
Harnessing Oxylipins and Inflammation Modulation for Prevention and Treatment of Colorectal Cancer

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. PUFAs and Enzymatically Formed Oxylipins
3. Increased Dietary Omega-3 PUFAs Might Lower Colorectal Cancer Risk
4. Nonsteroidal Anti-Inflammatory Drugs Prevent Colorectal Cancer
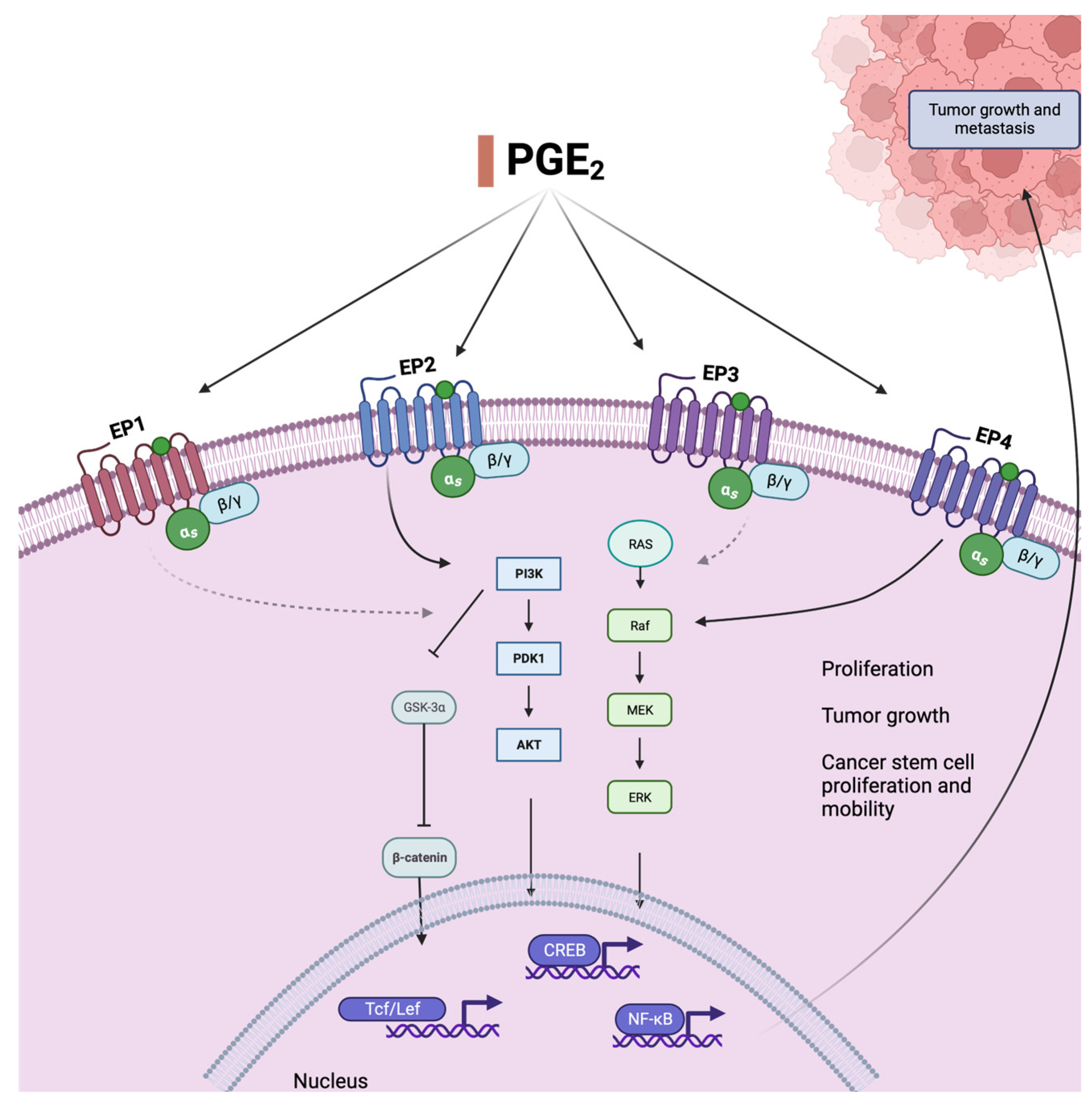
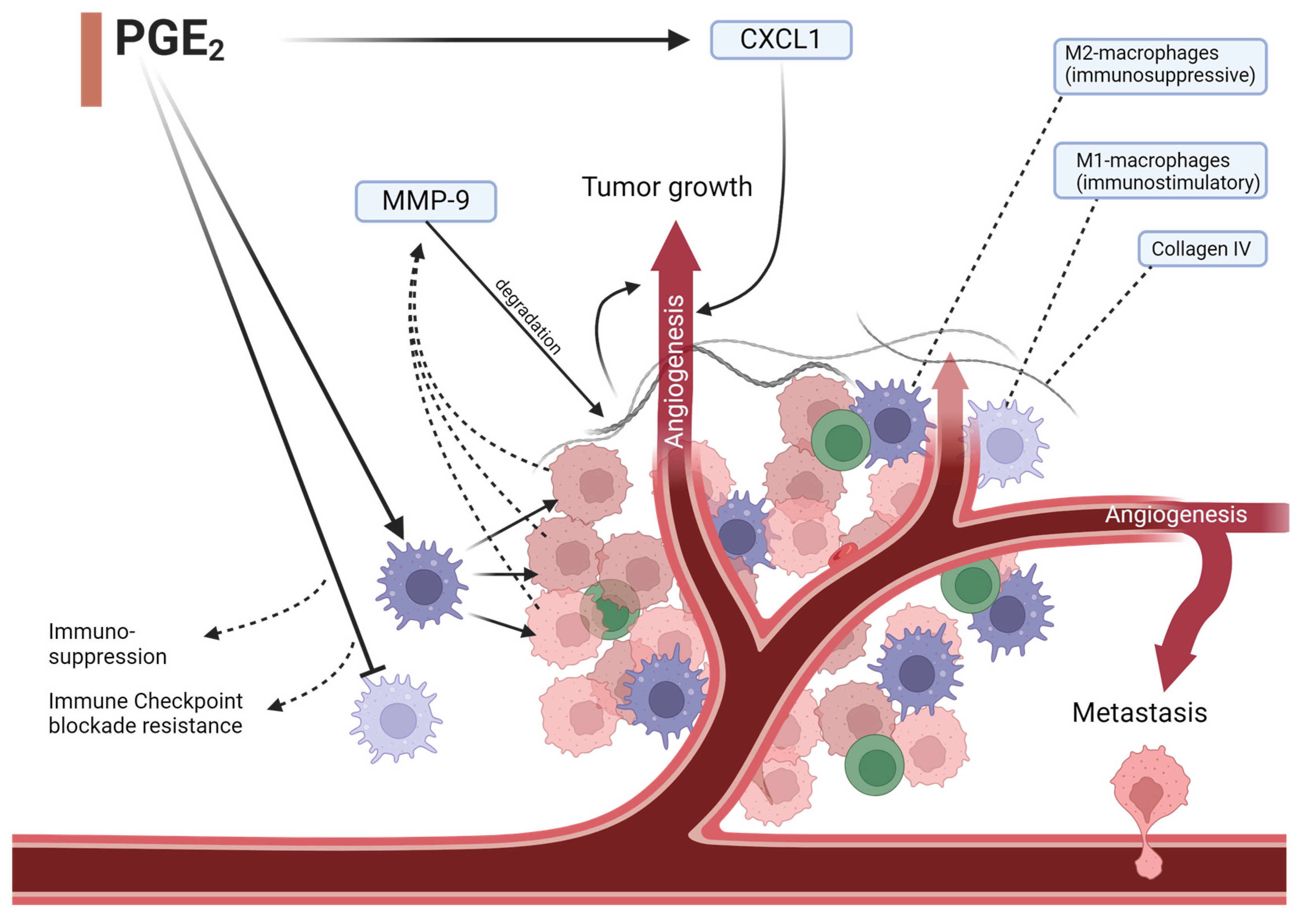
5. Lipid Mediators and Colorectal Cancer
6. Chemoprevention Strategies in Colorectal Cancer—Beyond NSAIDS
7. Concomitant Medications and Immune Therapy
8. Conclusions and Perspectives
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- Strum, W.B. Colorectal Adenomas. N. Engl. J. Med. 2016, 374, 1065–1075. [Google Scholar] [CrossRef]
- Lichtenstern, C.R.; Ngu, R.K.; Shalapour, S.; Karin, M. Immunotherapy, Inflammation and Colorectal Cancer. Cells 2020, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Calder, P.C. Eicosanoids. Essays Biochem. 2020, 64, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Hawley, M.; Holinstat, M. The expansive role of oxylipins on platelet biology. J. Mol. Med. 2017, 95, 575–588. [Google Scholar] [CrossRef]
- Kulkarni, P.S.; Srinivasan, B.D. Eicosapentaenoic acid metabolism in human and rabbit anterior uvea. Prostaglandins 1986, 31, 1159–1164. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta 2015, 1851, 397–413. [Google Scholar] [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.A. The 1982 Nobel Prize in Physiology or Medicine. Science 1982, 218, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Bartram, H.P.; Gostner, A.; Scheppach, W.; Reddy, B.S.; Rao, C.V.; Dusel, G.; Richter, F.; Richter, A.; Kasper, H. Effects of fish oil on rectal cell proliferation, mucosal fatty acids, and prostaglandin E2 release in healthy subjects. Gastroenterology 1993, 105, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Anti, M.; Marra, G.; Armelao, F.; Bartoli, G.M.; Ficarelli, R.; Percesepe, A.; De Vitis, I.; Maria, G.; Sofo, L.; Rapaccini, G.L.; et al. Effect of ω-3 fatty acids on rectal mucosal cell proliferation in subjects at risk for colon cancer. Gastroenterology 1992, 103, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N.; Chavarro, J.E.; Lee, I.M.; Willett, W.C.; Ma, J. A 22-year prospective study of fish, n-3 fatty acid intake, and colorectal cancer risk in men. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wu, K.; Ogino, S.; Giovannucci, E.L.; Chan, A.T.; Song, M. Association Between Risk Factors for Colorectal Cancer and Risk of Serrated Polyps and Conventional Adenomas. Gastroenterology 2018, 155, 355–373.e318. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hancock, R.L.; Mohammadpour, H.; McGregor, B.; Manalo, P.; Khaiboullina, S.; Hall, M.R.; Pardini, L.; Pardini, R.S. Influence of omega-3 fatty acids on the growth of human colon carcinoma in nude mice. Cancer Lett. 2002, 187, 169–177. [Google Scholar] [CrossRef]
- Piazzi, G.; D’Argenio, G.; Prossomariti, A.; Lembo, V.; Mazzone, G.; Candela, M.; Biagi, E.; Brigidi, P.; Vitaglione, P.; Fogliano, V.; et al. Eicosapentaenoic acid free fatty acid prevents and suppresses colonic neoplasia in colitis-associated colorectal cancer acting on Notch signaling and gut microbiota. Int. J. Cancer 2014, 135, 2004–2013. [Google Scholar] [CrossRef]
- Nowak, J.; Weylandt, K.H.; Habbel, P.; Wang, J.; Dignass, A.; Glickman, J.N.; Kang, J.X. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis 2007, 28, 1991–1995. [Google Scholar] [CrossRef]
- Tu, M.; Wang, W.; Zhang, G.; Hammock, B.D. ω-3 Polyunsaturated Fatty Acids on Colonic Inflammation and Colon Cancer: Roles of Lipid-Metabolizing Enzymes Involved. Nutrients 2020, 12, 3301. [Google Scholar] [CrossRef]
- Hull, M.A.; Ow, P.L.; Ruddock, S.; Brend, T.; Smith, A.F.; Marshall, H.; Song, M.; Chan, A.T.; Garrett, W.S.; Yilmaz, O.; et al. Randomised, placebo-controlled, phase 3 trial of the effect of the omega-3 polyunsaturated fatty acid eicosapentaenoic acid (EPA) on colorectal cancer recurrence and survival after surgery for resectable liver metastases: EPA for Metastasis Trial 2 (EMT2) study protocol. BMJ Open 2023, 13, e077427. [Google Scholar] [CrossRef]
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: Case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Nan, H.; Hutter, C.M.; Lin, Y.; Jacobs, E.J.; Ulrich, C.M.; White, E.; Baron, J.A.; Berndt, S.I.; Brenner, H.; Butterbach, K.; et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA 2015, 313, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Fowkes, F.G.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Friis, S.; Riis, A.H.; Erichsen, R.; Baron, J.A.; Sørensen, H.T. Low-Dose Aspirin or Nonsteroidal Anti-inflammatory Drug Use and Colorectal Cancer Risk: A Population-Based, Case-Control Study. Ann. Intern. Med. 2015, 163, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Similar reductions in the risk of human colon cancer by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer 2008, 8, 237. [Google Scholar] [CrossRef]
- Bambace, N.M.; Holmes, C.E. The platelet contribution to cancer progression. J. Thromb. Haemost. 2011, 9, 237–249. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Endogenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S200–S215. [Google Scholar] [CrossRef]
- Weylandt, K.H.; Chiu, C.Y.; Gomolka, B.; Waechter, S.F.; Wiedenmann, B. Omega-3 fatty acids and their lipid mediators: Towards an understanding of resolvin and protectin formation. Prostaglandins Other Lipid Mediat. 2012, 97, 73–82. [Google Scholar] [CrossRef]
- Fuller, H.; Race, A.D.; Fenton, H.; Burke, L.; Downing, A.; Williams, E.A.; Rees, C.J.; Brown, L.C.; Loadman, P.M.; Hull, M.A. Plasma and rectal mucosal oxylipin levels during aspirin and eicosapentaenoic acid treatment in the seAFOod polyp prevention trial. Prostaglandins Leukot. Essent. Fat. Acids 2023, 192, 102570. [Google Scholar] [CrossRef]
- Rohwer, N.; Kühl, A.A.; Ostermann, A.I.; Hartung, N.M.; Schebb, N.H.; Zopf, D.; McDonald, F.M.; Weylandt, K.H. Effects of chronic low-dose aspirin treatment on tumor prevention in three mouse models of intestinal tumorigenesis. Cancer Med. 2020, 9, 2535–2550. [Google Scholar] [CrossRef]
- Bibbins-Domingo, K. Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2016, 164, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, S.P.; O’Keefe, L.R.; Grossman, E.S.; Maciosek, M.V. U.S. Preventive Services Task Force Evidence Syntheses, formerly Systematic Evidence Reviews. In Aspirin Use to Prevent Cardiovascular Disease and Colorectal Cancer: An Updated Decision Analysis for the U.S. Preventive Services Task Force; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2022. [Google Scholar]
- Chan, A.T. Aspirin and the USPSTF-What About Cancer? JAMA Oncol. 2022, 8, 1392–1394. [Google Scholar] [CrossRef]
- He, Y.; Van’t Veer, L.J.; Mikolajewska-Hanclich, I.; van Velthuysen, M.L.; Zeestraten, E.C.; Nagtegaal, I.D.; van de Velde, C.J.; Marijnen, C.A. PIK3CA mutations predict local recurrences in rectal cancer patients. Clin. Cancer Res. 2009, 15, 6956–6962. [Google Scholar] [CrossRef]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Qian, Z.R.; Baba, Y.; Shima, K.; et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Yen, J.-H.; Kocieda, V.P.; Jing, H.; Ganea, D. Prostaglandin E2 Induces Matrix Metalloproteinase 9 Expression in Dendritic Cells through Two Independent Signaling Pathways Leading to Activator Protein 1 (AP-1) Activation. J. Biol. Chem. 2011, 286, 38913–38923. [Google Scholar] [CrossRef] [PubMed]
- De Keijzer, S.; Meddens, M.B.M.; Torensma, R.; Cambi, A. The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle. Int. J. Mol. Sci. 2013, 14, 6542–6555. [Google Scholar] [CrossRef]
- Hsu, H.H.; Lin, Y.M.; Shen, C.Y.; Shibu, M.A.; Li, S.Y.; Chang, S.H.; Lin, C.C.; Chen, R.J.; Viswanadha, V.P.; Shih, H.N.; et al. Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1132. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H.; Brown, J.; Daikoku, T.; Ning, W.; Shi, Q.; Richmond, A.; Strieter, R.; Dey, S.K.; DuBois, R.N. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med. 2006, 203, 941–951. [Google Scholar] [CrossRef]
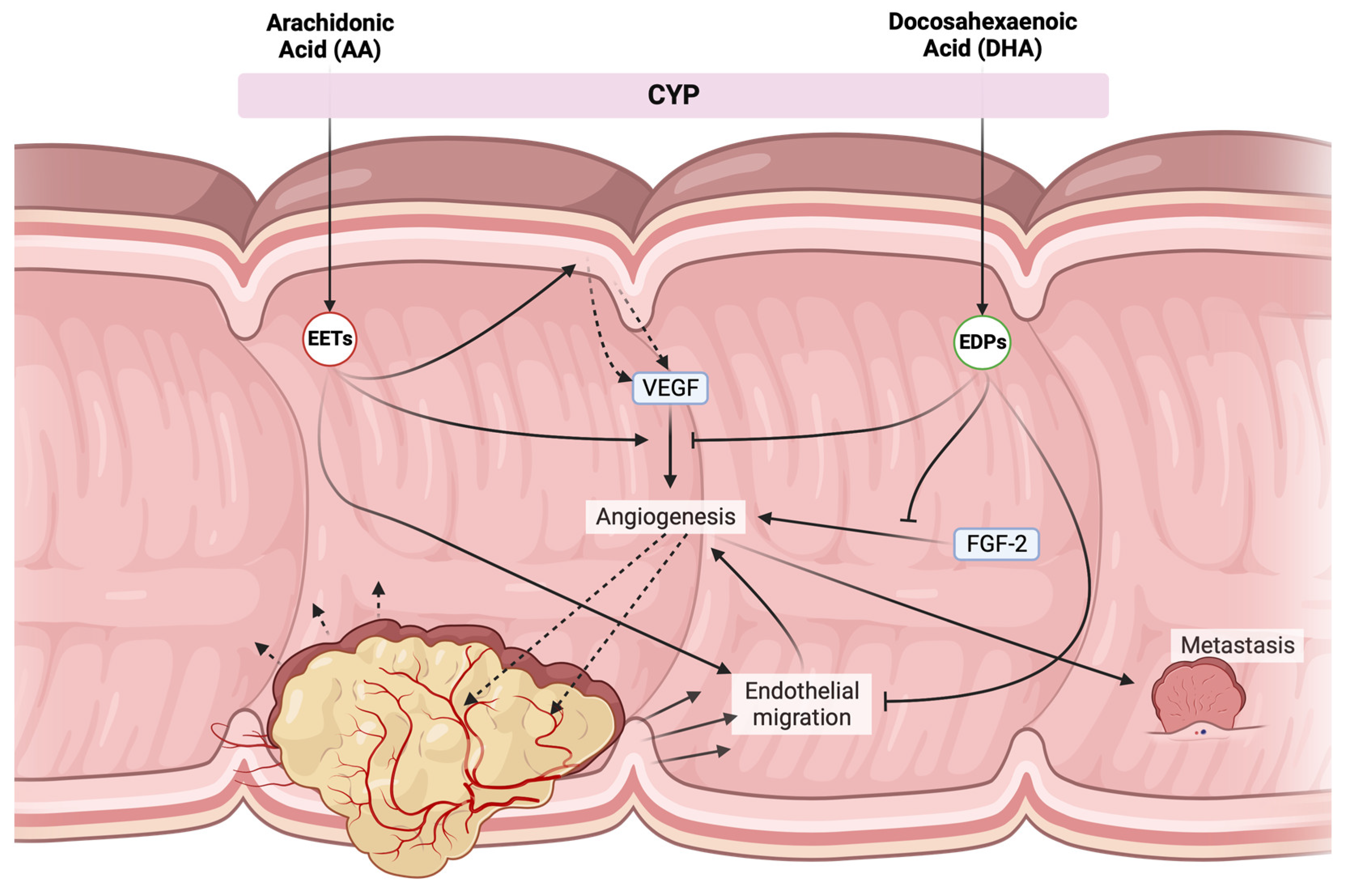
- Szymczak, M.; Murray, M.; Petrovic, N. Modulation of angiogenesis by ω-3 polyunsaturated fatty acids is mediated by cyclooxygenases. Blood 2008, 111, 3514–3521. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Yu, W.; He, J.; Liu, W.; Yang, J.; Lin, X.; Zhang, Y.; Wang, X.; Jiang, W.; Luo, J.; et al. Reprogramming immunosuppressive myeloid cells facilitates immunotherapy for colorectal cancer. EMBO Mol. Med. 2021, 13, e12798. [Google Scholar] [CrossRef] [PubMed]
- Grabocka, E.; Bar-Sagi, D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 2016, 167, 1803–1813.e12. [Google Scholar] [CrossRef]
- Dinu, D.; Dobre, M.; Panaitescu, E.; Birla, R.; Iosif, C.; Hoara, P.; Caragui, A.; Boeriu, M.; Constantinoiu, S.; Ardeleanu, C. Prognostic significance of KRAS gene mutations in colorectal cancer—Preliminary study. J. Med. Life 2014, 7, 581–587. [Google Scholar]
- Li, H.; Liu, K.; Boardman, L.A.; Zhao, Y.; Wang, L.; Sheng, Y.; Oi, N.; Limburg, P.J.; Bode, A.M.; Dong, Z. Circulating Prostaglandin Biosynthesis in Colorectal Cancer and Potential Clinical Significance. EBioMedicine 2015, 2, 165–171. [Google Scholar] [CrossRef]
- Sakai, H.; Suzuki, T.; Takahashi, Y.; Ukai, M.; Tauchi, K.; Fujii, T.; Horikawa, N.; Minamimura, T.; Tabuchi, Y.; Morii, M.; et al. Upregulation of thromboxane synthase in human colorectal carcinoma and the cancer cell proliferation by thromboxane A2. FEBS Lett. 2006, 580, 3368–3374. [Google Scholar] [CrossRef]
- Shimizu, T.; Fujii, T.; Takahashi, Y.; Takahashi, Y.; Suzuki, T.; Ukai, M.; Tauchi, K.; Horikawa, N.; Tsukada, K.; Sakai, H. Up-regulation of Kv7.1 channels in thromboxane A2-induced colonic cancer cell proliferation. Pflugers Arch. 2014, 466, 541–548. [Google Scholar] [CrossRef]
- Stadler, S.; Nguyen, C.H.; Schachner, H.; Milovanovic, D.; Holzner, S.; Brenner, S.; Eichsteininger, J.; Stadler, M.; Senfter, D.; Krenn, L.; et al. Colon cancer cell-derived 12(S)-HETE induces the retraction of cancer-associated fibroblast via MLC2, RHO/ROCK and Ca2+ signalling. Cell Mol. Life Sci. 2017, 74, 1907–1921. [Google Scholar] [CrossRef]
- Dong, T.; Dave, P.; Yoo, E.; Ebright, B.; Ahluwalia, K.; Zhou, E.; Asante, I.; Salimova, M.; Pei, H.; Lin, T.; et al. NAP1051, a Lipoxin A4 Biomimetic Analogue, Demonstrates Antitumor Activity Against the Tumor Microenvironment. Mol. Cancer Ther. 2021, 20, 2384–2397. [Google Scholar] [CrossRef]
- Liu, H.; Zeng, J.; Huang, W.; Xu, Q.; Ye, D.; Sun, R.; Zhang, D. Colorectal Cancer Is Associated with a Deficiency of Lipoxin A4, an Endogenous Anti-inflammatory Mediator. J. Cancer 2019, 10, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Bodduluri, S.R.; Mathis, S.; Maturu, P.; Krishnan, E.; Satpathy, S.R.; Chilton, P.M.; Mitchell, T.C.; Lira, S.; Locati, M.; Mantovani, A.; et al. Mast Cell-Dependent CD8+ T-cell Recruitment Mediates Immune Surveillance of Intestinal Tumors in ApcMin/+ Mice. Cancer Immunol. Res. 2018, 6, 332–347. [Google Scholar] [CrossRef]
- Satapathy, S.R.; Topi, G.; Osman, J.; Hellman, K.; Ek, F.; Olsson, R.; Sime, W.; Mehdawi, L.M.; Sjolander, A. Tumour suppressor 15-hydroxyprostaglandin dehydrogenase induces differentiation in colon cancer via GLI1 inhibition. Oncogenesis 2020, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Shureiqi, I.; Wojno, K.J.; Poore, J.A.; Reddy, R.G.; Moussalli, M.J.; Spindler, S.A.; Greenson, J.K.; Normolle, D.; Hasan, A.A.; Lawrence, T.S.; et al. Decreased 13-S-hydroxyoctadecadienoic acid levels and 15-lipoxygenase-1 expression in human colon cancers. Carcinogenesis 1999, 20, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Nixon, J.B.; Kim, K.S.; Lamb, P.W.; Bottone, F.G.; Eling, T.E. 15-Lipoxygenase-1 has anti-tumorigenic effects in colorectal cancer. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 7–15. [Google Scholar] [CrossRef]
- Lee, H.N.; Choi, Y.S.; Kim, S.H.; Zhong, X.; Kim, W.; Park, J.S.; Saeidi, S.; Han, B.W.; Kim, N.; Lee, H.S.; et al. Resolvin D1 suppresses inflammation-associated tumorigenesis in the colon by inhibiting IL-6-induced mitotic spindle abnormality. FASEB J. 2021, 35, e21432. [Google Scholar] [CrossRef]
- Zhong, X.; Lee, H.N.; Surh, Y.J. RvD1 inhibits TNFα-induced c-Myc expression in normal intestinal epithelial cells and destabilizes hyper-expressed c-Myc in colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 316–323. [Google Scholar] [CrossRef]
- Zhang, G.; Panigrahy, D.; Mahakian, L.M.; Yang, J.; Liu, J.Y.; Stephen Lee, K.S.; Wettersten, H.I.; Ulu, A.; Hu, X.; Tam, S.; et al. Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 6530–6535. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Edin, M.L.; Wang, Y.; Luo, Y.; Wan, D.; Yang, H.; Song, C.Q.; Xue, W.; Sanidad, K.Z.; et al. Targeted Metabolomics Identifies the Cytochrome P450 Monooxygenase Eicosanoid Pathway as a Novel Therapeutic Target of Colon Tumorigenesis. Cancer Res. 2019, 79, 1822–1830. [Google Scholar] [CrossRef]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnés, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 2012, 122, 178–191. [Google Scholar] [CrossRef]
- Matyori, A.; Brown, C.P.; Ali, A.; Sherbeny, F. Statins utilization trends and expenditures in the U.S. before and after the implementation of the 2013 ACC/AHA guidelines. Saudi Pharm. J. 2023, 31, 795–800. [Google Scholar] [CrossRef]
- Poynter, J.N.; Gruber, S.B.; Higgins, P.D.; Almog, R.; Bonner, J.D.; Rennert, H.S.; Low, M.; Greenson, J.K.; Rennert, G. Statins and the risk of colorectal cancer. N. Engl. J. Med. 2005, 352, 2184–2192. [Google Scholar] [CrossRef]
- Farwell, W.R.; Scranton, R.E.; Lawler, E.V.; Lew, R.A.; Brophy, M.T.; Fiore, L.D.; Gaziano, J.M. The association between statins and cancer incidence in a veterans population. J. Natl. Cancer Inst. 2008, 100, 134–139. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Napoli, C.; Lerman, A. Statin effects beyond lipid lowering—Are they clinically relevant? Eur. Heart J. 2003, 24, 225–248. [Google Scholar] [CrossRef]
- Gazzerro, P.; Proto, M.C.; Gangemi, G.; Malfitano, A.M.; Ciaglia, E.; Pisanti, S.; Santoro, A.; Laezza, C.; Bifulco, M. Pharmacological actions of statins: A critical appraisal in the management of cancer. Pharmacol. Rev. 2012, 64, 102–146. [Google Scholar] [CrossRef] [PubMed]
- Demierre, M.F.; Higgins, P.D.; Gruber, S.B.; Hawk, E.; Lippman, S.M. Statins and cancer prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Rathinam, R.; Berrier, A.; Alahari, S.K. Role of Rho GTPases and their regulators in cancer progression. Front. Biosci. (Landmark Ed.) 2011, 16, 2561–2571. [Google Scholar] [CrossRef]
- Duncan, R.E.; El-Sohemy, A.; Archer, M.C. Mevalonate promotes the growth of tumors derived from human cancer cells in vivo and stimulates proliferation in vitro with enhanced cyclin-dependent kinase-2 activity. J. Biol. Chem. 2004, 279, 33079–33084. [Google Scholar] [CrossRef] [PubMed]
- Ascer, E.; Bertolami, M.C.; Venturinelli, M.L.; Buccheri, V.; Souza, J.; Nicolau, J.C.; Ramires, J.A.; Serrano, C.V., Jr. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis 2004, 177, 161–166. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Mausner-Fainberg, K.; Luboshits, G.; Mor, A.; Maysel-Auslender, S.; Rubinstein, A.; Keren, G.; George, J. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis 2008, 197, 829–839. [Google Scholar] [CrossRef]
- Mosheimer, B.A.; Kaneider, N.C.; Feistritzer, C.; Djanani, A.; Sturn, D.H.; Patsch, J.R.; Wiedermann, C.J. CD40-ligand-dependent induction of COX-2 gene expression in endothelial cells by activated platelets: Inhibitory effects of atorvastatin. Blood Coagul. Fibrinolysis 2005, 16, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Margaritis, M.; Lee, R.; Channon, K.; Antoniades, C. Statins as anti-inflammatory agents in atherogenesis: Molecular mechanisms and lessons from the recent clinical trials. Curr. Pharm. Des. 2012, 18, 1519–1530. [Google Scholar] [CrossRef]
- Davignon, J.; Jacob, R.F.; Mason, R.P. The antioxidant effects of statins. Coron. Artery Dis. 2004, 15, 251–258. [Google Scholar] [CrossRef]
- Elewa, H.F.; El-Remessy, A.B.; Somanath, P.R.; Fagan, S.C. Diverse effects of statins on angiogenesis: New therapeutic avenues. Pharmacotherapy 2010, 30, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.J.; Lob, S.; Lindau, D.; Horzer, H.; Guckel, B.; Klein, G.; Glatzle, J.; Rammensee, H.G.; Brucher, B.L.; Konigsrainer, A. Simvastatin reduces tumor cell adhesion to human peritoneal mesothelial cells by decreased expression of VCAM-1 and β1 integrin. Int. J. Oncol. 2011, 39, 1593–1600. [Google Scholar] [CrossRef]
- Tanaka, S.; Ishihara, N.; Suzuki, S.; Watanabe, Y.; Nagayama, D.; Yamaguchi, T.; Ohira, M.; Saiki, A.; Tanaka, T.; Tatsuno, I. Fatty acid desaturase 2 is up-regulated by the treatment with statin through geranylgeranyl pyrophosphate-dependent Rho kinase pathway in HepG2 cells. Sci. Rep. 2019, 9, 10009. [Google Scholar] [CrossRef]
- Wang, C.; Enssle, J.; Pietzner, A.; Schmöcker, C.; Weiland, L.; Ritter, O.; Jaensch, M.; Elbelt, U.; Pagonas, N.; Weylandt, K.H. Essential Polyunsaturated Fatty Acids in Blood from Patients with and without Catheter-Proven Coronary Artery Disease. Int. J. Mol. Sci. 2022, 23, 766. [Google Scholar] [CrossRef] [PubMed]
- Garshick, M.S.; Block, R.; Drenkova, K.; Tawil, M.; James, G.; Brenna, J.T. Statin therapy upregulates arachidonic acid status via enhanced endogenous synthesis in patients with plaque psoriasis. Prostaglandins Leukot. Essent. Fat. Acids 2022, 180, 102428. [Google Scholar] [CrossRef] [PubMed]
- Gottschall, H.; Schmocker, C.; Hartmann, D.; Rohwer, N.; Rund, K.; Kutzner, L.; Nolte, F.; Ostermann, A.I.; Schebb, N.H.; Weylandt, K.H. Aspirin alone and combined with a statin suppresses eicosanoid formation in human colon tissue. J. Lipid Res. 2018, 59, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Jara-Gutiérrez, Á.; Baladrón, V. The Role of Prostaglandins in Different Types of Cancer. Cells 2021, 10, 1487. [Google Scholar] [CrossRef]
- Larsson, S.C.; Orsini, N.; Wolk, A. Diabetes mellitus and risk of colorectal cancer: A meta-analysis. J. Natl. Cancer Inst. 2005, 97, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.W.; Jiang, A.A.; Toh, E.M.S.; Ng, C.H.; Ong, Z.H.; Peng, S.; Tham, H.Y.; Sundar, R.; Chong, C.S.; Khoo, C.M. Metformin and colorectal cancer: A systematic review, meta-analysis and meta-regression. Int. J. Colorectal Dis. 2020, 35, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-w.; Choi, E.-A.; Kim, Y.-S.; Kim, Y.; You, H.-S.; Han, Y.-E.; Kim, H.-S.; Bae, Y.-J.; Kim, J.; Kang, H.-T. Metformin usage and the risk of colorectal cancer: A national cohort study. Int. J. Color. Dis. 2021, 36, 303–310. [Google Scholar] [CrossRef]
- Liu, Q.; Yuan, W.; Tong, D.; Liu, G.; Lan, W.; Zhang, D.; Xiao, H.; Zhang, Y.; Huang, Z.; Yang, J.; et al. Metformin represses bladder cancer progression by inhibiting stem cell repopulation via COX2/PGE2/STAT3 axis. Oncotarget 2016, 7, 28235–28246. [Google Scholar] [CrossRef] [PubMed]
- Dahabiyeh, L.A.; Mujammami, M.; Arafat, T.; Benabdelkamel, H.; Alfadda, A.A.; Abdel Rahman, A.M. A Metabolic Pattern in Healthy Subjects Given a Single Dose of Metformin: A Metabolomics Approach. Front. Pharmacol. 2021, 12, 705932. [Google Scholar] [CrossRef]
- Dahabiyeh, L.A.; Mujammami, M.; AlMalki, R.H.; Arafat, T.; Benabdelkamel, H.; Alfadda, A.A.; Abdel Rahman, A.M. Lipids Alterations Associated with Metformin in Healthy Subjects: An Investigation Using Mass Spectrometry Shotgun Approach. Int. J. Mol. Sci. 2022, 23, 11478. [Google Scholar] [CrossRef]
- Guo, Z.; Sevrioukova, I.F.; Denisov, I.G.; Zhang, X.; Chiu, T.L.; Thomas, D.G.; Hanse, E.A.; Cuellar, R.A.D.; Grinkova, Y.V.; Langenfeld, V.W.; et al. Heme Binding Biguanides Target Cytochrome P450-Dependent Cancer Cell Mitochondria. Cell Chem. Biol. 2017, 24, 1259–1275.e1256. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Johnson, V.; Barrera, J.; Porras, M.; Hinojosa, D.; Hernández, I.; McGarrah, P.; Potter, D.A. Targeting cytochrome P450-dependent cancer cell mitochondria: Cancer associated CYPs and where to find them. Cancer Metastasis Rev. 2018, 37, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lu, C.; Zhang, K.; Lin, C.; Wu, F.; Tang, X.; Wu, D.; Dou, Y.; Han, R.; Wang, Y.; et al. Metformin Combining PD-1 Inhibitor Enhanced Anti-Tumor Efficacy in STK11 Mutant Lung Cancer Through AXIN-1-Dependent Inhibition of STING Ubiquitination. Front. Mol. Biosci. 2022, 9, 780200. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e607. [Google Scholar] [CrossRef] [PubMed]
- Choe, E.J.; Lee, C.H.; Bae, J.H.; Park, J.M.; Park, S.S.; Baek, M.C. Atorvastatin Enhances the Efficacy of Immune Checkpoint Therapy and Suppresses the Cellular and Extracellular Vesicle PD-L1. Pharmaceutics 2022, 14, 1660. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.G.; Wang, W.; Rothenberger, E.; Yang, J.; Gilligan, M.M.; Kipper, F.C.; Attaya, A.; Gartung, A.; Hwang, S.H.; Gillespie, M.J.; et al. Enhancing cancer immunotherapy via inhibition of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2024, 121, e2314085121. [Google Scholar] [CrossRef] [PubMed]
- Cortellini, A.; Tucci, M.; Adamo, V.; Stucci, L.S.; Russo, A.; Tanda, E.T.; Spagnolo, F.; Rastelli, F.; Bisonni, R.; Santini, D.; et al. Integrated analysis of concomitant medications and oncological outcomes from PD-1/PD-L1 checkpoint inhibitors in clinical practice. J. Immunother. Cancer 2020, 8, e001361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Chen, S.; Li, Z.; Chen, J.; Li, W. The effect of concomitant use of statins, NSAIDs, low-dose aspirin, metformin and beta-blockers on outcomes in patients receiving immune checkpoint inhibitors: A systematic review and meta-analysis. Oncoimmunology 2021, 10, 1957605. [Google Scholar] [CrossRef]
- Araki, T.; Kanda, S.; Ide, T.; Sonehara, K.; Komatsu, M.; Tateishi, K.; Minagawa, T.; Kiniwa, Y.; Kawakami, S.; Nomura, S.; et al. Antiplatelet drugs may increase the risk for checkpoint inhibitor-related pneumonitis in advanced cancer patients. ESMO Open 2023, 8, 102030. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Z.; Li, R.; Huang, R.; Peng, X. The association between aspirin use and immune-related adverse events in specific cancer patients receiving ICIs therapy: Analysis of the FAERS database. Front. Pharmacol. 2023, 14, 1259628. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Dragnev, K.; Sarwar, T.; Shirai, K. Clinical outcomes in non-small-cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Manag. 2019, 8, Lmt11. [Google Scholar] [CrossRef]
- Yang, J.; Kim, S.H.; Jung, E.H.; Kim, S.-A.; Suh, K.J.; Lee, J.Y.; Kim, J.-W.; Kim, J.W.; Lee, J.-O.; Kim, Y.J.; et al. The effect of metformin or dipeptidyl peptidase 4 inhibitors on clinical outcomes in metastatic non-small cell lung cancer treated with immune checkpoint inhibitors. Thoracic Cancer 2023, 14, 52–60. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gretschel, J.; El Hage, R.; Wang, R.; Chen, Y.; Pietzner, A.; Loew, A.; Leineweber, C.G.; Wördemann, J.; Rohwer, N.; Weylandt, K.H.; et al. Harnessing Oxylipins and Inflammation Modulation for Prevention and Treatment of Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 5408. https://doi.org/10.3390/ijms25105408
Gretschel J, El Hage R, Wang R, Chen Y, Pietzner A, Loew A, Leineweber CG, Wördemann J, Rohwer N, Weylandt KH, et al. Harnessing Oxylipins and Inflammation Modulation for Prevention and Treatment of Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(10):5408. https://doi.org/10.3390/ijms25105408
Chicago/Turabian StyleGretschel, Julius, Racha El Hage, Ruirui Wang, Yifang Chen, Anne Pietzner, Andreas Loew, Can G. Leineweber, Jonas Wördemann, Nadine Rohwer, Karsten H. Weylandt, and et al. 2024. "Harnessing Oxylipins and Inflammation Modulation for Prevention and Treatment of Colorectal Cancer" International Journal of Molecular Sciences 25, no. 10: 5408. https://doi.org/10.3390/ijms25105408
APA StyleGretschel, J., El Hage, R., Wang, R., Chen, Y., Pietzner, A., Loew, A., Leineweber, C. G., Wördemann, J., Rohwer, N., Weylandt, K. H., & Schmöcker, C. (2024). Harnessing Oxylipins and Inflammation Modulation for Prevention and Treatment of Colorectal Cancer. International Journal of Molecular Sciences, 25(10), 5408. https://doi.org/10.3390/ijms25105408