

Humanization of the Reaction Specificity of Mouse Alox15b Inversely Modified the Susceptibility of Corresponding Knock-In Mice in Two Different Animal Inflammation Models
 and
and
Abstract
1. Introduction
2. Results
2.1. Alox15b Knock-in Mice Express an Arachidonic Acid 15-Lipoxygenating Alox15b Mutant Instead of the Arachidonic Acid 8-Lipoxygenating Wildtype Enzyme
2.2. In DSS Colitis Alox15b-KI Mice Experience a More Severe Loss of Body Weight and Recovered Less Rapidly
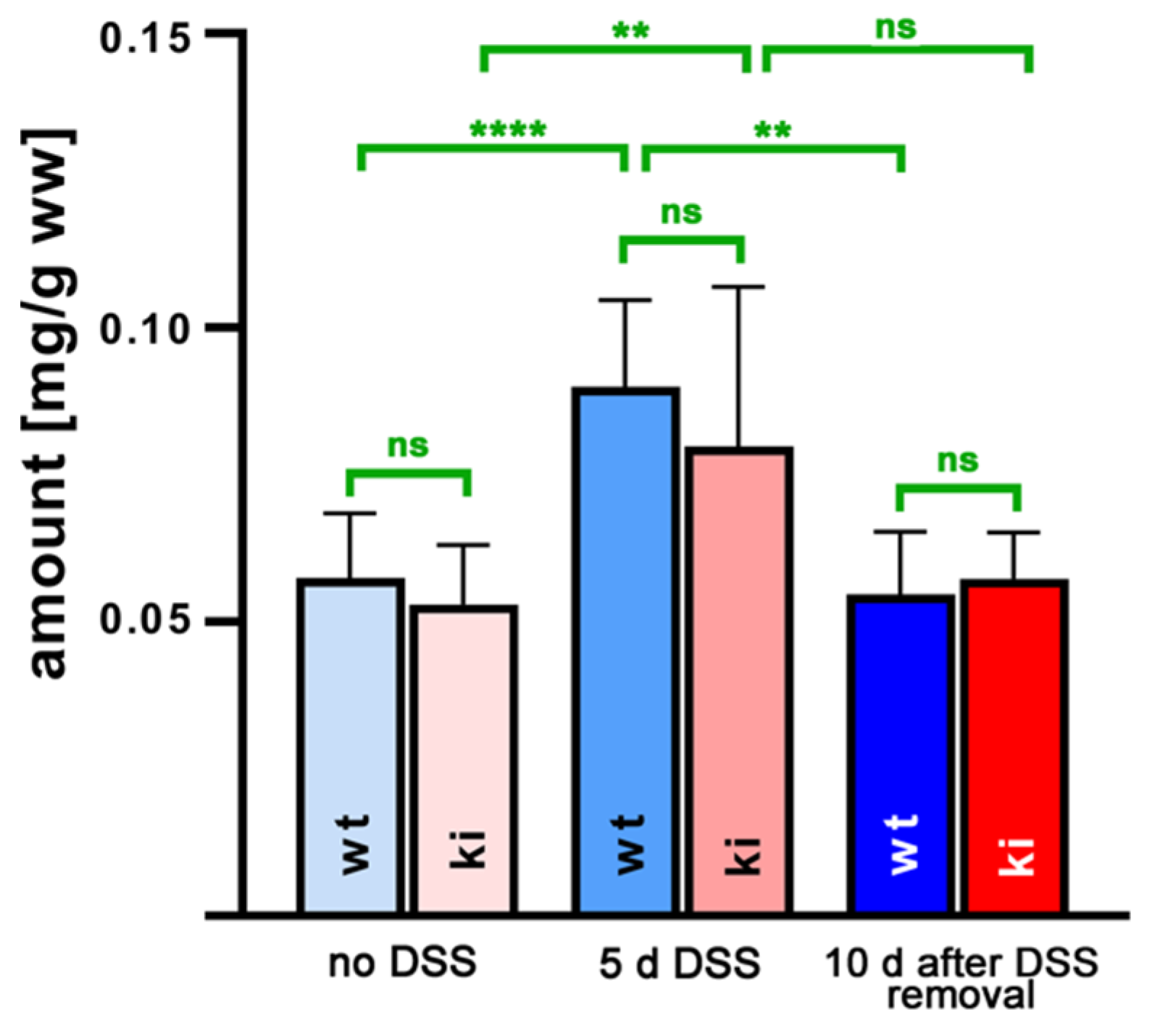
2.3. Oxilipin Profiles in Colon Tissues of Alox15b-KI Mice and Wildtype Controls during the Time-Course of DSS-Induced Colitis
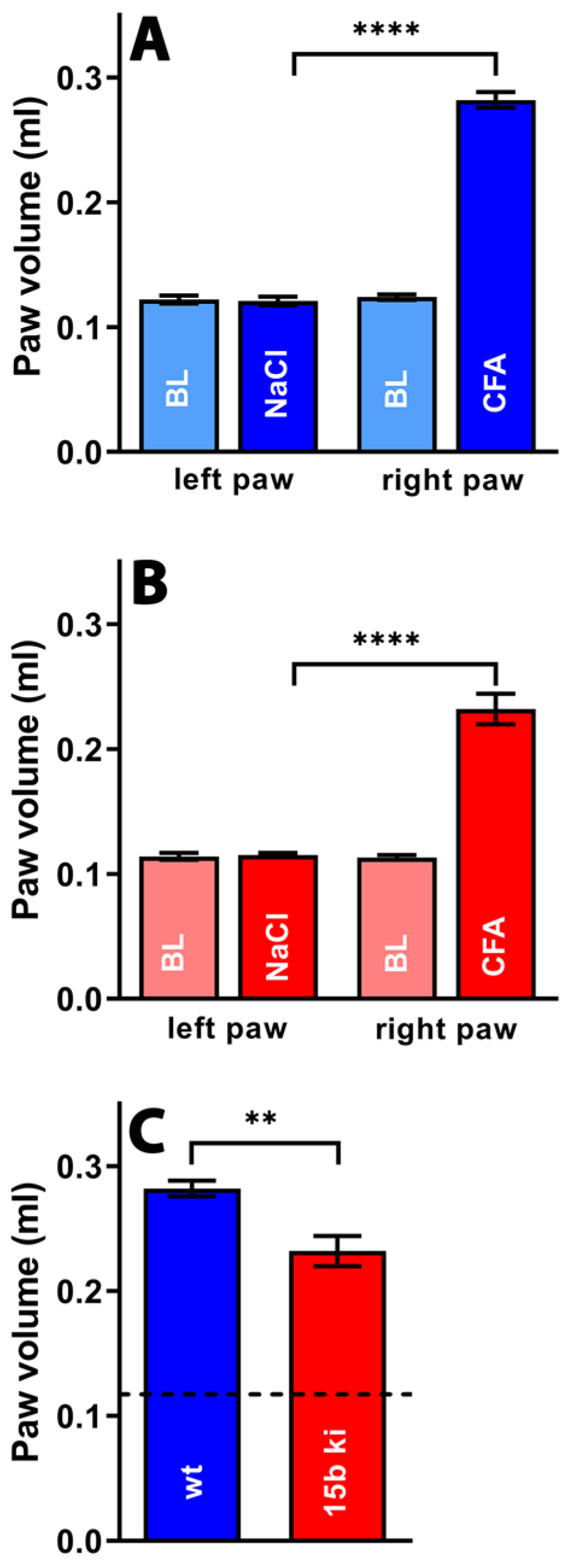
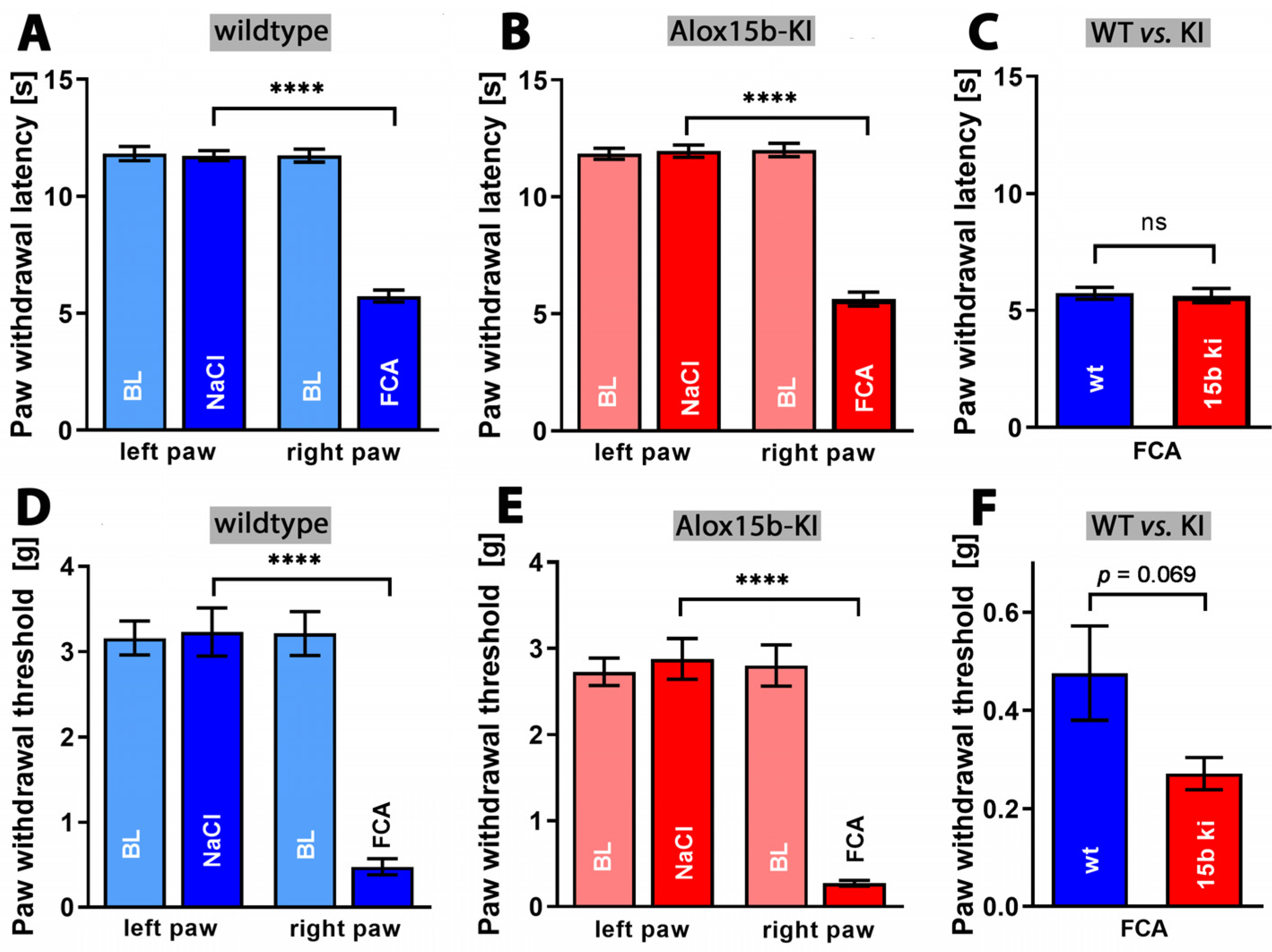
2.4. Alox15-Knock-in Mice Show Reduced Paw Swelling in the CFA-Induced Paw Inflammation Model
3. Discussion
3.1. Alox15-KI Mice Lose Significantly More Body Weight during the Time Course of DSS-Induced Colitis
3.2. Humanization of Alox15b Reaction Specificity Does Not Induce Pronounced Alterations in the Oxylipin Profile in Normal and Inflamed Colon Tissues
3.3. Humanization of Alox15b Reaction Specificity of Alox15b Partly Protected Mice in the Paw Edema Inflammation Model but Does Not Impact Pain Perception
4. Materials and Methods
4.1. Chemicals
4.2. Animals
4.3. Ex Vivo Activity Assay of Mouse Alox15b
4.4. Dextran Sulfate Sodium (DSS) Induced Experimental Colitis
4.5. Solid Phase Tissue Lipid Extraction
4.6. LC-MS/MS Based Oxylipidome Analyses
4.7. Complete Freund’s Adjuvant Induced Paw Inflammation Model
4.8. Statistics and Data Presentation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M. A calculated response: Control of inflammation by the innate immune system. J. Clin. Investig. 2008, 118, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Feehan, K.T.; Gilroy, D.W. Is Resolution the End of Inflammation? Trends Mol. Med. 2019, 25, 198–214. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2012, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Ebert, R.; Cumbana, R.; Lehmann, C.; Kutzner, L.; Toewe, A.; Ferreirós, N.; Parnham, M.J.; Schebb, N.H.; Steinhilber, D.; Kahnt, A.S. Long-term stimulation of toll-like receptor-2 and -4 upregulates 5-LO and 15-LO-2 expression thereby inducing a lipid mediator shift in human monocyte-derived macrophages. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158702. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Kuhn, H.; Heydeck, D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015, 573, 1–32. [Google Scholar] [CrossRef]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA 1997, 94, 6148–6152. [Google Scholar] [CrossRef]
- Funk, C.D.; Furci, L.; FitzGerald, G.A. Molecular cloning, primary structure, and expression of the human platelet/erythroleukemia cell 12-lipoxygenase. Proc. Natl. Acad. Sci. USA 1990, 87, 5638–5642. [Google Scholar] [CrossRef]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Biringer, R.G. The enzymology of human eicosanoid pathways: The lipoxygenase branches. Mol. Biol. Rep. 2020, 47, 7189–7207. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2015, 1851, 308–330. [Google Scholar] [CrossRef]
- Andreou, A.; Feussner, I. Lipoxygenases—Structure and reaction mechanism. Phytochemistry 2009, 70, 1504–1510. [Google Scholar] [CrossRef]
- Liu, M.; Yokomizo, T. The role of leukotrienes in allergic diseases. Allergol. Int. 2014, 64, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Clària, J.; Serhan, C.N. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2013, 19, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D.; Chen, X.-S.; Johnson, E.N.; Zhao, L. Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediat. 2002, 68–69, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Humeniuk, L.; Kozlov, N.; Roigas, S.; Adel, S.; Heydeck, D. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog. Lipid Res. 2018, 72, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Freire-Moar, J.; Alavi-Nassab, A.; Ng, M.; Mulkins, M.; Sigal, E. Cloning and characterization of a murine macrophage lipoxygenase. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1995, 1254, 112–116. [Google Scholar] [CrossRef]
- Sloane, D.L.; Dixon, R.A.; Craik, C.S.; Sigal, E. Expression of cloned human 15-lipoxygenase in eukaryotic and prokaryotic systems. Adv. Prostaglandin Thromboxane Leukot. Res. 1991, 21A, 5–8. [Google Scholar]
- Sloane, D.L.; Leung, R.; Craik, C.S.; Sigal, E. A primary determinant for lipoxygenase positional specificity. Nature 1991, 354, 149–152. [Google Scholar] [CrossRef]
- Vogel, R.; Jansen, C.; Roffeis, J.; Reddanna, P.; Forsell, P.; Claesson, H.-E.; Kuhn, H.; Walther, M. Applicability of the triad concept for the positional specificity of mammalian lipoxygenases. J. Biol. Chem. 2010, 285, 5369–5376. [Google Scholar] [CrossRef]
- Jisaka, M.; Kim, R.B.; Boeglin, W.E.; Brash, A.R. Identification of amino acid determinants of the positional specificity of mouse 8S-lipoxygenase and human 15S-lipoxygenase-2. J. Biol. Chem. 2000, 275, 1287–1293. [Google Scholar] [CrossRef]
- Schäfer, M.; Kakularam, K.R.; Reisch, F.; Rothe, M.; Stehling, S.; Heydeck, D.; Püschel, G.P.; Kuhn, H. Male Knock-in Mice Expressing an Arachidonic Acid Lipoxygenase 15B (Alox15B) with Humanized Reaction Specificity Are Prematurely Growth Arrested When Aging. Biomedicines 2022, 10, 1379. [Google Scholar] [CrossRef] [PubMed]
- Jisaka, M.; Kim, R.B.; Boeglin, W.E.; Nanney, L.B.; Brash, A.R. Molecular cloning and functional expression of a phorbol ester-inducible 8S-lipoxygenase from mouse skin. J. Biol. Chem. 1997, 272, 24410–24416. [Google Scholar] [CrossRef] [PubMed]
- Furstenberger, G.; Hagedorn, H.; Jacobi, T.; Besemfelder, E.; Stephan, M.; Lehmann, W.D.; Marks, F. Characterization of an 8-lipoxygenase activity induced by the phorbol ester tumor promoter 12-O-tetradecanoylphorbol-13-acetate in mouse skin in vivo. J. Biol. Chem. 1991, 266, 15738–15745. [Google Scholar] [CrossRef] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Kroschwald, S.; Chiu, C.-Y.; Heydeck, D.; Rohwer, N.; Gehring, T.; Seifert, U.; Lux, A.; Rothe, M.; Weylandt, K.-H.; Kuhn, H. Female mice carrying a defective Alox15 gene are protected from experimental colitis via sustained maintenance of the intestinal epithelial barrier function. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2018, 1863, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Chiu, C.; Huang, D.; Smyl, C.; Rothe, M.; Rund, K.M.; Schebb, N.H.; Kühn, H.; Weylandt, K. Omega-3 fatty acids protect from colitis via an Alox15-derived eicosanoid. FASEB J. 2021, 35, e21491. [Google Scholar] [CrossRef]
- Perše, M.; Cerar, A. Dextran sodium sulphate colitis mouse model: Traps and tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kühl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar]
- Endo, J.; Sano, M.; Isobe, Y.; Fukuda, K.; Kang, J.X.; Arai, H.; Arita, M. 18-HEPE, an n-3 fatty acid metabolite released by macrophages, prevents pressure overload–induced maladaptive cardiac remodeling. J. Exp. Med. 2014, 211, 1673–1687. [Google Scholar] [CrossRef]
- Kutzner, L.; Goloshchapova, K.; Rund, K.M.; Jübermann, M.; Blum, M.; Rothe, M.; Kirsch, S.F.; Schunck, W.-H.; Kühn, H.; Schebb, N.H. Human lipoxygenase isoforms form complex patterns of double and triple oxygenated compounds from eicosapentaenoic acid. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158806. [Google Scholar] [CrossRef]
- Lagarde, M. Editorial: Health Benefits of Docosahexaenoic Acid (DHA). Pharmacol. Res. 1999, 40, 205–206. [Google Scholar] [CrossRef]
- Li, J.; Pora, B.L.R.; Dong, K.; Hasjim, J. Health benefits of docosahexaenoic acid and its bioavailability: A review. Food Sci. Nutr. 2021, 9, 5229–5243. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J. Lipid Res. 2009, 50, S400–S405. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-K.; Lü, N.; Xu, Z.-Z.; Liu, T.; Serhan, C.N.; Ji, R.-R. Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J. Neurosci. 2011, 31, 15072–15085. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2014, 1851, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Pillinger, M.H. 15d-PGJ2: The anti-inflammatory prostaglandin? Clin. Immunol. 2005, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, C.; Wu, J. 15-Deoxy-∆-12,14-Prostaglandin J2 (15d-PGJ2), an Endogenous Ligand of PPAR-γ: Function and Mechanism. PPAR Res. 2019, 2019, 7242030. [Google Scholar] [CrossRef]
- Soares, A.; Nosjean, O.; Cozzone, D.; D’orazio, D.; Becchi, M.; Guichardant, M.; Ferry, G.; Boutin, J.; Lagarde, M.; Géloën, A. Covalent binding of 15-deoxy-delta12,14-prostaglandin J2 to PPARγ. Biochem. Biophys. Res. Commun. 2005, 337, 521–525. [Google Scholar] [CrossRef]
- Morris, C.J. Carrageenan-induced paw edema in the rat and mouse. Methods Mol. Biol. 2003, 225, 115–121. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Cheah, M.; Fawcett, J.W.; Andrews, M.R. Assessment of Thermal Pain Sensation in Rats and Mice Using the Hargreaves Test. Bio-Protocol 2017, 7, e2506. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.L.; Hansen, R.B.; Storm, M.A.; Olesen, J.; Hansen, T.F.; Ossipov, M.; Izarzugaza, J.M.G.; Porreca, F.; Kristensen, D.M. Von Frey testing revisited: Provision of an online algorithm for improved accuracy of 50% thresholds. Eur. J. Pain 2019, 24, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Blum, M.; Hoff, U.; Wesser, T.; Fechner, M.; Westphal, C.; Gürgen, D.; Catar, R.; Philippe, A.; Wu, K.; et al. Renal Ischemia/Reperfusion Injury in Soluble Epoxide Hydrolase-Deficient Mice. PLoS ONE 2016, 11, e0145645. [Google Scholar] [CrossRef]
- Claesson, H.-E.; Odlander, B.; Jakobsson, P.-J. Leukotriene B4 in the immune system. Int. J. Immunopharmacol. 1992, 14, 441–449. [Google Scholar] [CrossRef]
- Ford-Hutchinson, A.W. Leukotriene B4 in inflammation. Crit. Rev. Immunol. 1990, 10, 1–12. [Google Scholar]
- Haeggström, J.; Wetterholm, A. Enzymes and receptors in the leukotriene cascade. Cell. Mol. Life Sci. 2002, 59, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Lim, J.Y.; Hwang, S.W. Resolvins: Endogenously-Generated Potent Painkilling Substances and their Therapeutic Perspectives. Curr. Neuropharmacol. 2013, 11, 664–676. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef]
- Chen, X.-S.; Shelter, J.R.; Johnson, E.N.; Funk, C.D. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature 1994, 372, 179–182. [Google Scholar] [CrossRef]
- Marbach-Breitrück, E.; Rohwer, N.; Infante-Duarte, C.; Romero-Suarez, S.; Labuz, D.; Machelska, H.; Kutzner, L.; Schebb, N.H.; Rothe, M.; Reddanna, P.; et al. Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice. Metabolites 2021, 11, 698. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Group | Metabolite (ng Metabolite/g Tissue Protein) | ||
|---|---|---|---|---|
| 5S,12S-diHETE | 8S,15S-diHETE | 10S,17S-diHDHA | ||
| Wildtype (WT) | Day 0 | 38.3 ± 8.3 | 8.6 ± 1.2 | 24.3 ± 4.9 |
| Day 5 | 99.7 ± 32.7 | 16.4 ± 4.2 | 87.2 ± 41.3 | |
| Day 15 | 31.3 ± 8.5 | 9.1 ± 3.0 | 16.4 ± 7.7 | |
| Knock-in (KI) | Day 0 | 35.2 ± 10.4 | 6.8 ± 1.0 | 14.6 ± 2.5 |
| Day 5 | 109.3 ± 79.3 | 18.9 ± 6.8 | 62.1 ± 33.4 | |
| Day 15 | 32.2 ± 11.3 | 11.0 ± 0.7 | 12.4 ± 2.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schäfer, M.; Reisch, F.; Labuz, D.; Machelska, H.; Stehling, S.; Püschel, G.P.; Rothe, M.; Heydeck, D.; Kuhn, H. Humanization of the Reaction Specificity of Mouse Alox15b Inversely Modified the Susceptibility of Corresponding Knock-In Mice in Two Different Animal Inflammation Models. Int. J. Mol. Sci. 2023, 24, 11034. https://doi.org/10.3390/ijms241311034
Schäfer M, Reisch F, Labuz D, Machelska H, Stehling S, Püschel GP, Rothe M, Heydeck D, Kuhn H. Humanization of the Reaction Specificity of Mouse Alox15b Inversely Modified the Susceptibility of Corresponding Knock-In Mice in Two Different Animal Inflammation Models. International Journal of Molecular Sciences. 2023; 24(13):11034. https://doi.org/10.3390/ijms241311034
Chicago/Turabian StyleSchäfer, Marjann, Florian Reisch, Dominika Labuz, Halina Machelska, Sabine Stehling, Gerhard P. Püschel, Michael Rothe, Dagmar Heydeck, and Hartmut Kuhn. 2023. "Humanization of the Reaction Specificity of Mouse Alox15b Inversely Modified the Susceptibility of Corresponding Knock-In Mice in Two Different Animal Inflammation Models" International Journal of Molecular Sciences 24, no. 13: 11034. https://doi.org/10.3390/ijms241311034
APA StyleSchäfer, M., Reisch, F., Labuz, D., Machelska, H., Stehling, S., Püschel, G. P., Rothe, M., Heydeck, D., & Kuhn, H. (2023). Humanization of the Reaction Specificity of Mouse Alox15b Inversely Modified the Susceptibility of Corresponding Knock-In Mice in Two Different Animal Inflammation Models. International Journal of Molecular Sciences, 24(13), 11034. https://doi.org/10.3390/ijms241311034