
Effects of Diabetes Mellitus-Related Dysglycemia on the Functions of Blood–Brain Barrier and the Risk of Dementia



Abstract
1. Diabetes Mellitus—A Short Resume
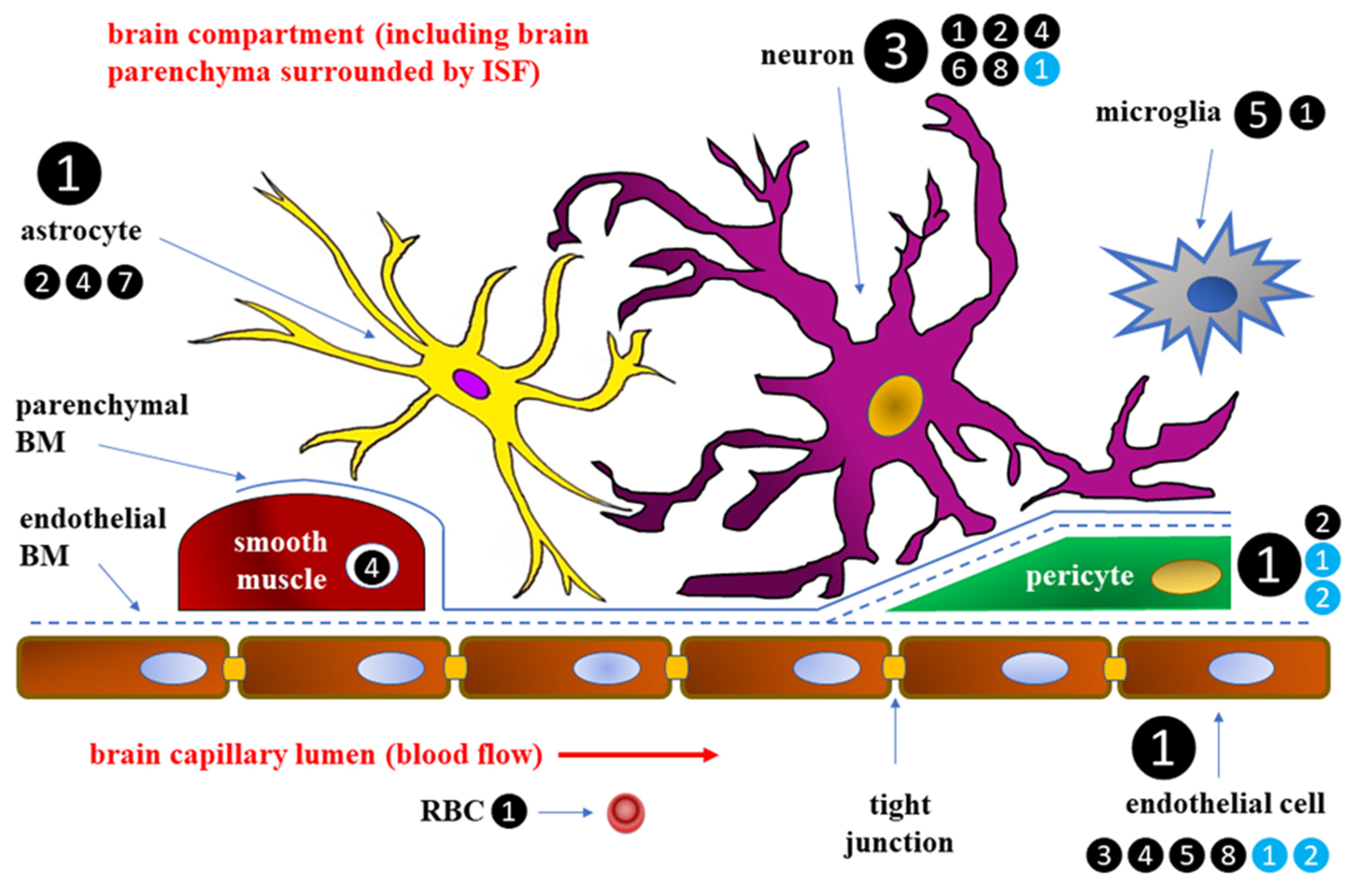
2. Blood–Brain Barrier
2.1. Glucose Transport across BBB
2.1.1. Hyperglycemia and Its Effect on Glucose Transport across the BBB
2.1.2. Hypoglycemia and Its Effect on Glucose Transport across the BBB
2.2. Alterations in Amino Acid Transport across the BBB in the Course of DM
2.3. Effects of DM on BBB Integrity and Permeability
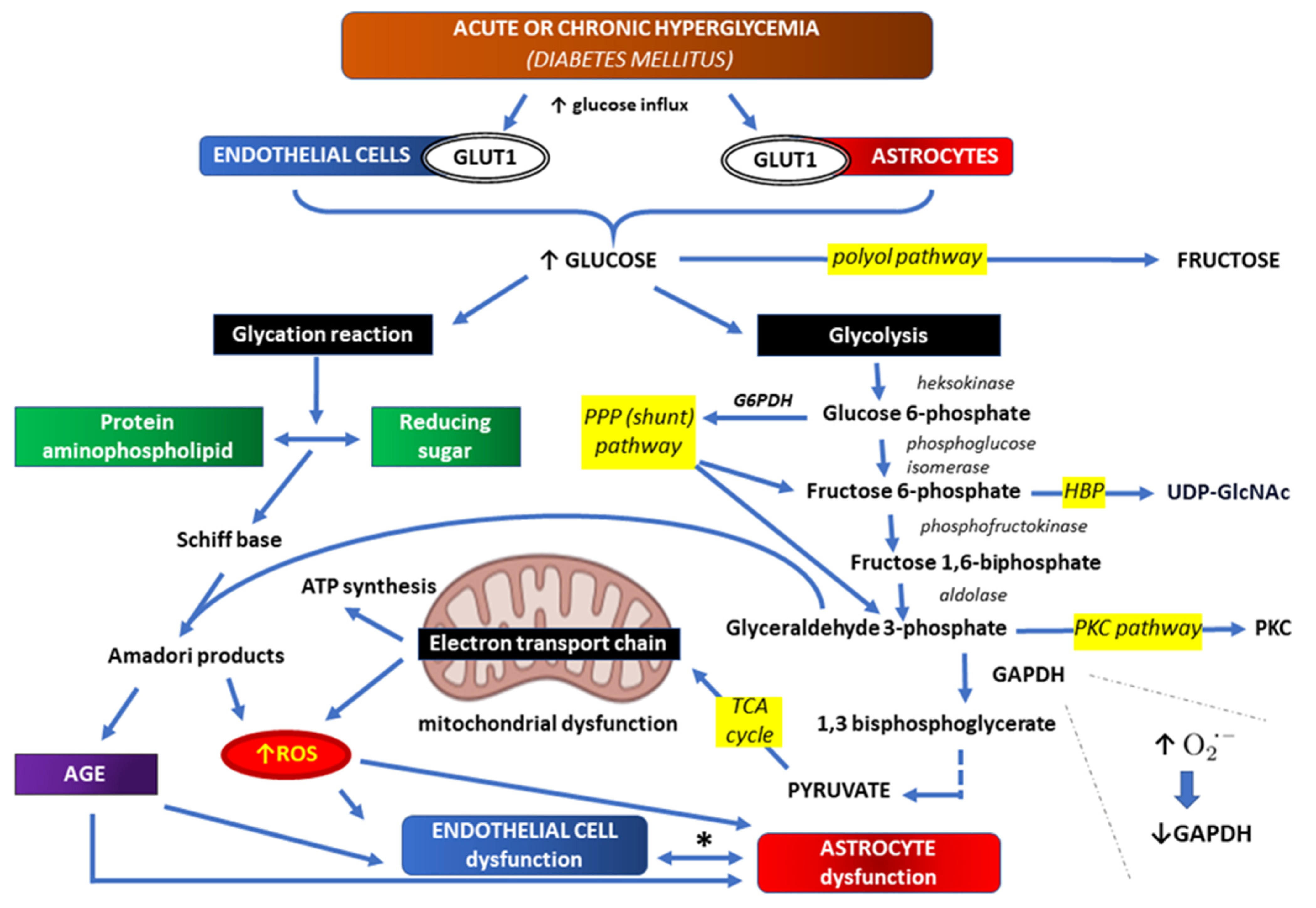
3. Correlations between Hyperglycemia and Oxidative Stress within BBB
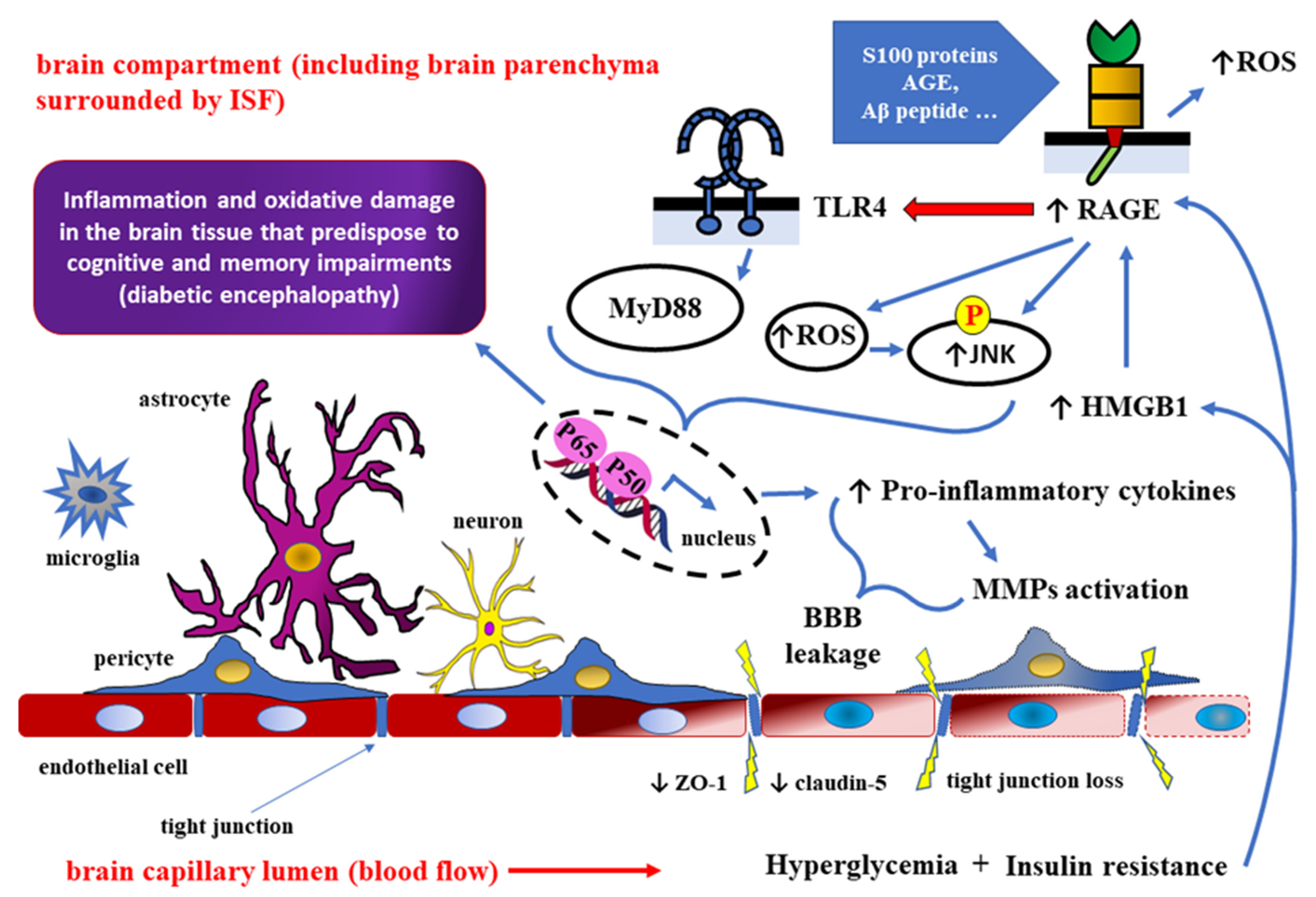
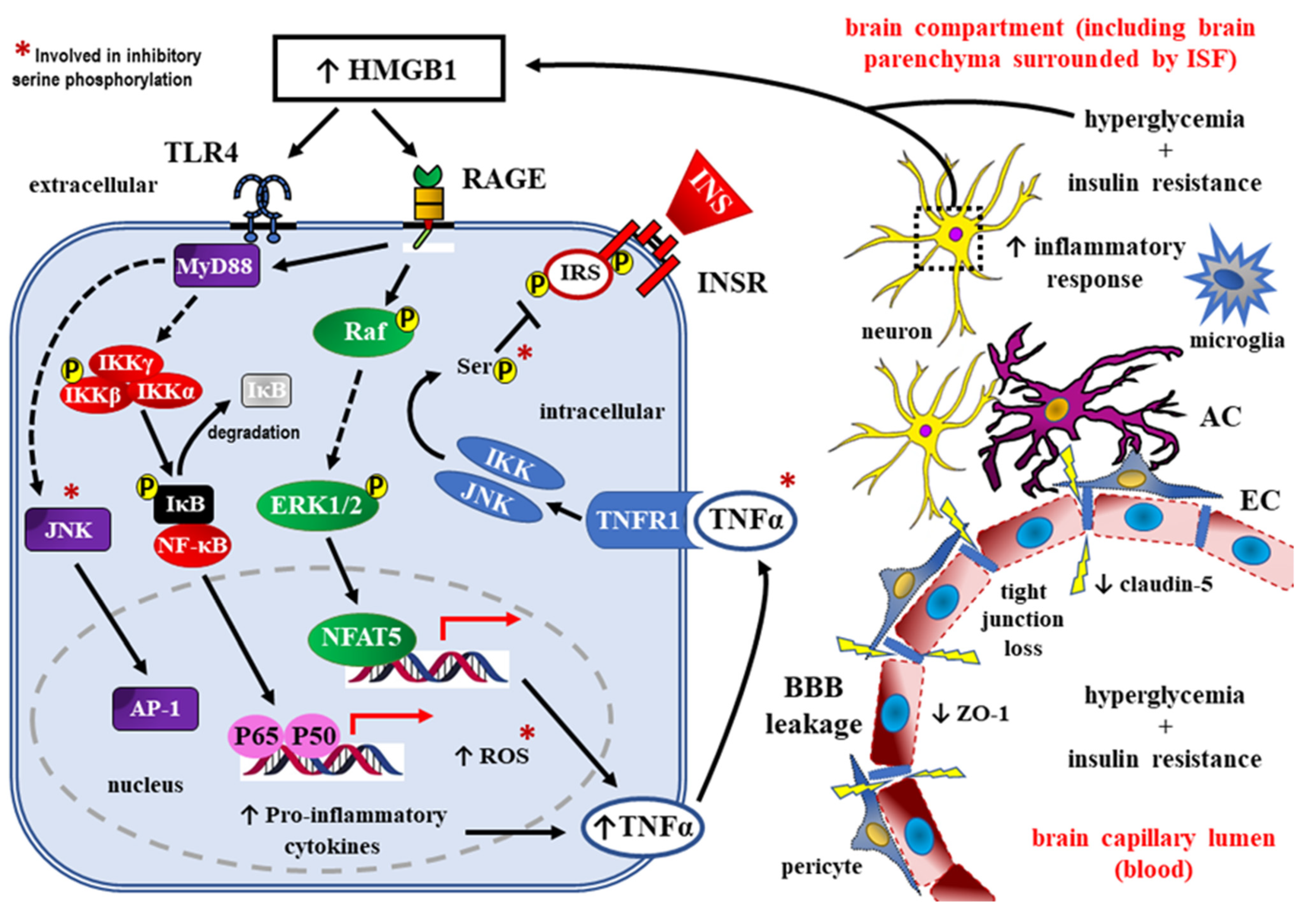
4. Role of HMGB1 as RAGE Ligand in Detrimental Effects of DM towards the CNS
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | beta-amyloid |
| ABC-transporter | ATP-binding cassette transporter (P-glycoprotein—P-gp) |
| AD | Alzheimer’s disease |
| AGE | advanced glycation end products |
| ATP | adenosine triphosphate |
| BBB | blood–brain barrier |
| BiP | binding immunoglobulin protein |
| BRCP | breast cancer resistance protein |
| CCL-2 | C-C motif chemokine ligand 2 (also known as MCP-1—monocyte chemoattractant protein-1) |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DHA | dehydroascorbic acid |
| DM | diabetes mellitus |
| FAD | flavin adenine dinucleotide |
| FADH | flavin adenine dinucleotide (reduced form) |
| GADPH | glyceraldehyde 3 phosphate dehydrogenase |
| GLUT-1, GLUT-3, GLUT-4 | glucose transporter 1, 3, and 4, respectively |
| GLUTs | glucose transporters |
| GSH | reduced L-Glutathione |
| GSK3 | glycogen synthase kinase-3 |
| HAAF | hypoglycemia-associated autonomic failure |
| HBP | hexosamine biosynthetic pathway |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HMGB1 | high mobility group box 1 |
| ICAM-1 | intercellular adhesion molecule 1 |
| IL-1, IL-4, IL-6 | interleukin 1, 4, and 6, respectively |
| iNOS | inducible nitric oxide synthase |
| IRS-1 | insulin receptor substrate 1 |
| JAM-1 | junctional adhesion molecule-1 |
| JNK | c-Jun N-terminal kinase |
| MAPK phosphatase-1 | mitogen activated protein kinase phosphatase 1 |
| MARCKS | myristoylated alanine rich protein kinase C substrate |
| MMP-1, MMP-2, MMP-9 | matrix metalloproteinase-2 |
| MMPs | matrix metalloproteinases |
| MRP-4 | multidrug resistance protein |
| MyD88 | myeloid differentiation primary response 88 (adapter protein) |
| NAD | nicotinamide-adenine dinucleotide |
| NADH | nicotinamide adenine dinucleotide (reduced form) |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 inflammasome | leucine-rich repeat (LRR)-containing proteins (NLR) family member 3 inflammasome |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| p50/p65 | NF-κB heterodimer, member of the Rel family of transcription factors |
| P-gp | P-glycoprotein |
| PKC | protein kinase C |
| PKCβ | β isoform of protein kinase C |
| PPP | pentose phosphate pathway |
| PRR | pattern-recognition receptors |
| RAGE | receptors for advanced glycation end-products |
| ROS | reactive oxygen species |
| SGLTs | sodium-glucose co-transporters |
| s-RAGE | soluble receptor for advanced glycation end-products |
| STAT-3 | signal transducer and activator of transcription 3 |
| TCA | tricarboxylic acid |
| TIMP-1, TIMP-2 | tissue inhibitor of matrix metalloproteinase 1 and 2, respectively |
| TLR4 | Toll-like receptor 4 |
| TLRs | Toll-like receptors |
| TNF-α | tumor necrosis factor alpha |
| UDP-GlcNAc | uridine diphosphate N-acetylglucosamine |
| VEGF | vascular endothelial growth factor |
| ZO-1 | zonula occludens-1 (also known as tight junction protein-1) |
References
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, C.H. The internal secretion of the pancreas. 1922. Indian J. Med. Res. 2007, 125, 251–266. [Google Scholar] [PubMed]
- Krolewski, A.S.; Warram, J.H. Natural history of diabetes mellitus. In Principles and Practice of Endocrinology and Metabolism; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 1320–1327. [Google Scholar]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Banks, W.A. The Blood-Brain Barrier Interface in Diabetes Mellitus: Dysfunctions, Mechanisms and Approaches to Treatment. Curr. Pharm. Des. 2020, 26, 1438–1447. [Google Scholar] [CrossRef]
- Bradbury, M.W. The blood-brain barrier. Exp. Physiol. 1993, 78, 453–472. [Google Scholar] [CrossRef]
- Emmi, A.; Wenzel, H.J.; Schwartzkroin, P.A.; Taglialatela, M.; Castaldo, P.; Bianchi, L.; Nerbonne, J.; Robertson, G.A.; Janigro, D. Do Glia Have Heart? Expression and Functional Role for Ether-A-Go-Go Currents in Hippocampal Astrocytes. J. Neurosci. 2000, 20, 3915–3925. [Google Scholar] [CrossRef] [PubMed]
- Davson, H.; Segal, M.B. Physiology of the CSF and Blood-Brain Barriers; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Hawkins, B.; Davis, T. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Pooja Naik, L.C. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M.; Rogers, R.C.; McDougal, D.H.; Ritter, S.; Qualls-Creekmore, E.; Hermann, G.E.; Zhang, J.; Jiang, S.; Wei, J.; et al. Glucose transporters in the 21st Century. Am. J. Physiol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Zhao, F.-Q. Functional Properties and Genomics of Glucose Transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012, 100, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Duelli, R.; Kuschinsky, W. Brain Glucose Transporters: Relationship to Local Energy Demand. News Physiol. Sci. 2001, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Vemula, S.; Roder, K.E.; Yang, T.; Bhat, G.J.; Thekkumkara, T.J.; Abbruscato, T.J. A Functional Role for Sodium-Dependent Glucose Transport across the Blood-Brain Barrier during Oxygen Glucose Deprivation. J. Pharmacol. Exp. Ther. 2009, 328, 487–495. [Google Scholar] [CrossRef]
- McAllister, M.S.; Krizanac-Bengez, L.; Macchia, F.; Naftalin, R.; Pedley, K.C.; Mayberg, M.; Marroni, M.; Leaman, S.; Stanness, K.A.; Janigro, D. Mechanisms of glucose transport at the blood–brain barrier: An in vitro study. Brain Res. 2001, 904, 20–30. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S.; Swartz, B.E. The Human Brain GLUT1 Glucose Transporter: Ultrastructural Localization to the Blood—Brain Barrier Endothelia. J. Cereb. Blood Flow Metab. 1994, 14, 106–112. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S. Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. Neurorx 2005, 2, 27–43. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Frank, H.J.L.; Pardridge, W.M.; Jankovic-Vokes, T.; Vinters, H.V.; Morris, W.L. Insulin Binding to the Blood-Brain Barrier in the Streptozotocin Diabetic Rat. J. Neurochem. 1986, 47, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Insulin and the brain. Arch. Physiol. Biochem. 2009, 115, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human Blood? Brain Barrier Insulin Receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jaspan, J.B.; Huang, W.; Kastin, A.J. Transport of Insulin Across the Blood-Brain Barrier: Saturability at Euglycemic Doses of Insulin. Peptides 1997, 18, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nirwane, A.; Yao, Y. Basement membrane and blood–brain barrier. Stroke Vasc. Neurol. 2018, 4, 78–82. [Google Scholar] [CrossRef]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef]
- Li, X.-H.; Lv, B.-L.; Xie, J.-Z.; Liu, J.; Zhou, X.-W.; Wang, J.-Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef]
- Lam, J.K.Y.; Wang, Y.; Shiu, S.W.M.; Wong, Y.; Betteridge, D.J.; Tan, K.C.B. Effect of insulin on the soluble receptor for advanced glycation end products (RAGE). Diabet. Med. 2013, 30, 702–709. [Google Scholar] [CrossRef]
- Thomas, M.C.; on behalf of the FinnDiane Study Group; Söderlund, J.; Lehto, M.; Mäkinen, V.-P.; Moran, J.L.; Cooper, M.E.; Forsblom, C.; Groop, P.-H. Soluble receptor for AGE (RAGE) is a novel independent predictor of all-cause and cardiovascular mortality in type 1 diabetes. Diabetologia 2011, 54, 2669–2677. [Google Scholar] [CrossRef]
- Fujisawa, K.; Katakami, N.; Kaneto, H.; Naka, T.; Takahara, M.; Sakamoto, F.; Irie, Y.; Miyashita, K.; Kubo, F.; Yasuda, T.; et al. Circulating soluble RAGE as a predictive biomarker of cardiovascular event risk in patients with type 2 diabetes. Atherosclerosis 2013, 227, 425–428. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Imaizumi, T. Serum Levels of Soluble Form of Receptor for Advanced Glycation End Products (sRAGE) May Reflect Tissue RAGE Expression In Diabetes. Arter. Thromb. Vasc. Biol. 2007, 27, e32, author reply e33-4. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.-I. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Yamagishi, S.-I.; Matsui, T.; Nakamura, K.; Imaizumi, T. Role of Advanced Glycation End Products (AGEs) in Thrombogenic Abnormalities in Diabetes. Curr. Neurovascular Res. 2006, 3, 73–77. [Google Scholar] [CrossRef]
- Duelli, R.; Maurer, M.; Staudt, R.; Heiland, S.; Duembgen, L.; Kuschinsky, W. Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res. 2000, 858, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.-K.; Xian, Y.-X.; Zhang, L.; Lai, H.; Hou, X.-G.; Xu, Y.-X.; Yu, T.; Xu, F.-Y.; Song, J.; Fu, C.-L.; et al. Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin. Med. J. 2007, 120, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Triguero, D.; Farrell, C.R. Downregulation of Blood-Brain Barrier Glucose Transporter in Experimental Diabetes. Diabetes 1990, 39, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Appel, N.M.; Hokari, M.; Oki, J.; Holman, G.D.; Maher, F.; Koehler-Stec, E.M.; Vannucci, S.J.; Smith, Q.R. Blood-Brain Barrier Glucose Transporter: Effects of hypo- and hyperglycemia revisited. J. Neurochem. 1999, 72, 238–247. [Google Scholar] [CrossRef]
- Gruetter, R.; Novotny, E.J.; Boulware, S.D.; Rothman, D.L.; Shulman, R.G. 1H NMR Studies of Glucose Transport in the Human Brain. J. Cereb. Blood Flow Metab. 1996, 16, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, S.G.; Knudsen, G.M.; Capaldo, B.; Postiglione, A.; Paulson, O.B. Blood-Brain Barrier Transport and Brain Metabolism of Glucose during Acute Hyperglycemia in Humans1. J. Clin. Endocrinol. Metab. 2001, 86, 1986–1990. [Google Scholar] [CrossRef]
- Nielsen, J.K.; Djurhuus, C.B.; Gravholt, C.H.; Carus, A.C.; Granild-Jensen, J.; Ørskov, H.; Christiansen, J.S. Continuous Glucose Monitoring in Interstitial Subcutaneous Adipose Tissue and Skeletal Muscle Reflects Excursions in Cerebral Cortex. Diabetes 2005, 54, 1635–1639. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013. [Google Scholar] [CrossRef]
- Leão, L.L.; Tangen, G.; Barca, M.L.; Engedal, K.; Santos, S.H.S.; Machado, F.S.M.; de Paula, A.M.B.; Monteiro-Junior, R.S. Does hyperglycemia downregulate glucose transporters in the brain? Med. Hypotheses 2020, 139, 109614. [Google Scholar] [CrossRef]
- Cryer, P.E. Mechanisms of Hypoglycemia-Associated Autonomic Failure in Diabetes. N. Engl. J. Med. 2013, 369, 362–372. [Google Scholar] [CrossRef]
- Bakatselos, S.O. Hypoglycemia unawareness. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S92–S96. [Google Scholar] [CrossRef]
- McCrimmon, R.J.; Jacob, R.J.; Fan, X.; McNay, E.C.; Sherwin, R.S. Effects of recurrent antecedent hypoglycaemia and chronic hyperglycaemia on brainstem extra-cellular glucose concentrations during acute hypoglycaemia in conscious diabetic BB rats. Diabetologia 2003, 46, 1658–1661. [Google Scholar] [CrossRef][Green Version]
- Kumagai, A.K.; Kang, Y.-S.; Boado, R.J.; Pardridge, W.M. Upregulation of Blood-Brain Barrier GLUT1 Glucose Transporter Protein and mRNA in Experimental Chronic Hypoglycemia. Diabetes 1995, 44, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- McCall, A.L.; Fixman, L.B.; Fleming, N.; Tornheim, K.; Chick, W.; Ruderman, N.B. Chronic hypoglycemia increases brain glucose transport. Am. J. Physiol. Metab. 1986, 251, E442–E447. [Google Scholar] [CrossRef]
- Mastaitis, J.W.; Wurmbach, E.; Cheng, H.; Sealfon, S.C.; Mobbs, C.V. Acute Induction of Gene Expression in Brain and Liver by Insulin-Induced Hypoglycemia. Diabetes 2005, 54, 952–958. [Google Scholar] [CrossRef]
- Katon, W.J.; Young, B.A.; Russo, J.; Lin, E.H.B.; Ciechanowski, P.; Ludman, E.J.; Von Korff, M.R. Association of Depression with Increased Risk of Severe Hypoglycemic Episodes in Patients with Diabetes. Ann. Fam. Med. 2013, 11, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Blood-Brain Barrier Choline Transport Is Reduced in Diabetic Rats. Diabetes 1987, 36, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Mans, A.M.; DeJoseph, M.R.; Davis, D.W.; Hawkins, R.A. Regional amino acid transport into brain during diabetes: Effect of plasma amino acids. Am. J. Physiol. Metab. 1987, 253, E575–E583. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Lundeen, T.F.; Norwood, K.M.; Brooks, H.L.; Egleton, R.D. Increased blood–brain barrier permeability and altered tight junctions in experimental diabetes in the rat: Contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 2006, 50, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.K.; Levin, E.C.; Clifford, P.M.; Han, M.; Tourtellotte, R.; Chamberlain, D.; Pollaro, M.; Coretti, N.J.; Kosciuk, M.C.; Nagele, E.P.; et al. Diabetes and Hypercholesterolemia Increase Blood-Brain Barrier Permeability and Brain Amyloid Deposition: Beneficial Effects of the LpPLA2 Inhibitor Darapladib. J. Alzheimer’s Dis. 2013, 35, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef]
- Huber, J.D.; VanGilder, R.L.; Houser, K.A. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am. J. Physiol. Circ. Physiol. 2006, 291, H2660–H2668. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood-brain barrier hyperpermeability. Diabetes, Obes. Metab. 2009, 11, 480–490. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Wang, Z.; Zhang, X.; Yao, L.; Wang, F.; Liu, S.; Yin, J.; Ling, E.-A.; Wang, L.; et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012, 202, 58–68. [Google Scholar] [CrossRef]
- Ball, K.K.; Harik, L.; Gandhi, G.K.; Cruz, N.F.; Dienel, G.A. Reduced gap junctional communication among astrocytes in experimental diabetes: Contributions of altered connexin protein levels and oxidative-nitrosative modifications. J. Neurosci. Res. 2011, 89, 2052–2067. [Google Scholar] [CrossRef]
- Gandhi, G.K.; Ball, K.K.; Cruz, N.F.; Dienel, G.A. Hyperglycaemia and Diabetes Impair Gap Junctional Communication among Astrocytes. ASN Neuro 2010, 2, e00030. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Tominaga, O.; Maeda, T.; Abe, M.-A.; Kanda, T. Advanced glycation end-products disrupt the blood–brain barrier by stimulating the release of transforming growth factor–β by pericytes and vascular endothelial growth factor and matrix metalloproteinase–2 by endothelial cells in vitro. Neurobiol. Aging 2013, 34, 1902–1912. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef]
- Vorbrodt, A.W.; Dobrogowska, D.H.; Tarnawski, M.; Meeker, H.C.; Carp, R.I. Immunogold study of altered expression of some interendothelial junctional molecules in the brain blood microvessels of diabetic scrapie-infected mice. J. Mol. Histol. 2006, 37, 27–35. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Z.; Shi, H. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell. Mol. Life Sci. 2012, 69, 115–128. [Google Scholar] [CrossRef]
- Oltmanns, K.M.; Melchert, U.H.; Scholand-Engler, H.G.; Schultes, B.; Schweiger, U.; Peters, A. Divergent effects of hyper- and hypoglycemia on circulating vascular endothelial growth factor in humans. Metabolism 2008, 57, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chehade, J.M.; Haas, M.J.; Mooradian, A.D. Diabetes-Related Changes in Rat Cerebral Occludin and Zonula Occludens-1 (ZO-1) Expression. Neurochem. Res. 2002, 27, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.H.; Stamatovic, S.M.; Andjelkovic, A.V. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 2009, 1254, 138–148. [Google Scholar] [CrossRef]
- Ding, C.; He, Q.; Li, P.-A. Diabetes increases expression of ICAM after a brief period of cerebral ischemia. J. Neuroimmunol. 2005, 161, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.R.; Keep, R.F. Effect of sustained-mild and transient-severe hyperglycemia on ischemia-induced blood-brain barrier opening. J. Cereb. Blood Flow Metab. 2007, 27, 1573–1582. [Google Scholar] [CrossRef]
- Ergul, A.; Elgebaly, M.M.; Middlemore, M.-L.; Li, W.; Elewa, H.; Switzer, J.A.; Hall, C.; Kozak, A.; Fagan, S.C. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC Neurol. 2007, 7, 33. [Google Scholar] [CrossRef]
- Vavilala, M.S.; Richards, T.L.; Roberts, J.S.; Chiu, H.; Pihoker, C.; Bradford, H.; Deeter, K.; Marro, K.I.; Shaw, D. Change in blood–brain barrier permeability during pediatric diabetic ketoacidosis treatment*. Pediatr. Crit. Care Med. 2009, 11, 332–338. [Google Scholar] [CrossRef]
- Liu, H.; Xu, X.; Yang, Z.; Deng, Y.; Liu, X.; Xie, L. Impaired function and expression of P-glycoprotein in blood–brain barrier of streptozotocin-induced diabetic rats. Brain Res. 2006, 1123, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, X.; Jia, L.; Liu, Y.; Yang, H.; Wang, G.; Xie, L. Insulin therapy restores impaired function and expression of P-glycoprotein in blood–brain barrier of experimental diabetes. Biochem. Pharmacol. 2008, 75, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.-J.; Kim, M.-H.; Jin, H.-E.; Shin, S.M.; Tsuruo, T.; Kim, S.G.; Kim, D.-D.; Shim, C.-K.; Chung, S.-J. Functional Induction of P-glycoprotein in the Blood-Brain Barrier of Streptozotocin-Induced Diabetic Rats: Evidence for the Involvement of Nuclear Factor-κB, a Nitrosative Stress-Sensitive Transcription Factor, in the Regulation. Drug Metab. Dispos. 2007, 35, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- McCuskey, P.A.; McCuskey, R.S. In vivo and electron microscopic study of the development of cerebral diabetic microangiography. Microcirc. Endothel. Lymphat. 1984, 1, 221–244. [Google Scholar]
- Prakash, R.; Johnson, M.; Fagan, S.C.; Ergul, A. Cerebral Neovascularization and Remodeling Patterns in Two Different Models of Type 2 Diabetes. PLoS ONE 2013, 8, e56264. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Naik, P.; Prasad, S.; Cucullo, L. Role and Function of Dehydrogenases in CNS and Blood-Brain Barrier Pathophysiology. In Dehydrogenases; Intech: London, UK, 2013. [Google Scholar]
- Takahashi, S.; Abe, T.; Izawa, Y.; Suzuki, N. Effects of fluctuating glucose concentrations on oxidative metabolism of glucose in cultured neurons and astroglia. J. Diabetes Mellit. 2012, 2, 19–26. [Google Scholar] [CrossRef]
- Okouchi, M.; Okayama, N.; Alexander, J.S.; Aw, T.Y. NRF2-Dependent Glutamate-L-Cysteine Ligase Catalytic Subunit Expression Mediates Insulin Protection Against Hyperglycemia-Induced Brain Endothelial Cell Apoptosis. Curr. Neurovascular Res. 2006, 3, 249–261. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood?brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astroglial Pentose Phosphate Pathway Rates in Response to High-Glucose Environments. ASN Neuro 2012, 4, e00078. [Google Scholar] [CrossRef]
- Devraj, K.; Klinger, M.E.; Myers, R.L.; Mokashi, A.; Hawkins, R.A.; Simpson, I.A. GLUT-1 glucose transporters in the blood-brain barrier: Differential phosphorylation. J. Neurosci. Res. 2011, 89, 1913–1925. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Challenger, S.; Kream, R. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur. J. Nutr. 2016, 55, 2339–2345. [Google Scholar] [CrossRef]
- Cipolla, M.J.; Huang, Q.; Sweet, J.G. Inhibition of Protein Kinase Cβ Reverses Increased Blood–Brain Barrier Permeability During Hyperglycemic Stroke and Prevents Edema Formation In Vivo. Stroke 2011, 42, 3252–3257. [Google Scholar] [CrossRef]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Son, M.; Hong, J.; Jeong, G.-B.; Paek, S.H.; Won, M.-H.; Lee, B. Activated microglial cells synthesize and secrete AGE-albumin. Anat. Cell Biol. 2012, 45, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Guerin-Dubourg, A.; Catan, A.; Bourdon, E.; Rondeau, P. Structural modifications of human albumin in diabetes. Diabetes Metab. 2012, 38, 171–178. [Google Scholar] [CrossRef]
- Lyons, T.J.; Basu, A. Biomarkers in diabetes: Hemoglobin A1c, vascular and tissue markers. Transl. Res. 2012, 159, 303–312. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and Basic Amino Acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous Danger Signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Min, H.J.; Ko, E.A.; Wu, J.; Kim, E.S.; Kwon, M.K.; Kwak, M.S.; Choi, J.E.; Lee, J.E.; Shin, J.-S. Chaperone-like Activity of High-Mobility Group Box 1 Protein and Its Role in Reducing the Formation of Polyglutamine Aggregates. J. Immunol. 2013, 190, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Štros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta 2010, 1799, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.R.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Nan, K.; Han, Y.; Fang, Q.; Huang, C.; Yu, L.; Ge, W.; Xiang, F.; Tao, Y.-X.; Cao, H.; Li, J. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-κB signaling in primary hippocampal neurons induced by Aβ25–35. Int. Immunopharmacol. 2019, 67, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.; Rauvala, H. Amphoterin as an extracellular regulator of cell motility: From discovery to disease. J. Intern. Med. 2004, 255, 351–366. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The Receptor for Advanced Glycation End Products (RAGE) Is a Cellular Binding Site for Amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-Induced Reactive Oxygen Species Increase Expression of the Receptor for Advanced Glycation End Products (RAGE) and RAGE Ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhong, J.; Zhang, X.; Liu, Z.; Yang, Y.; Gong, Q.; Ren, B. The Role of HMGB1 in the Pathogenesis of Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 2543268. [Google Scholar] [CrossRef]
- Gharaati, M.E.; Nahavandi, A.; Mojarad, T.B.; Roghani, M. Diabetic Encephalopathy Affecting Mitochondria and Axonal Transport Proteins. Basic Clin. Neurosci. J. 2020, 11, 781–794. [Google Scholar] [CrossRef]
- Gaikwad, S.; Puangmalai, N.; Bittar, A.; Montalbano, M.; Garcia, S.; McAllen, S.; Bhatt, N.; Sonawane, M.; Sengupta, U.; Kayed, R. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer’s disease and frontotemporal dementia. Cell Rep. 2021, 36, 109419. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, A.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Fernandes, A.; Aguilar-Pimentel, J.A.; de Angelis, M.H.; Guedes, J.R.; Brito, M.A.; Ortolano, S.; Pani, G.; Athanasopoulou, S.; Gonos, E.S.; et al. Towards frailty biomarkers: Candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res. Rev. 2018, 47, 214–277. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Fürnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; De Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1–nucleosome complexes: Implications for the pathogenesis of SLE. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Shi, Y.; Du, P.; Wang, J.; Han, Y.; Sun, B.; Feng, J. HMGB1/TLR4 promotes apoptosis and reduces autophagy of hippocampal neurons in diabetes combined with OSA. Life Sci. 2019, 239, 117020. [Google Scholar] [CrossRef] [PubMed]
- Famakin, B.M.; Tsymbalyuk, O.; Tsymbalyuk, N.; Ivanova, S.; Woo, S.K.; Kwon, M.S.; Gerzanich, V.; Simard, J.M. HMGB1 is a Potential Mediator of Astrocytic TLR4 Signaling Activation following Acute and Chronic Focal Cerebral Ischemia. Neurol. Res. Int. 2020, 2020, 3929438. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Jiang, Y.; Steinle, J.J. HMGB1 inhibits insulin signalling through TLR4 and RAGE in human retinal endothelial cells. Growth Factors 2018, 36, 164–171. [Google Scholar] [CrossRef]
- Wang, X.-X.; Tan, M.-S.; Yu, J.-T.; Tan, L. Matrix Metalloproteinases and Their Multiple Roles in Alzheimer’s Disease. BioMed. Res. Int. 2014, 2014, 908636. [Google Scholar] [CrossRef]
- Nagele, R.G.; Clifford, P.M.; Siu, G.; Levin, E.C.; Acharya, N.K.; Han, M.; Kosciuk, M.C.; Venkataraman, V.; Zavareh, S.; Zarrabi, S.; et al. Brain-Reactive Autoantibodies Prevalent in Human Sera Increase Intraneuronal Amyloid-β1-42 Deposition. J. Alzheimer’s Dis. 2011, 25, 605–622. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Lee, D.H.; Song, J. HMGB1 signaling pathway in diabetes-related dementia: Blood-brain barrier breakdown, brain insulin resistance, and Aβ accumulation. Biomed. Pharmacother. 2022, 150, 112933. [Google Scholar] [CrossRef]
- Skrha, J., Jr.; Kalousová, M.; Švarcová, J.; Muravská, A.; Kvasnička, J.; Landová, L.; Zima, T.; Škrha, J. Relationship of Soluble RAGE and RAGE Ligands HMGB1 and EN-RAGE to Endothelial Dysfunction in Type 1 and Type 2 Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2012, 120, 277–281. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased Toll-Like Receptor (TLR) Activation and TLR Ligands in Recently Diagnosed Type 2 Diabetic Subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Iwasaka, H.; Hasegawa, A.; Koga, H.; Noguchi, T. Effects of hyperglycemia and insulin therapy on high mobility group box 1 in endotoxin-induced acute lung injury in a rat model*. Crit. Care Med. 2008, 36, 2407–2413. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of Neurite Outgrowth and Cell Survival by Amphoterin and S100 Proteins through Receptor for Advanced Glycation End Products (RAGE) Activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef]
- de Souza, A.; Westra, J.; Limburg, P.; Bijl, M.; Kallenberg, C. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun. Rev. 2012, 11, 909–917. [Google Scholar] [CrossRef]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Larbi, A.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and Toll-Like Receptors 2 and 4 Are Required for Fibrillar Aβ-Stimulated Microglial Activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Kakimura, J.-I.; Shibagaki, K.; Tsuchiya, D.; Taniguchi, T.; Smith, M.A.; Perry, G.; Shimohama, S. Role of high mobility group protein-1 (HMG1) in amyloid-β homeostasis. Biochem. Biophys. Res. Commun. 2003, 301, 699–703. [Google Scholar] [CrossRef]
- Falcão, A.S.; Carvalho, L.A.R.; Lidónio, G.; Vaz, A.R.; Lucas, S.D.; Moreira, R.; Brites, D. Dipeptidyl Vinyl Sulfone as a Novel Chemical Tool to Inhibit HMGB1/NLRP3-Inflammasome and Inflamma-miRs in Aβ-Mediated Microglial Inflammation. ACS Chem. Neurosci. 2017, 8, 89–99. [Google Scholar] [CrossRef]
- Takata, K.; Takada, T.; Ito, A.; Asai, M.; Tawa, M.; Saito, Y.; Ashihara, E.; Tomimoto, H.; Kitamura, Y.; Shimohama, S. Microglial Amyloid-β1-40 Phagocytosis Dysfunction Is Caused by High-Mobility Group Box Protein-1: Implications for the Pathological Progression of Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012, 2012, 685739. [Google Scholar] [CrossRef]
- Lue, L.-F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogersa, J.; Schmidt, A.M.; Stern, D.M.; Du Yan, S. Involvement of Microglial Receptor for Advanced Glycation Endproducts (RAGE) in Alzheimer’s Disease: Identification of a Cellular Activation Mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Mazarati, A.; Maroso, M.; Iori, V.; Vezzani, A.; Carli, M. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and Receptor for Advanced Glycation End Products. Exp. Neurol. 2011, 232, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, Z.; Xie, J.; Kang, L.-N.; Wang, L.; Xu, B. High Mobility Group Box-1: A Missing Link between Diabetes and Its Complications. Mediat. Inflamm. 2016, 2016, 3896147. [Google Scholar] [CrossRef]
- Montes, V.N.; Subramanian, S.; Goodspeed, L.; Wang, S.A.; Omer, M.; Bobik, A.; Teshigawara, K.; Nishibori, M.; Chait, A. Anti-HMGB1 antibody reduces weight gain in mice fed a high-fat diet. Nutr. Diabetes 2015, 5, e161. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Ruiz, R.; Ortega, F.; Rodríguez, A.A.; Vázquez-Martínez, R.; Díaz-Ruiz, A.A.; Garcia-Navarro, S.; Giralt, M.; Garcia-Rios, A.A.; Cobo-Padilla, D.; Tinahones, F.J.; et al. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. Int. J. Obes. 2014, 38, 1545–1554. [Google Scholar] [CrossRef]
- Ghosh, G.; Wang, V.Y.-F.; Huang, D.-B.; Fusco, A. NF-κB regulation: Lessons from structures. Immunol. Rev. 2012, 246, 36–58. [Google Scholar] [CrossRef]
- Neuhofer, W. Role of NFAT5 in Inflammatory Disorders Associated with Osmotic Stress. Curr. Genom. 2010, 11, 584–590. [Google Scholar] [CrossRef]
- van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef]
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Häring, H.-U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Schultes, B.; Born, J.; Kern, W. Intranasal Insulin to Improve Memory Function in Humans. Neuroendocrinology 2007, 86, 136–142. [Google Scholar] [CrossRef]
- Moosavi, M.; Naghdi, N.; Choopani, S. Intra CA1 insulin microinjection improves memory consolidation and retrieval. Peptides 2007, 28, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in alzheimer’s disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Kanjwal, Y.; Mohanty, P.; Rao, S.; Sung, B.H.; Wilson, M.; Dandona, P. Insulin-induced vasodilatation of internal carotid artery. Metabolism 1999, 48, 1470–1473. [Google Scholar] [CrossRef]
- Hoscheidt, S.M.; Kellawan, J.M.; Berman, S.E.; Rivera-Rivera, L.A.; Krause, R.A.; Oh, J.M.; Beeri, M.S.; Rowley, H.A.; Wieben, O.; Carlsson, C.M.; et al. Insulin resistance is associated with lower arterial blood flow and reduced cortical perfusion in cognitively asymptomatic middle-aged adults. J. Cereb. Blood Flow Metab. 2016, 37, 2249–2261. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Watson, G.S.; Peskind, E.R.; Asthana, S.; Purganan, K.; Wait, C.; Chapman, D.; Schwartz, M.W.; Plymate, S.; Craft, S. Insulin increases CSF A 42 levels in normal older adults. Neurology 2003, 60, 1899–1903. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsubara, T.; Sobue, K.; Tanida, M.; Kasahara, R.; Naruse, K.; Taniura, H.; Sato, T.; Suzuki, K. Brain insulin resistance accelerates Aβ fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J. Neurochem. 2012, 121, 619–628. [Google Scholar] [CrossRef]
- de la Monte, S.M. Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimerʼs Disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 Perpetuates Metabolic Stress Induced by Aβ Peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, Inflammation, and Insulin Resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Nogueira-Machado, J.A.; Volpe, C.M.D.O.; Veloso, C.A.; Chaves, M.M. HMGB1, TLR and RAGE: A functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets 2011, 15, 1023–1035. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, F.; Yu, Z.; Guo, S.; Liu, N.; Jiang, Y.; Lo, E.H.; Xu, Y.; Wang, X. HDAC3 inhibition prevents blood-brain barrier permeability through Nrf2 activation in type 2 diabetes male mice. J. Neuroinflamm. 2019, 16, 103. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. Perivascular Drainage of Amyloid-β Peptides from the Brain and Its Failure in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef]
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K.-I. Role of toll-like receptor signalling in A uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kitamura, Y.; Tsuchiya, D.; Kawasaki, T.; Taniguchi, T.; Shimohama, S. High mobility group box protein-1 inhibits microglial A? clearance and enhances A? neurotoxicity. J. Neurosci. Res. 2004, 78, 880–891. [Google Scholar] [CrossRef]
- Schubert, M. The Correlation between Diabetes Mellitus and Neurodegenerative Diseases. Klin. Monbl. Augenheilkd. 2023, 240, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Chau, A.C.; Cheung, E.Y.; Chan, K.; Chow, W.; Shea, Y.; Chiu, P.K.; Mak, H.K. Impaired cerebral blood flow in type 2 diabetes mellitus A comparative study with subjective cognitive decline, vascular dementia and Alzheimer’s disease subjects. NeuroImage Clin. 2020, 27, 102302. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wątroba, M.; Grabowska, A.D.; Szukiewicz, D. Effects of Diabetes Mellitus-Related Dysglycemia on the Functions of Blood–Brain Barrier and the Risk of Dementia. Int. J. Mol. Sci. 2023, 24, 10069. https://doi.org/10.3390/ijms241210069
Wątroba M, Grabowska AD, Szukiewicz D. Effects of Diabetes Mellitus-Related Dysglycemia on the Functions of Blood–Brain Barrier and the Risk of Dementia. International Journal of Molecular Sciences. 2023; 24(12):10069. https://doi.org/10.3390/ijms241210069
Chicago/Turabian StyleWątroba, Mateusz, Anna D. Grabowska, and Dariusz Szukiewicz. 2023. "Effects of Diabetes Mellitus-Related Dysglycemia on the Functions of Blood–Brain Barrier and the Risk of Dementia" International Journal of Molecular Sciences 24, no. 12: 10069. https://doi.org/10.3390/ijms241210069
APA StyleWątroba, M., Grabowska, A. D., & Szukiewicz, D. (2023). Effects of Diabetes Mellitus-Related Dysglycemia on the Functions of Blood–Brain Barrier and the Risk of Dementia. International Journal of Molecular Sciences, 24(12), 10069. https://doi.org/10.3390/ijms241210069