chen, R.J.; Chang, C.C.; Lien, L.M.; Chiu, C.C.; Tseng, M.F.; Huang, L.T.; Lin, K.H. Licochalcone A attenuates glioma cell growth in vitro and in vivo through cell cycle arrest. Food Funct. 2018, 9, 4500–4507. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Row | Reference | Experimental Model | Main Findings |
|---|---|---|---|
| 1 | [56] | Molecular docking and Molecular dynamics simulations | Licochalcone A lead to a conformational disruption of the Aβ(1–42) protofibril. |
| 2 | [57] | SH-SY5Y cells | Licochalcone A shows a neuroprotective effect against Aβ-induced neurotoxicity, 5–35 inhibiting oxidative stress, mitochondrial dysfunction, and apoptosis. The proposed mechanism is through the activation of the PI3K/Akt/mTOR signaling pathway in SH-SY5Y cells. |
| 3 | [15] | Tet-On Aβ-GFP293/SH-SY5Y cells | Licochalcone A reduce oxidative stress, activate CREB-dependent BDNF/AKT/ERK signaling pathway involved in cell survival and CREB-dependent BCL2 for antiapoptosis. |
| 4 | [15,57,58,59] | Aβ-GFP 293/SH-SY5Y/RAW 264.6/BV-2 cells. | Licochalcone A ameliorate Aβ-induced aggregation, oxidative stress and promote neurite outgrowth in neuron like cells. Likewise, Licochalcone A prevents microglia-mediated inflammation. |
| 5 | [60] | Kainic Acid (model of temporal lobe epilepsy) | Inhibition of JNK1 by Licochalcone A can prevent neuronal degeneration in a mice experimental model of temporal lobe epilepsy TLE. |
| 6 | [61] | Rat primary cortical neurons culture | Neuroprotective properties of Licochalcone A against oxygen-glucose deprivation/reperfusion in cortical neurons could be explained trough the activation of the SIRT1/Nrf2 signaling and the inhibition of downstream NF-κB signaling pathway |
| 7 | [62] | Rat primary microglia culture | Licochalcone A exerts anti-neuroinflammatory and anti-oxidative effects in primary rat microglia mainly dependent on the arachidonic acid/COX-2/PGE2 pathway. |
| 8 | [63] | Lipopolysaccharide (LPS)-induced PD models in vivo and in vitro | Licochalcone A inhibits LPS-induced microglial activation via downregulation the activation of ERK1/2 and NF-κB p65 pathways. Licochalcone A treatment prevents dopaminergic neurodegeneration by inhibiting microglia-mediated neuroinflammation. |
| 9 | [64] | Neuropathic pain in a rat model. | Licochalcone A exerts protective effects against CCI-evoked neuropathic pain in rat model, through inhibiting microglia activation, p38 phosphorylation and inflammatory response. |
| Row | Reference | Clinical Trial | Main Findings |
|---|---|---|---|
| 1 | [136] | NCT04002024 | The study demonstrated the efficacy and safety of moisturizing cream containing licochalcone A, decanediol, L-carnitine, and salicylic acid as maintenance therapy in patients with acne of mild to moderate severity. |
| 2 | [135] | NCT02173054 | The use of a moisturizer containing licochalcone A, L-carnitine, and 1,2-decanediol in addition to adapalene gel might be superior to placebo in reducing skin irritations and improving the efficacy of patient adherence to pain medications. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olloquequi, J.; Ettcheto, M.; Cano, A.; Fortuna, A.; Bicker, J.; Sánchez-Lopez, E.; Paz, C.; Ureña, J.; Verdaguer, E.; Auladell, C.; et al. Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2023, 24, 14177. https://doi.org/10.3390/ijms241814177
Olloquequi J, Ettcheto M, Cano A, Fortuna A, Bicker J, Sánchez-Lopez E, Paz C, Ureña J, Verdaguer E, Auladell C, et al. Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment. International Journal of Molecular Sciences. 2023; 24(18):14177. https://doi.org/10.3390/ijms241814177
Chicago/Turabian StyleOlloquequi, Jordi, Miren Ettcheto, Amanda Cano, Ana Fortuna, Joana Bicker, Elena Sánchez-Lopez, Cristian Paz, Jesús Ureña, Ester Verdaguer, Carme Auladell, and et al. 2023. "Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment" International Journal of Molecular Sciences 24, no. 18: 14177. https://doi.org/10.3390/ijms241814177
APA StyleOlloquequi, J., Ettcheto, M., Cano, A., Fortuna, A., Bicker, J., Sánchez-Lopez, E., Paz, C., Ureña, J., Verdaguer, E., Auladell, C., & Camins, A. (2023). Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment. International Journal of Molecular Sciences, 24(18), 14177. https://doi.org/10.3390/ijms241814177