
Role of Estrogen and Estrogen Receptor in GH-Secreting Adenomas

 ,
,  ,
,
Abstract
1. Somatotroph Adenomas and Acromegaly
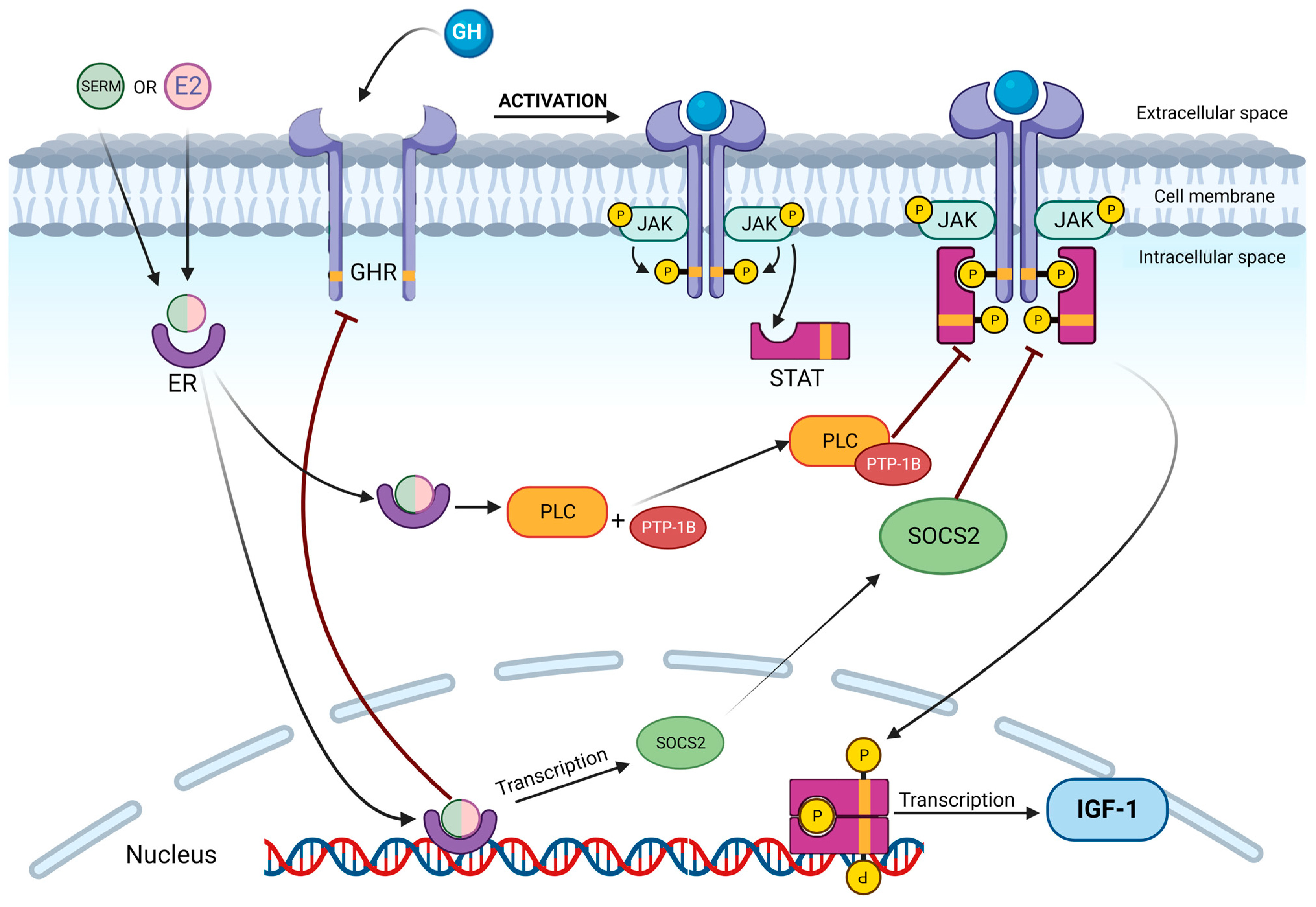
2. The GH-IGF1 Axis and Estrogens: How They Work
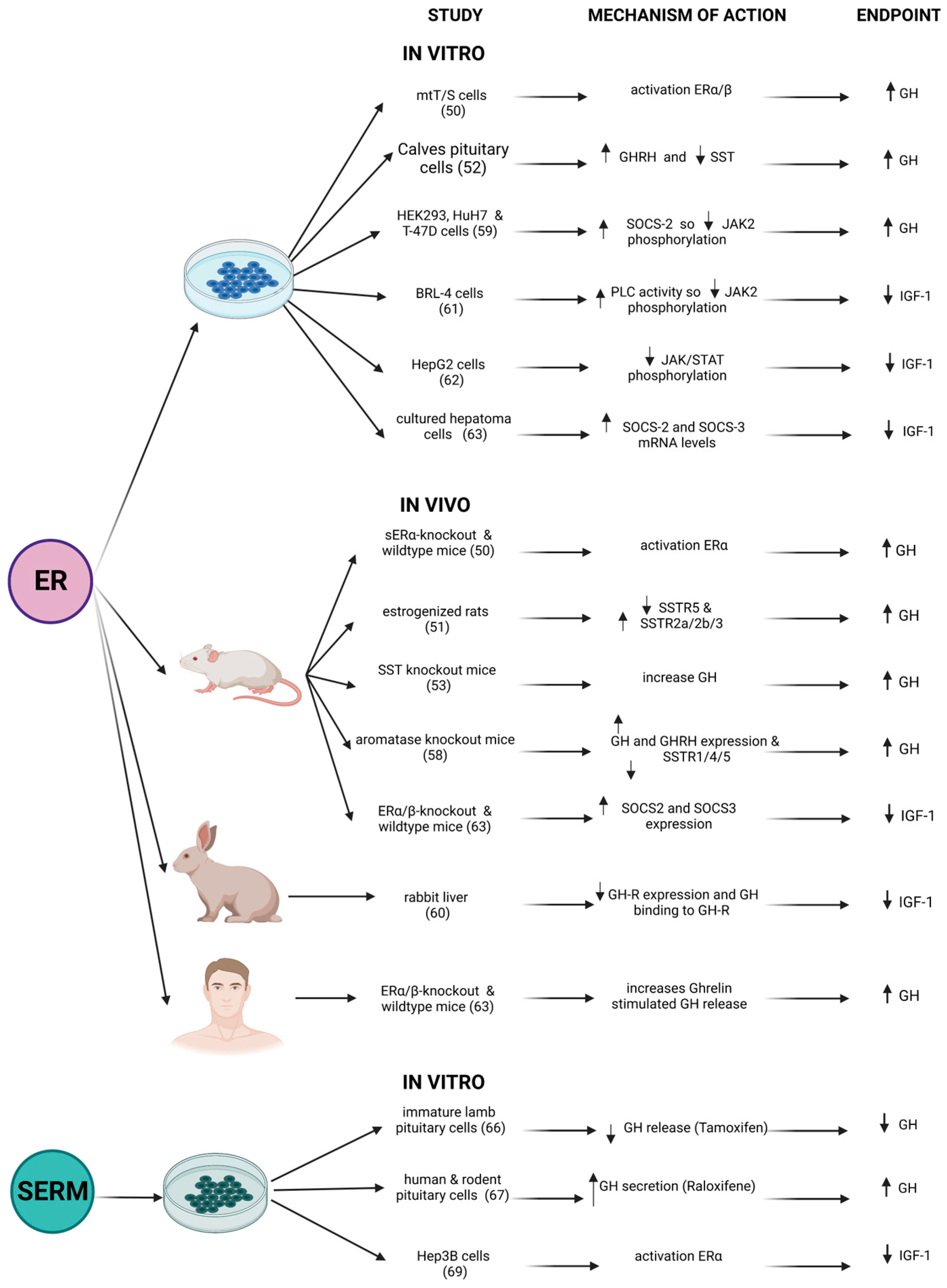
3. Effects of Estrogens on GH-IGF1 Axis: What In Vitro and In Vivo Models Show Us
4. Clinical Use of Estrogens and/or SERMs in Patients with Acromegaly
4.1. The Use of Oral Estrogens in Acromegaly
4.2. Targeting the Estrogen Receptor with SERMs in Acromegaly

{kind=link}
{kind=link}
{kind=link}
| Studies | N of Patients | Male/ Female | Drug Used | Concomitant Therapy | GH Effect | IGF1 Relative Reduction | IGF1 Normalization |
|---|---|---|---|---|---|---|---|
| Oral Estrogen | |||||||
| Cozzi et al. (2003) [77] | 8 | 0/8 | Ethinylestradiol 30–40–30 mcg/day + desogestrel 50–70–100 mcg/day | OCT + CAB (2/8), OCT (3/8) | = | 45% in 6/8 patients | 4/8 (50%) |
| Vallette et al. (2010) [78] | 11 | 0/11 | Ethinylestradiol 20 mcg + levonorgestrel 100 mcg | OCT (7/11) | = | 56.8% | 8/11 (73%) |
| Shimon and Barkan (2012) [79] | 4 | 0/4 | ethinylestradiol 20 mcg + Gestodene 75 μg or ethinyl-estradiol 0.035 mg + cyproterone acetate 2 mg or Transdermal estrogen | PEG (1/4), OCT (2/4) | = | 34 to 68% | 3/4 (75%) |
| Magalhães et al. (2022) [82] | 8 | 0/8 | ethinylestradiol 0.03 mg and levonorgestrel 0.15 mg | OCT or LAN (6/8) | =/↑ | 21 to 54% | 3/8 (37%) |
| SERMs | |||||||
| Cozzi et al. (1997) [95] | 19 | 6/13 | Tamoxifen 40 mg/die | none | ↑ | 18% to 60% | 4/19 (21%) |
| Balili et al. (2014) [31] | 17 | 15/2 | Tamoxifen 20–40 mg/die | OCT + PEG (1/17) or CAB (1/17) or OCT alone (1/17) | = | 17.5% | 8/17 (47%) |
| Mirfakhraee et al. (2021) [96] | 1 | 0/1 | Tamoxifen | anastrazole | ↑ | 60% | 1/1 (100%) |
| Attanasio et al. (2003) [97] | 13 | 0/13 | Raloxifene 60 mg/die | OCT (3/13), CAB (1/13) | = | 35% | 7/13 (54%) |
| Dimaraki et al. (2004) [34] | 8 | 8/0 | Raloxifene 60 mg twice a day | OCT (2/8) | = | 16% | 2/8 (25%) |
| Duarte et al. (2016) [35] | 16 | 16/0 | Clomiphene citrate 50 mg/die | OCT alone (4/16), OCT + CAB (7/16), CAB alone (5/16) | = | 41% | 7/16 (44%) |
| Koroglu et al. (2022) [98] | 1 | 1/0 | Clomiphene citrate 25 mg/die | LAN | No data | 51% | 1/1 (100%) |
5. GH-IGF1 Axis and Tumor Development: The Role of Estrogens
| Drug Name | Molecular Features | Drug Action | Side Effects | Indication |
|---|---|---|---|---|
| SERMs | ||||
| Tamoxifene | Mixed agonist/antagonist action on ER | Breast, brain → − Bone, endometrium, cardiovascular → + Vagina → +/− | Endometrial cancer, VTE, hot flushes, atrophic vaginitis | Adjuvant treatment in ER+ breast cancer |
| Raloxifene | Brest, brain → − Cardiovascular, bone → + Endometrium, vagina → = | VTE, hot flushes, leg cramps | Prevention and treatment of post-menopausal osteoporosis | |
| Clomiphene | Breast, brain, endometrium, vagina → − | Headache, hot flushes, GI disturbance, ovarian enlargement | Treatment of ovulatory dysfunction in infertile women | |
| Toremifene | Breast, brain → − Cardiovascular, bone → + Endometrium, vagina → +/− | VTE, hot flushes, atrophic vaginitis | Treatment of metastatic ER+ breast cancer | |
| SERDs | ||||
| Fulvestrant | Pure ER antagonism | ER degradation, no estrogenic activity | Hot flushes, GI disturbances | Treatment of locally advanced/metastatic ER+ breast cancer |
| AI | ||||
| Anastrozole | Nonsteroidal | Reversible aromatase inhibition | Bone loss, nausea, hot flushes | Adjuvant treatment in ER+ breast cancer |
| Letrozole | Nonsteroidal | Reversible aromatase inhibition | Bone loss, nausea, hot flushes | Adjuvant treatment in ER+ breast cancer |
| Exemestane | Steroidal | Irreversible aromatase inhibition | Bone loss, Hypertension, atrophic vaginitis | Adjuvant treatment in ER+ breast cancer |
| Estrogens | ||||
| EE | Synthetic estrogen | Strong ER binding | VTE, breast cancer, endometrial cancer, ovarian cancer | Contraception, HRT |
| E2V | Natural Estrogen | Weak ER binding | ||
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoskuldsdottir, G.T.; Fjalldal, S.B.; Sigurjonsdottir, H.A. The incidence and prevalence of acromegaly, a nationwide study from 1955 through 2013. Pituitary 2015, 18, 803–807. [Google Scholar] [CrossRef]
- Lavrentaki, A.; Paluzzi, A.; Wass, J.A.H.; Karavitaki, N. Epidemiology of acromegaly: Review of population studies. Pituitary 2017, 20, 4–9. [Google Scholar] [CrossRef]
- Agustsson, T.T.; Baldvinsdottir, T.; Jonasson, J.G.; Olafsdottir, E.; Steinthorsdottir, V.; Sigurdsson, G.; Thorsson, A.V.; Carroll, P.V.; Korbonits, M.; Benediktsson, R. The epidemiology of pituitary adenomas in Iceland, 1955–2012: A nationwide population-based study. Eur. J. Endocrinol. 2015, 173, 655–664. [Google Scholar] [CrossRef]
- Gruppetta, M.; Mercieca, C.; Vassallo, J. Prevalence and incidence of pituitary adenomas: A population based study in Malta. Pituitary 2013, 16, 545–553. [Google Scholar] [CrossRef]
- Dal, J.; Skov, B.G.; Andersen, M.; Feldt-Rasmussen, U.; Feltoft, C.L.; Karmisholt, J.; Nielsen, E.H.; Dekkers, O.M.; Jørgensen, J.O.L. Sex differences in acromegaly at diagnosis: A nationwide cohort study and meta-analysis of the literature. Clin. Endocrinol. 2021, 94, 625–635. [Google Scholar] [CrossRef]
- Lenders, N.F.; McCormack, A.I.; Ho, K.K.Y. Management of Endocrine Disease: Does gender matter in the management of acromegaly? Eur. J. Endocrinol. 2020, 182, R67–R82. [Google Scholar] [CrossRef]
- Colao, A.; Ferone, D.; Marzullo, P.; Lombardi, G. Systemic Complications of Acromegaly: Epidemiology, Pathogenesis, and Management. Endocr. Rev. 2004, 25, 102–152. [Google Scholar] [CrossRef]
- Zendran, I.; Gut, G.; Kałużny, M.; Zawadzka, K.; Bolanowski, M. Acromegaly Caused by Ectopic Growth Hormone Releasing Hormone Secretion: A Review. Front. Endocrinol. 2022, 13, 867965. [Google Scholar] [CrossRef]
- Melmed, S.; Kaiser, U.B.; Lopes, M.B.; Bertherat, J.; Syro, L.V.; Raverot, G.; Reincke, M.; Johannsson, G.; Beckers, A.; Fleseriu, M.; et al. Clinical Biology of the Pituitary Adenoma. Endocr. Rev. 2022, 43, 1003–1037. [Google Scholar] [CrossRef]
- Fleseriu, M.; Langlois, F.; Lim, D.S.T.; Varlamov, E.V.; Melmed, S. Acromegaly: Pathogenesis, diagnosis, and management. Lancet Diabetes Endocrinol. 2022, 10, 804–826. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef]
- Regazzo, D.; Losa, M.; Albiger, N.M.; Terreni, M.R.; Vazza, G.; Ceccato, F.; Emanuelli, E.; Denaro, L.; Scaroni, C.; Occhi, G. The GIP/GIPR axis is functionally linked to GH-secretion increase in a significant proportion of gsp-somatotropinomas. Eur. J. Endocrinol. 2017, 176, 543–553. [Google Scholar] [CrossRef]
- Chang, M.; Yang, C.; Bao, X.; Wang, R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front. Endocrinol. 2020, 11, 596554. [Google Scholar] [CrossRef]
- Katznelson, L.; Laws, E.R.; Melmed, S.; Molitch, M.E.; Murad, M.H.; Utz, A.; Wass, J.A. Acromegaly: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 3933–3951. [Google Scholar] [CrossRef]
- Fleseriu, M.; Biller, B.M.K.; Freda, P.U.; Gadelha, M.R.; Giustina, A.; Katznelson, L.; Molitch, M.E.; Samson, S.L.; Strasburger, C.J.; van der Lely, A.J.; et al. A Pituitary Society update to acromegaly management guidelines. Pituitary 2021, 24, 1–13. [Google Scholar] [CrossRef]
- Giustina, A.; Barkan, A.; Beckers, A.; Biermasz, N.; Biller, B.M.K.; Boguszewski, C.; Bolanowski, M.; Bonert, V.; Bronstein, M.D.; Casanueva, F.F.; et al. A Consensus on the Diagnosis and Treatment of Acromegaly Comorbidities: An Update. J. Clin. Endocrinol. Metab. 2019, 105, e937–e946. [Google Scholar] [CrossRef]
- Casanueva, F.F.; Barkan, A.L.; Buchfelder, M.; Klibanski, A.; Laws, E.R.; Loeffler, J.S.; Melmed, S.; Mortini, P.; Wass, J.; Giustina, A.L. Criteria for the definition of Pituitary Tumor Centers of Excellence (PTCOE): A Pituitary Society Statement. Pituitary 2017, 20, 489–498. [Google Scholar] [CrossRef]
- Barbot, M.; Padova Pituitary Club; Ceccato, F.; Lizzul, L.; Daniele, A.; Zilio, M.; Gardiman, M.P.; Denaro, L.; Emanuelli, E.; Vianello, F.; et al. Perioperative multidisciplinary management of endoscopic transsphenoidal surgery for sellar lesions: Practical suggestions from the Padova model. Neurosurg. Rev. 2020, 43, 1109–1116. [Google Scholar] [CrossRef]
- Chen, C.-J.; Ironside, N.; Pomeraniec, I.J.; Chivukula, S.; Buell, T.J.; Ding, D.; Taylor, D.G.; Dallapiazza, R.F.; Lee, C.-C.; Bergsneider, M. Microsurgical versus endoscopic transsphenoidal resection for acromegaly: A systematic review of outcomes and complications. Acta Neurochir. 2017, 159, 2193–2207. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Wildemberg, L.E.; Bronstein, M.D.; Gatto, F.; Ferone, D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary 2017, 20, 100–108. [Google Scholar] [CrossRef]
- Colao, A.; Grasso, L.F.S.; Giustina, A.; Melmed, S.; Chanson, P.; Pereira, A.M.; Pivonello, R. Acromegaly. Nat. Rev. Dis. Prim. 2019, 5, 20. [Google Scholar] [CrossRef]
- Caron, P.J.; Bevan, J.S.; Petersenn, S.; Flanagan, D.; Tabarin, A.; Prévost, G.; Maisonobe, P.; Clermont, A.; on behalf of the PRIMARYS. Tumor shrinkage with lanreotide autogel 120 mg as primary therapy in acromegaly: Results of a prospective multicenter clinical trial. J. Clin. Endocrinol. Metab. 2014, 99, 1282–1290. [Google Scholar] [CrossRef]
- Mondin, A.; Manara, R.; Voltan, G.; Tizianel, I.; Denaro, L.; Ferrari, M.; Barbot, M.; Scaroni, C.; Ceccato, F. Pasireotide-Induced Shrinkage in GH and ACTH Secreting Pituitary Adenoma: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2022, 13, 935759. [Google Scholar] [CrossRef]
- Carmichael, J.D.; Bonert, V.S.; Nuño, M.; Ly, D.; Melmed, S. Acromegaly clinical trial methodology impact on reported biochemical efficacy rates of somatostatin receptor ligand treatments: A meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, 1825–1833. [Google Scholar] [CrossRef]
- Chiloiro, S.; Bianchi, A.; Giampietro, A.; Pontecorvi, A.; Raverot, G.; De Marinis, L. Second line treatment of acromegaly: Pasireotide or Pegvisomant? Best Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101684. [Google Scholar] [CrossRef]
- Bernabeu, I.; Rodriguez-Gomez, I.A.; Ramos-Levi, A.M.; Marazuela, M. Profile of pegvisomant in the management of acromegaly: An evidence based review of its place in therapy. Res. Rep. Endocr. Disord. 2015, 5, 47–58. [Google Scholar] [CrossRef]
- Bollerslev, J.; Heck, A.; Olarescu, N.C. Management of Endocrine Disease: Individualised management of acromegaly. Eur. J. Endocrinol. 2019, 181, R57–R71. [Google Scholar] [CrossRef]
- Cocchiara, F.; Campana, C.; Nista, F.; Corica, G.; Ceraudo, M.; Milioto, A.; Rossi, D.C.; Zona, G.; Ferone, D.; Gatto, F. Evaluation of acromegaly treatment direct costs with respect to biochemical control and follow-up length. Pituitary 2022, 25, 246–257. [Google Scholar] [CrossRef]
- Albright, F.; Reifenstein, E.G. Effect of estrogens in acromegaly. Trans. Conf. Metab. Asp. Conval. 1946, 14, 102–122. [Google Scholar]
- Jørgensen, J.O.L.; Christensen, J.J.; Krag, M.; Fisker, S.; Ovesen, P.; Christiansen, J.S. Serum insulin-like growth factor I levels in growth hormone-deficient adults: Influence of sex steroids. Horm Res. 2004, 62, 73–76. [Google Scholar] [CrossRef]
- Balili, I.; Barkan, A. Tamoxifen as a therapeutic agent in acromegaly. Pituitary 2014, 17, 500–504. [Google Scholar] [CrossRef]
- Stone, J.C.; Clark, J.; Cuneo, R.; Russell, A.W.; Doi, S.A.R. Estrogen and selective estrogen receptor modulators (SERMs) for the treatment of acromegaly: A meta-analysis of published observational studies. Pituitary 2014, 17, 284–295. [Google Scholar] [CrossRef]
- Duarte, F.H.; Jallad, R.S.; Bronstein, M.D. Clomiphene citrate for treatment of acromegaly not controlled by conventional therapies. J. Clin. Endocrinol. Metab. 2015, 100, 1863–1869. [Google Scholar] [CrossRef]
- Dimaraki, E.V.; Symons, K.V.; Barkan, A.L. Raloxifene decreases serum IGF-I in male patients with active acromegaly. Eur. J. Endocrinol. 2004, 150, 481–487. [Google Scholar] [CrossRef]
- Duarte, F.H.; Jallad, R.S.; Bronstein, M.D. Estrogens and selective estrogen receptor modulators in acromegaly. Endocrine 2016, 54, 306–314. [Google Scholar] [CrossRef]
- Birzniece, V.; Ho, K.K.Y. Sex steroids and the GH axis: Implications for the management of hypopituitarism. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 59–69. [Google Scholar] [CrossRef]
- Müller, E.E.; Locatelli, V.; Cocchi, D. Neuroendocrine Control of Growth Hormone Secretion. Physiol. Rev. 1999, 79, 511–607. [Google Scholar] [CrossRef]
- Giustina, A.; Veldhuis, J.D. Pathophysiology of the Neuroregulation of Growth Hormone Secretion in Experimental Animals and the Human. Endocr. Rev. 1998, 19, 717–797. [Google Scholar]
- Carter-Su, C.; Schwartz, J.; Smit, L.J. Molecular Mechanism of Growth Hormone Action. Annu. Rev. Physiol. 1996, 58, 187–207. [Google Scholar] [CrossRef]
- Argetsinger, L.S.; Campbell, G.S.; Yang, X.; Witthuhn, B.A.; Silvennoinen, O.; Ihle, J.N.; Carter-Su, C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 1993, 74, 237–244. [Google Scholar] [CrossRef]
- Ram, P.A.; Park, S.H.; Choi, H.K.; Waxman, D.J. Growth hormone activation of Stat 1, Stat 3, and Stat 5 in rat liver: Differential kinetics of hormone desensitization and growth hormone stimulation of both tyrosine phosphorylation and serine/threonine phosphorylation. J. Biol. Chem. 1996, 271, 5929–5940. [Google Scholar] [CrossRef]
- Herrington, J.; Smit, L.S.; Schwartz, J.; Carter-Su, C. The role of STAT proteins in growth hormone signaling. Oncogene 2000, 19, 2585–2597. [Google Scholar] [CrossRef]
- Vidal, O.M.; Merino, R.; Rico-Bautista, E.; Fernandez-Perez, L.; Chia, D.J.; Woelfle, J.; Ono, M.; Lenhard, B.; Norstedt, G.; Rotwein, P.; et al. In vivo transcript profiling and phylogenetic analysis identifies suppressor of cytokine signaling 2 as a direct signal transducer and activator of transcription 5b target in liver. Mol. Endocrinol. 2007, 21, 293–311. [Google Scholar] [CrossRef]
- Ram, P.A.; Waxman, D.J. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J. Biol. Chem. 1999, 274, 35553–35561. [Google Scholar] [CrossRef]
- Greenhalgh, C.J.; Bertolino, P.; Asa, S.L.; Metcalf, D.; Corbin, J.E.; Adams, T.E.; Davey, H.W.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Growth enhancement in suppressor of cytokine signaling 2 (SOCS-2)-deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b). Mol. Endocrinol. 2002, 16, 1394–1406. [Google Scholar] [CrossRef]
- Arao, Y.; Korach, K.S. The physiological role of estrogen receptor functional domains. Essays Biochem. 2021, 65, 867–875. [Google Scholar] [CrossRef]
- Matthews, J. Estrogen Signaling: A Subtle Balance Between ER and ER. Mol. Interv. 2003, 3, 281–292. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen Receptors Alpha and Beta Subtype-Selective Ligands and Clinical Potential. Steroids 2011, 18, 1492–1501. [Google Scholar] [CrossRef]
- Shupnik, M.A.; Pitt, L.K.; Soh, A.Y.; Anderson, A.; Lopes, M.B.; Laws, E.R. Selective expression of estrogen receptor and β isoforms in human pituitary tumors. J. Clin. Endocrinol. Metab. 1998, 83, 3965–3972. [Google Scholar] [CrossRef]
- Avtanski, D.; Novaira, H.J.; Wu, S.; Romero, C.J.; Kineman, R.; Luque, R.M.; Wondisford, F.; Radovick, S. Both estrogen receptor α and β stimulate pituitary GH gene expression. Mol. Endocrinol. 2014, 28, 40–52. [Google Scholar] [CrossRef]
- Kimura, N.; Tomizawa, S.; Arai, K.N.; Kimura, N. Chronic treatment with estrogen up-regulates expression of sst2 messenger ribonucleic acid (mRNA) but down-regulates expression of sst5 mRNA in rat pituitaries. Endocrinology 1998, 139, 1573–1580. [Google Scholar] [CrossRef]
- Hassan, H.A.; Enright, W.J.; Tucker, H.A.; Merkel, R.A. Estrogen and androgen elicit growth hormone release via dissimilar patterns of hypothalamic neuropeptide secretion. Steroids 2001, 66, 71–80. [Google Scholar] [CrossRef]
- Adams, J.M.; Otero-Corchon, V.; Hammond, G.L.; Veldhuis, J.D.; Qi, N.; Low, M.J. Somatostatin Is Essential for the Sexual Dimorphism of GH Secretion, Corticosteroid-Binding Globulin Production, and Corticosterone Levels in Mice. Endocrinology 2015, 156, 1052–1065. [Google Scholar] [CrossRef]
- Lanfranco, F.; Motta, G.; Baldi, M.; Gasco, V.; Grottoli, S.; Benso, A.; Broglio, F.; Ghigo, E. Ghrelin and Anterior Pituitary Function. Front. Horm. Res. 2010, 38, 206–211. [Google Scholar]
- Kok, P.; Paulo, R.C.; Cosma, M.; Mielke, K.L.; Miles, J.M.; Bowers, C.Y.; Veldhuis, J.D. Estrogen Supplementation Selectively Enhances Hypothalamo-Pituitary Sensitivity to Ghrelin in Post-menopausal Women. J. Clin. Endocrinol. Metab. 2008, 93, 4020–4026. [Google Scholar] [CrossRef]
- Veldhuis, J.D.; Patrie, J.T.; Brill, K.T.; Weltman, J.Y.; Mueller, E.E.; Bowers, C.Y.; Weltman, A. Contributions of Gender and Systemic Estradiol and Testosterone Concentrations to Maximal Secretagogue Drive of Burst-Like Growth Hormone Secretion in Healthy Middle-Aged and Older Adults. J. Clin. Endocrinol. Metab. 2004, 89, 6291–6296. [Google Scholar] [CrossRef]
- Caglar, A.S.; Kapucu, A.; Dar, K.A.; Ozkaya, H.M.; Caglar, E.; Ince, H.; Kadioglu, P. Localization of the aromatase enzyme expression in the human pituitary gland and its effect on growth hormone, prolactin, and thyroid stimulating hormone axis. Endocrine 2015, 49, 761–768. [Google Scholar] [CrossRef]
- Yan, M.; Jones, M.E.E.; Hernandez, M.; Liu, D.; Simpson, E.R.; Chen, C. Functional modification of pituitary somatotropes in the aromatase knockout mouse and the effect of estrogen replacement. Endocrinology 2004, 145, 604–612. [Google Scholar] [CrossRef]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Sjogren, K.; Watts, C.K.W.; Low, T.H.; Leong, G.M.; Ross, R.J.M.; Ho, K.K.Y. Estrogen inhibits GH signaling by suppressing GH-induced JAK2 phosphorylation, an effect mediated by SOCS-2. Proc. Natl. Acad. Sci. USA 2003, 100, 1016–1021. [Google Scholar] [CrossRef]
- Domene, H.M.; Marín, G.; Sztein, J.; Yu, Y.M.; Baron, J.; Cassorla, F.G. Estradiol inhibits growth hormone receptor gene expression in rabbit liver. Mol. Cell. Endocrinol. 1994, 103, 81–87. [Google Scholar] [CrossRef]
- Fernández, L.; Flores-Morales, A.; Lahuna, O.; Sliva, D.; Norstedt, G.; Haldosén, L.A.; Mode, A.; Gustafsson, J.A. Desensitization of the growth hormone-induced Janus kinase 2 (Jak 2)/signal transducer and activator of transcription 5 (Stat5)-signaling pathway requires protein synthesis and phospholipase C. Endocrinology 1998, 139, 1815–1824. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, H.S.; Kim, S.H.; Yang, Y.R.; Bae, Y.S.; Chang, J.S.; Kwon, H.M.; Ryu, S.H.; Suh, P.-G. Phospholipase Cγ1 negatively regulates growth hormone signalling by forming a ternary complex with Jak2 and protein tyrosine phosphatase-1B. Nat. Cell Biol. 2006, 8, 1389–1397. [Google Scholar] [CrossRef]
- Leong, G.M.; Moverare, S.; Brce, J.; Doyle, N.; Sjögren, K.; Dahlman-Wright, K.; Gustafsson, J.-A.; Ho, K.K.Y.; Ohlsson, C.; Leung, K.-C. Estrogen up-regulates hepatic expression of suppressors of cytokine signaling-2 and -3 in vivo and in vitro. Endocrinology 2004, 145, 5525–5531. [Google Scholar] [CrossRef]
- Jelinsky, S.A.; Harris, H.A.; Brown, E.L.; Flanagan, K.; Zhang, X.; Tunkey, C.; Lai, K.; Lane, M.V.; Simcoe, D.K.; Evans, M.J. Global transcription profiling of estrogen activity: Estrogen receptor alpha regulates gene expression in the kidney. Endocrinology 2003, 144, 701–710. [Google Scholar] [CrossRef]
- Clemmons, D.R.; Underwood, L.E.; Ridgway, E.C.; Kliman, B.; Kjellberg, R.N.; Van Wyk, J.J. Estradiol treatment of acromegaly. Reduction of immunoreactive somatomedin-C and improvement in metabolic status. Am. J. Med. 1980, 69, 571–575. [Google Scholar] [CrossRef]
- Malaab, S.A.; Pollak, M.N.; Goodyer, C.G. Direct effects of tamoxifen on growth hormone secretion by pituitary cells in vitro. Eur. J. Cancer 1992, 28, 788–793. [Google Scholar] [CrossRef]
- Tulipano, G.; Bonfanti, C.; Poiesi, C.; Burattin, A.; Turazzi, S.; Barone, G.; Cozzi, R.; Bollati, A.; Valle, D.; Giustina, A. Effects of the selective estrogen receptor modulator LY117018 on growth hormone secretion: In Vitro studies. Metabolism 2004, 53, 563–570. [Google Scholar] [CrossRef]
- Blair, R.M.; Fang, H.; Branham, W.S.; Hass, B.S.; Dial, S.L.; Moland, C.L.; Tong, W.; Shi, L.; Perkins, R.; Sheehan, D.M. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: Structural diversity of ligands. Toxicol. Sci. 2000, 54, 138–153. [Google Scholar] [CrossRef]
- Fournier, B.; Gutzwiller, S.; Dittmar, T.; Matthias, G.; Steenbergh, P.; Matthias, P. Estrogen receptor (ER)-alpha, but not ER-beta, mediates regulation of the insulin-like growth factor I gene by antiestrogens. J. Biol. Chem. 2001, 276, 35444–35449. [Google Scholar] [CrossRef]
- Weissberger, A.J.; Ho, K.K. Activation of the somatotropic axis by testosterone in adult males: Evidence for the role of aromatization. J. Clin. Endocrinol. Metab. 1993, 76, 1407–1412. [Google Scholar]
- Veldhuis, J.D.; Metzger, D.L.; Martha, P.M., Jr.; Mauras, N.; Kerrigan, J.R.; Keenan, B.; Rogol, A.D.; Pincus, S.M. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: Evidence from pubertal pathophysiology and sex-steroid hormone replacement. J. Clin. Endocrinol. Metab. 1997, 82, 3414–3420. [Google Scholar]
- Veldhuis, J.D.; Mielke, K.L.; Cosma, M.; Soares-Welch, C.; Paulo, R.; Miles, J.M.; Bowers, C.Y. Aromatase and 5alpha-reductase inhibition during an exogenous testosterone clamp unveils selective sex steroid modulation of somatostatin and growth hormone secretagogue actions in healthy older men. J. Clin. Endocrinol. Metab. 2009, 94, 973–981. [Google Scholar] [CrossRef]
- Roelfsema, F.; Yang, R.J.; Bowers, C.Y.; Veldhuis, J.D. Modulating Effects of Progesterone on Spontaneous Nocturnal and Ghrelin-Induced GH Secretion in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2019, 104, 2385–2394. [Google Scholar] [CrossRef]
- Mc Cullagh, E.P.; Beck, J.C.; Schaffenburg, C.A. Control of diabetes and other features of acromegaly following treatment with estrogens. Diabetes 1955, 4, 13–23. [Google Scholar] [CrossRef]
- Shoung, N.; Ho, K.K.Y. Managing Estrogen Therapy in the Pituitary Patient. J. Endocr. Soc. 2023, 7, bvad051. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar]
- Cozzi, R.; Barausse, M.; Lodrini, S.; Lasio, G.; Attanasio, R. Estroprogestinic pill normalizes IGF-I levels in acromegalic women. J. Endocrinol. Investig. 2003, 26, 347–352. [Google Scholar] [CrossRef]
- Vallette, S.; Serri, O. Oral estroprogestin: An alternative low cost therapy for women with postoperative persistent acromegaly? Pituitary 2010, 13, 311–314. [Google Scholar] [CrossRef]
- Shimon, I.; Barkan, A. Estrogen treatment for acromegaly. Pituitary 2012, 15, 601–607. [Google Scholar] [CrossRef]
- Weissberger, A.J.; Ho, K.K.; Lazarus, L. Contrasting effects of oral and transdermal routes of estrogen replacement therapy on 24-hour growth hormone (GH) secretion, insulin-like growth factor I, and GH-binding protein in post-menopausal women. J. Clin. Endocrinol. Metab. 1991, 72, 374–381. [Google Scholar] [CrossRef]
- Friend, K.E.; Hartman, M.L.; Pezzoli, S.S.; Clasey, J.L.; Thorner, M.O. Both oral and transdermal estrogen increase growth hormone release in post-menopausal women—A clinical research center study. J. Clin. Endocrinol. Metab. 1996, 81, 2250–2256. [Google Scholar] [PubMed]
- Magalhães, J.; Ventura, N.; Lamback, E.B.; Da Silva, D.; Camacho, A.H.; Chimelli, L.; Gadelha, M.R.; Kasuki, L. A prospective study on the efficacy of oral estrogen in female patients with acromegaly. Pituitary 2022, 25, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Chaidarun, S.S.; Klibanski, A.; Alexander, J.M. Tumor-specific expression of alternatively spliced estrogen receptor messenger ribonucleic acid variants in human pituitary adenomas. J. Clin. Endocrinol. Metab. 1997, 82, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Ferraù, F.; Romeo, P.D.; Puglisi, S.; Ragonese, M.; Spagnolo, F.; Salpietro, C.; Ientile, R.; Currò, M.; Visalli, G.; Alibrandi, A.; et al. GSTP1 gene methylation and AHR rs2066853 variant predict resistance to first generation somatostatin analogs in patients with acromegaly. J. Endocrinol. Investig. 2019, 42, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Than, N.N.; Tomlinson, C.L.; Haldar, D.; King, A.L.; Moore, D.; Newsome, P.N. Clinical effectiveness of cell therapies in patients with chronic liver disease and acute-on-chronic liver failure: A systematic review protocol. Syst. Rev. 2016, 5, 100. [Google Scholar] [CrossRef]
- Chan, K.K.-L.; Leung, T.H.-Y.; Chan, D.W.; Wei, N.; Lau, G.T.-Y.; Liu, S.S.; Siu, M.K.-Y.; Ngan, H.Y.-S. Targeting estrogen receptor subtypes (ERα and ERβ) with selective ER modulators in ovarian cancer. J. Endocrinol. 2014, 221, 325–336. [Google Scholar] [CrossRef]
- Banerjee, A.; Cai, S.; Xie, G.; Li, N.; Bai, X.; Lavudi, K.; Wang, K.; Zhang, X.; Zhang, J.; Patnaik, S.; et al. A Novel Estrogen Receptor β Agonist Diminishes Ovarian Cancer Stem Cells via Suppressing the Epithelial-To-Mesenchymal Transition. Cancers 2022, 14, 2311. [Google Scholar] [CrossRef]
- García-Malpartida, K.; Martín-Gorgojo, A.; Rocha, M.; Gómez-Balaguer, M.; Hernández-Mijares, A. Prolactinoma induced by estrogen and cyproterone acetate in a male-to-female transsexual. Fertil. Steril. 2010, 94, 1097.e13–1097.e15. [Google Scholar] [CrossRef]
- Šošić-Jurjević, B.; Ajdžanović, V.; Miljić, D.; Trifunović, S.; Filipović, B.; Stanković, S.; Bolevich, S.; Jakovljević, V.; Milošević, V. Pituitary Hyperplasia, Hormonal Changes and Prolactinoma Development in Males Exposed to Estrogens-An Insight from Translational Studies. Int. J. Mol. Sci. 2020, 16, 2024. [Google Scholar] [CrossRef]
- García-Barrado, M.J.; Blanco, E.J.; Iglesias-Osma, M.C.; Carretero-Hernández, M.; Catalano-Iniesta, L.; Sanchez-Robledo, V.; Carretero, M.; Herrero, J.J.; Carrero, S.; Carretero, J. Relation among aromatase P450 and tumoral growth in human prolactinomas. Int. J. Mol. Sci. 2017, 18, 2299. [Google Scholar] [CrossRef]
- Ceccato, F.; Lizzul, L.; Voltan, G.; Barbot, M.; Scaroni, C. Anastrozole as add-on therapy for cabergoline-resistant prolactin-secreting pituitary adenomas: Real-life experience in male patients. Pituitary 2021, 24, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H. Selective estrogen receptor modulators (SERMS): Keys to understanding their function. Menopause 2020, 27, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Howell, S.J. Tamoxifen evolution. Br. J. Cancer 2023, 128, 421–425. [Google Scholar] [CrossRef]
- Birzniece, V.; Sutanto, S.; Ho, K.K.Y. Gender difference in the neuroendocrine regulation of growth hormone axis by selective estrogen receptor modulators. J. Clin. Endocrinol. Metab. 2012, 97, E521–E527. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, R.; Attanasio, R.; Oppizzi, G.; Orlandi, P.; Giustina, A.; Lodrini, S.; Da Re, N.; Dallabonzana, D. Effects of tamoxifen on GH and IGF-1 levels in acromegaly. J. Endocrinol. Investig. 1997, 20, 445–451. [Google Scholar] [CrossRef]
- Mirfakhraee, S.; Chan, A.V.C.; Ganji, N.; Abramowitz, J. Dual treatment of acromegaly and hormone-receptor-positive breast cancer with tamoxifen: A case report. J. Med. Case Rep. 2021, 15, 207. [Google Scholar] [CrossRef]
- Attanasio, R.; Barausse, M.; Cozzi, R. Raloxifene lowers IGF-1 levels in acromegalic women. Eur. J. Endocrinol. 2003, 148, 443–448. [Google Scholar] [CrossRef]
- Koroglu, E.P.; Soyaltin, U.E.; Yeral, S.; Yurekli, B.S. An acromegaly case treated with clomiphene citrate: Add-on treatment in primary medical therapy. Hormones 2023, 22, 139–142. [Google Scholar] [CrossRef]
- Ramsey, M.M.; Ingram, R.L.; Cashion, A.B.; Ng, A.H.; Cline, J.M.; Parlow, A.F.; Sonntag, W.E. Growth Hormone-Deficient Dwarf Animals Are Resistant to Dimethylbenzanthracine (DMBA)-Induced Mammary Carcinogenesis. Endocrinology 2002, 143, 4139–4142. [Google Scholar] [CrossRef]
- Swanson, S.M.; Unterman, T.G. The growth hormone-deficient Spontaneous Dwarf rat is resistant to chemically induced mammary carcinogenesis. Carcinogenesis 2002, 23, 977–982. [Google Scholar] [CrossRef]
- Zhang, X.; Mehta, R.G.; Lantvit, D.D.; Coschigano, K.T.; Kopchick, J.J.; Green, J.E.; Hedayat, S.; Christov, K.T.; Ray, V.H.; Unterman, T.G.; et al. Inhibition of estrogen-independent mammary carcinogenesis by disruption of growth hormone signaling. Carcinogenesis 2007, 28, 143–150. [Google Scholar] [CrossRef]
- Dworakowska, D.; Gueorguiev, M.; Kelly, P.; Monson, J.P.; Besser, G.M.; Chew, S.L.; A Akker, S.; Drake, W.M.; Fairclough, P.D.; Grossman, A.B.; et al. Repeated colonoscopic screening of patients with acromegaly: 15-year experience identifies those at risk of new colonic neoplasia and allows for effective screening guidelines. Eur. J. Endocrinol. 2010, 163, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Terzolo, M.; Puglisi, S.; Reimondo, G.; Dimopoulou, C.; Stalla, G.K. Thyroid and colorectal cancer screening in acromegaly patients: Should it be different from that in the general population? Eur. J. Endocrinol. 2020, 183, D1–D13. [Google Scholar] [CrossRef]
- Orme, S.M.; McNally, R.J.Q.; Cartwright, R.A.; Belchetz, P.E. Mortality and Cancer Incidence in Acromegaly: A Retrospective Cohort Study 1. J. Clin. Endocrinol. Metab. 1998, 83, 2730–2734. [Google Scholar] [CrossRef]
- Falch, C.M.; Olarescu, N.C.; Bollerslev, J.; Dekkers, O.M.; Heck, A. Trends in incidence and mortality risk for acromegaly in Norway: A cohort study. Endocrine 2023, 80, 152–159. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Kasuki, L.; Lim, D.S.T.; Fleseriu, M. Systemic Complications of Acromegaly and the Impact of the Current Treatment Landscape: An Update. Endocr. Rev. 2019, 40, 268–332. [Google Scholar] [CrossRef]
- Fleseriu, M.; Barkan, A.; Schneider, M.D.P.; Darhi, Y.; de Pierrefeu, A.; Ribeiro-Oliveira, A.; Petersenn, S.; Neggers, S.; Melmed, S. Prevalence of comorbidities and concomitant medication use in acromegaly: Analysis of real-world data from the United States. Pituitary 2022, 25, 296–307. [Google Scholar] [CrossRef]
- Maione, L.; Brue, T.; Beckers, A.; Delemer, B.; Petrossians, P.; Borson-Chazot, F.; Chabre, O.; François, P.; Bertherat, J.; Cortet-Rudelli, C.; et al. Changes in the management and comorbidities of acromegaly over three decades: The French Acromegaly Registry. Eur. J. Endocrinol. 2017, 176, 645–655. [Google Scholar] [CrossRef]
- Terzolo, M.; Reimondo, G.; Berchialla, P.; Ferrante, E.; Malchiodi, E.; De Marinis, L.; Pivonello, R.; Grottoli, S.; Losa, M.; Cannavo, S.; et al. Acromegaly is associated with increased cancer risk: A survey in Italy. Endocr. Relat. Cancer 2017, 24, 495–504. [Google Scholar] [CrossRef]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma Insulin-Like Growth Factor-I and Prostate Cancer Risk: A Prospective Study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- E Hankinson, S.; Willett, W.C.; A Colditz, G.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; E Speizer, F.; Pollak, M. Circulating concentrations of insulin-like growth factor I and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Spitz, M.R.; Mistry, J.; Gu, J.; Hong, W.K.; Wu, X. Plasma Levels of Insulin-Like Growth Factor-I and Lung Cancer Risk: A Case-Control Analysis. J. Natl. Cancer Inst. 1999, 91, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Pollak, M.N.; Giovannucci, E.; Chan, J.; Tao, Y.; Hennekens, C.H.; Stampfer, M.J. Prospective Study of Colorectal Cancer Risk in Men and Plasma Levels of Insulin-Like Growth Factor (IGF)-I and IGF-Binding Protein-3. J. Natl. Cancer Inst. 1999, 91, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, N.; Hazra, S.; Thareja, S. Selective Estrogen receptor degraders (SERDs) for the treatment of breast cancer: An overview. Eur. J. Med. Chem. 2023, 256, 115422. [Google Scholar] [CrossRef]
- Ellis, A.J.; Hendrick, V.M.; Williams, R.; Komm, B.S. Selective estrogen receptor modulators in clinical practice: A safety overview. Expert Opin. Drug Saf. 2015, 14, 921–934. [Google Scholar] [CrossRef]
- Kelly, C.M.; Buzdar, A.U. Anastrozole. Expert Opin. Drug Saf. 2010, 9, 995–1003. [Google Scholar] [CrossRef]
- Sobral, A.F.; Amaral, C.; Correia-da-Silva, G.; Teixeira, N. Unravelling exemestane: From biology to clinical prospects. J. Steroid Biochem. Mol. Biol. 2016, 163, 1–11. [Google Scholar] [CrossRef]
- Subramani, R.; Nandy, S.B.; Pedroza, D.A.; Lakshmanaswamy, R. Role of Growth Hormone in Breast Cancer. Endocrinology 2017, 158, 1543–1555. [Google Scholar] [CrossRef]
- Felice, D.L.; El-Shennawy, L.; Zhao, S.; Lantvit, D.L.; Shen, Q.; Unterman, T.G.; Swanson, S.M.; Frasor, J. Growth Hormone Potentiates 17β-Estradiol-Dependent Breast Cancer Cell Proliferation Independently of IGF-I Receptor Signaling. Endocrinology 2013, 154, 3219–3227. [Google Scholar] [CrossRef]
- Annunziata, M.; Granata, R.; Ghigo, E. The IGF system. Acta Diabetol. 2011, 48, 1–9. [Google Scholar] [CrossRef]
- Moromisato, D.Y.; Moromisato, M.Y.; Zanconato, S.; Roberts, J.; Brasel, J.A.; Cooper, D.M. Effect of hypoxia on lung, heart, and liver insulin-like growth factor-I gene and receptor expression in the newborn rat. Crit. Care Med. 1996, 24, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, E. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha /ARNT. EMBO J. 1998, 17, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Basu, R.; Mathes, S.C.; Arnett, N.A.; Duran-Ortiz, S.; Funk, K.R.; Brittain, A.L.; Kulkarni, P.; Terry, J.C.; Davis, E.; et al. Growth Hormone Upregulates Mediators of Melanoma Drug Efflux and Epithelial-to-Mesenchymal Transition In Vitro and In Vivo. Cancers 2020, 12, 3640. [Google Scholar] [CrossRef]
- Boguszewski, C.L.; Boguszewski, M.C.D.S. Growth Hormone’s Links to Cancer. Endocr. Rev. 2019, 40, 558–574. [Google Scholar] [CrossRef]
- Nakonechnaya, A.O.; Jefferson, H.S.; Chen, X.; Shewchuk, B.M. Differential effects of exogenous and autocrine growth hormone on LNCaP prostate cancer cell proliferation and survival. J. Cell Biochem. 2013, 114, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Nakonechnaya, A.O.; Shewchuk, B.M. Growth hormone enhances LNCaP prostate cancer cell motility. Endocr. Res. 2015, 40, 97–105. [Google Scholar] [CrossRef]
- Firth, S.M.; Baxter, R.C. Cellular Actions of the Insulin-Like Growth Factor Binding Proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef]
- Cohen, P.; Graves, H.C.; Peehl, D.M.; Kamarei, M.; Giudice, L.C.; Rosenfeld, R.G. Prostate-specific antigen (PSA) is an insulin-like growth factor binding protein-3 protease found in seminal plasma. J. Clin. Endocrinol. Metab. 1992, 75, 1046–1053. [Google Scholar]
- Bidosee, M.; Karry, R.; Weiss-Messer, E.; Barkey, R.J. Growth hormone affects gene expression and proliferation in human prostate cancer cells. Int. J. Androl. 2011, 34, 124–137. [Google Scholar] [CrossRef]
- Recouvreux, M.V.; Wu, J.B.; Gao, A.C.; Zonis, S.; Chesnokova, V.; Bhowmick, N.; Chung, L.W.; Melmed, S. Androgen Receptor Regulation of Local Growth Hormone in Prostate Cancer Cells. Endocrinology 2017, 158, 2255–2268. [Google Scholar] [CrossRef]
- Wang, J.-J.; Chong, Q.-Y.; Sun, X.-B.; You, M.-L.; Pandey, V.; Chen, Y.-J.; Zhuang, Q.-S.; Liu, D.-X.; Ma, L.; Wu, Z.-S.; et al. Autocrine hGH stimulates oncogenicity, epithelial-mesenchymal transition and cancer stem cell-like behavior in human colorectal carcinoma. Oncotarget 2017, 8, 103900–103918. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, V.; Zonis, S.; Zhou, C.; Recouvreux, M.V.; Ben-Shlomo, A.; Araki, T.; Barrett, R.; Workman, M.; Wawrowsky, K.; Ljubimov, V.A.; et al. Growth hormone is permissive for neoplastic colon growth. Proc. Natl. Acad. Sci. USA 2016, 113, E3250–E3259. [Google Scholar] [CrossRef] [PubMed]
- Belizon, A.; Balik, E.; Kirman, I.; Remotti, H.; Ciau, N.; Jain, S.; Whelan, R.L. Insulin-Like Growth Factor Binding Protein-3 Inhibits Colitis-Induced Carcinogenesis. Dis. Colon Rectum 2007, 50, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.F.; Webb, E.L.; Matakidou, A.; Sellick, G.S.; Williams, R.D.; Bridle, H.; Eisen, T.; Houlston, R.S.; GELCAPS Consortium. Variants in the GH-IGF axis confer susceptibilityto lung cancer. Genome Res. 2006, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Lu, H.; Feng, J.; Shu, J.; Zheng, D.; Hou, Y. Lung Cancer Risk Associated with Thr495Pro Polymorphism of GHR in Chinese Population. Jpn. J. Clin. Oncol. 2008, 38, 308–316. [Google Scholar] [CrossRef]
- Van Dyke, A.L.; Cote, M.L.; Wenzlaff, A.S.; Abrams, J.; Land, S.; Iyer, P.; Schwartz, A.G. Chromosome 5p Region SNPs Are Associated with Risk of NSCLC among Women. J. Cancer Epidemiol. 2009, 2009, 242151. [Google Scholar] [CrossRef]



| Sample | GH Effect | Mechanisms of Action | IGF1 Effect | Mechanisms of Action |
|---|---|---|---|---|
| Estrogens | ↑ | Increased Ghrelin sensivity | ↓ | Decrease GH receptor expression on target cells |
| Increased GH mRNA production | PLC activation → inhibition of JAK/STAT signaling pathway | |||
| Loss of negative feedback after IGF1 decrease | Upregulation of SOCS2 → impairment of JAK/STAT pathway | |||
| SERMs | ↓ | Anti-estrogenic effect at hypothalamus and pituitary level (in vitro) * | ↓ | Decrease GH receptor expression on target cells |
| PLC activation → inhibition of JAK/STAT signaling pathway | ||||
| Upregulation of SOCS2 → impairment of JAK/STAT pathway |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voltan, G.; Mazzeo, P.; Regazzo, D.; Scaroni, C.; Ceccato, F. Role of Estrogen and Estrogen Receptor in GH-Secreting Adenomas. Int. J. Mol. Sci. 2023, 24, 9920. https://doi.org/10.3390/ijms24129920
Voltan G, Mazzeo P, Regazzo D, Scaroni C, Ceccato F. Role of Estrogen and Estrogen Receptor in GH-Secreting Adenomas. International Journal of Molecular Sciences. 2023; 24(12):9920. https://doi.org/10.3390/ijms24129920
Chicago/Turabian StyleVoltan, Giacomo, Pierluigi Mazzeo, Daniela Regazzo, Carla Scaroni, and Filippo Ceccato. 2023. "Role of Estrogen and Estrogen Receptor in GH-Secreting Adenomas" International Journal of Molecular Sciences 24, no. 12: 9920. https://doi.org/10.3390/ijms24129920
APA StyleVoltan, G., Mazzeo, P., Regazzo, D., Scaroni, C., & Ceccato, F. (2023). Role of Estrogen and Estrogen Receptor in GH-Secreting Adenomas. International Journal of Molecular Sciences, 24(12), 9920. https://doi.org/10.3390/ijms24129920