
Asymmetric Dimethylation of Ribosomal S6 Kinase 2 Regulates Its Cellular Localisation and Pro-Survival Function

 ,
,  , ,
, ,  and
and 
Abstract
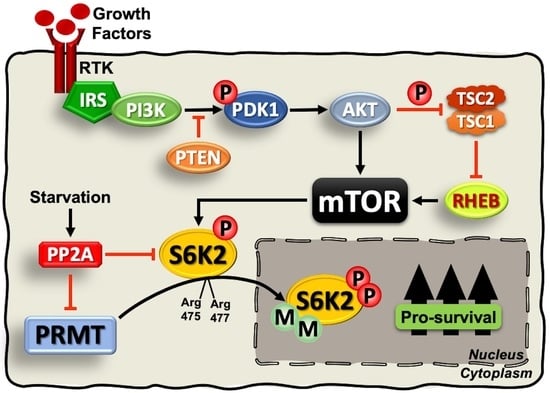
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
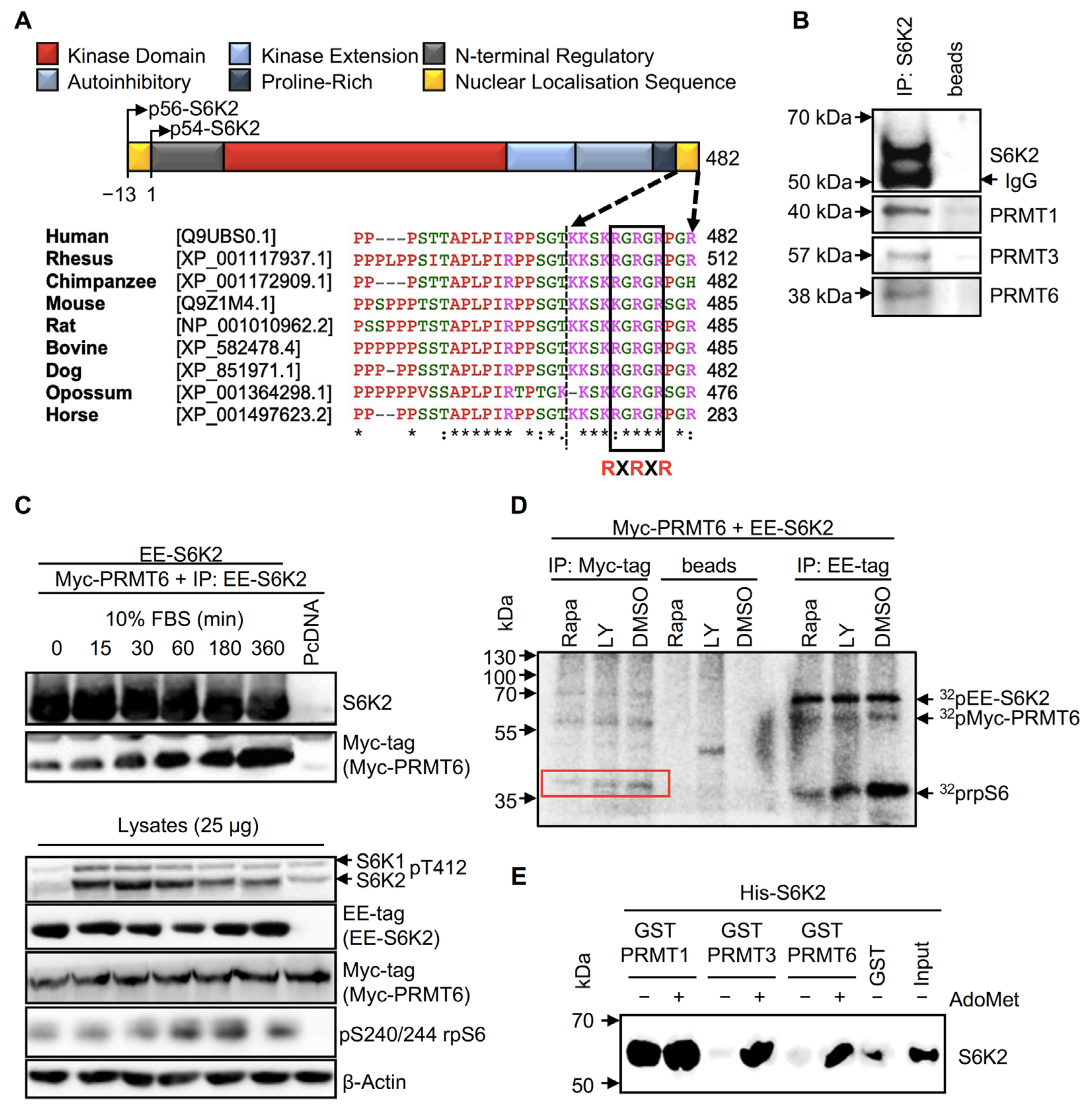
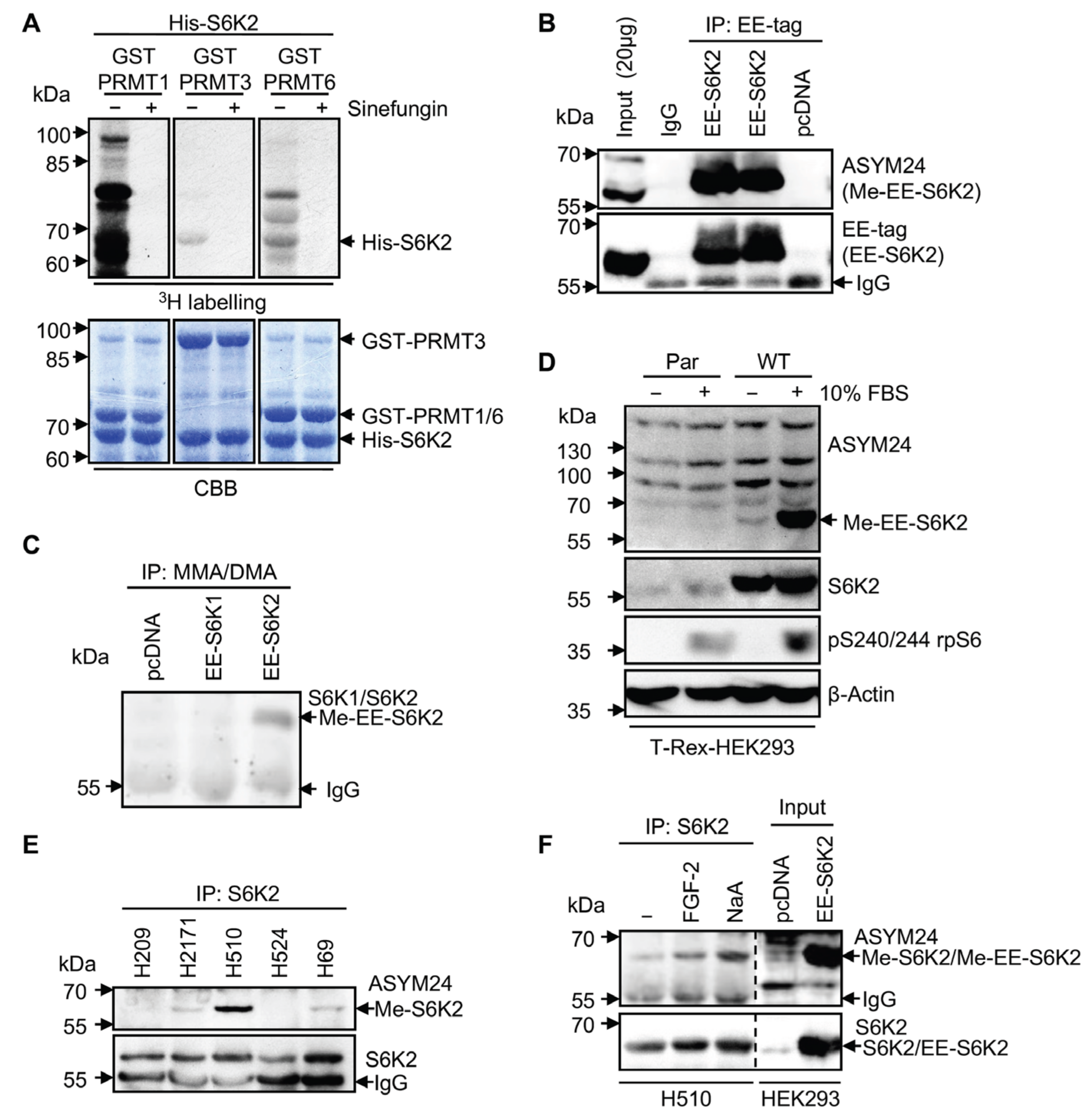
2.1. S6K2 Interacts with PRMT1, PRMT3, and PRMT6 In Vivo and In Vitro
2.2. S6K2 Is Methylated In Vitro and in Cells
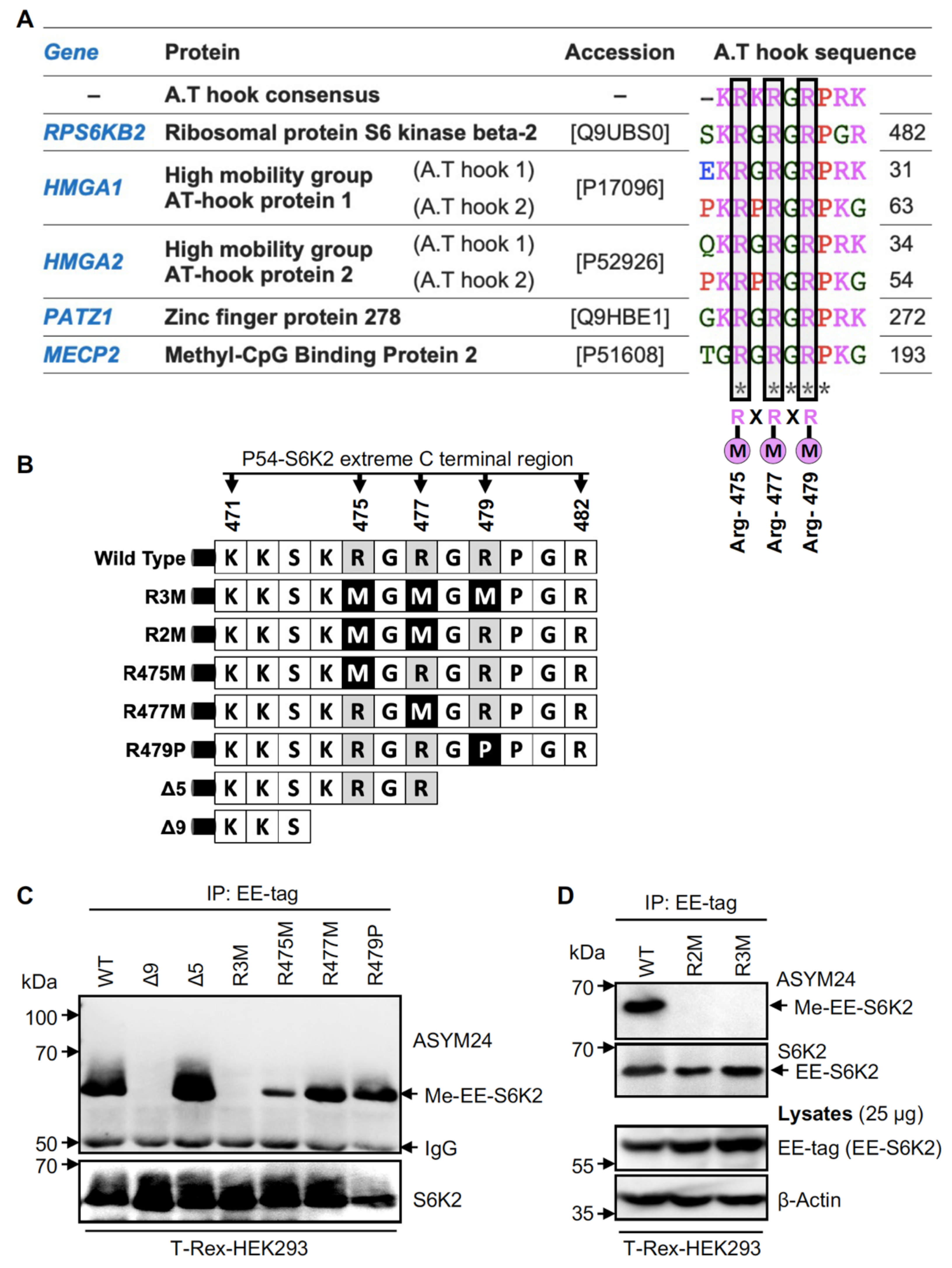
2.3. Methylation of S6K2 Occurs at Arg-475 and Arg-477
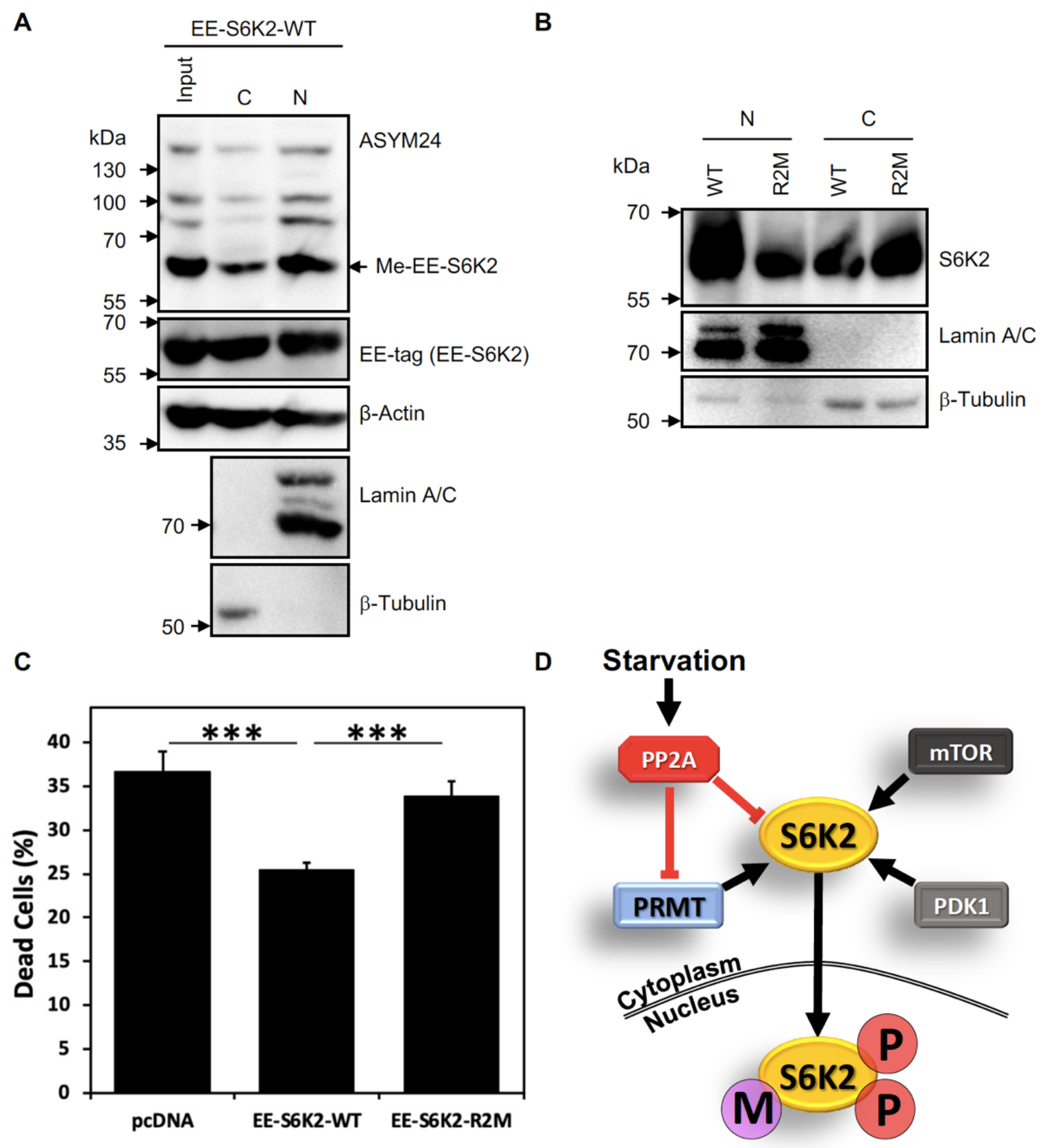
2.4. S6K2 Methylation Modulates Its Subcellular Localisation and Pro-Survival Effects
3. Materials and Methods
3.1. General Reagents
3.2. Reagents Used in the Cell Treatments
3.3. Expression Constructs and Recombinant Proteins
3.4. Antibodies
3.5. Cell Culture
3.6. Establishing Tetracycline-Inducible p54-S6K2 Cell Lines
3.7. DNA Transfections
3.8. Starvation-Induced Cell Death Assay
3.9. Cell Lysis and Subcellular Fractionation
3.10. Immunoprecipitation
3.11. Immunoblotting
3.12. Expression of GST-Fusion Proteins
3.13. GST Pull-Down Assay
3.14. In Vitro Methylation Assay
3.15. In Vitro Kinase Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Wellbrock, C.; Khanzada, U.K.; Aubert, M.; Arozarena, I.; Davidson, S.; Bowen, F.; Parker, P.J.; Filonenko, V.V.; Gout, I.T.; et al. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCepsilon, B-Raf and S6K2. EMBO J. 2006, 25, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Seckl, M.J. S6K2: The Neglected S6 Kinase Family Member. Front. Oncol. 2013, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.S.; Myronova, O.; Tsuchiya, Y.; Niewiarowski, A.; Tsaneva, I.; Gout, I. Identification of the general transcription factor Yin Yang 1 as a novel and specific binding partner for S6 Kinase 2. Cell Signal. 2013, 25, 1054–1063. [Google Scholar] [CrossRef]
- Valovka, T.; Verdier, F.; Cramer, R.; Zhyvoloup, A.; Fenton, T.; Rebholz, H.; Wang, M.-L.; Gzhegotsky, M.; Lutsyk, A.; Matsuka, G.; et al. Protein kinase C phosphorylates ribosomal protein S6 kinase betaII and regulates its subcellular localization. Mol. Cell Biol. 2003, 23, 852–863. [Google Scholar] [CrossRef]
- Gout, I.; Minami, T.; Hara, K.; Tsujishita, Y.; Filonenko, V.; Waterfield, M.D.; Yonezawa, K. Molecular Cloning and Characterization of a Novel p70 S6 Kinase, p70 S6 Kinase β Containing a Proline-rich Region. J. Biol. Chem. 1998, 273, 30061–30064. [Google Scholar] [CrossRef]
- Filonenko, V.V.; Tytarenko, R.; Azatjan, S.K.; Savinska, L.O.; Gaydar, Y.A.; Gout, I.T.; Usenko, V.S.; Lyzogubov, V.V. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp. Oncol. 2004, 26, 294–299. [Google Scholar]
- Boyer, D.; Quintanilla, R.; Lee-Fruman, K.K. Regulation of catalytic activity of S6 kinase 2 during cell cycle. Mol. Cell Biochem. 2008, 307, 59–64. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Zhang, F.; Kerbl-Knapp, J.; Rodriguez Colman, M.J.; Meinitzer, A.; Macher, T.; Vujić, N.; Fasching, S.; Jany-Luig, E.; Korbelius, M.; Kuentzel, K.B.; et al. Global analysis of protein arginine methylation. Cell Rep. Methods 2021, 1, 100016. [Google Scholar] [CrossRef]
- Islam, M.S.; McDonough, M.A.; Chowdhury, R.; Gault, J.; Khan, A.; Pires, E.; Schofield, C.J. Biochemical and structural investigations clarify the substrate selectivity of the 2-oxoglutarate oxygenase JMJD6. J. Biol. Chem. 2019, 294, 11637–11652. [Google Scholar] [CrossRef] [PubMed]
- Amaral, C.L.; Freitas, L.B.; Tamura, R.E.; Tavares, M.R.; Pavan, I.C.B.; Bajgelman, M.C.; Simabuco, F.M. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016, 16, 602. [Google Scholar] [CrossRef]
- Carmo, C.R.; Lyons-Lewis, J.; Seckl, M.J.; Costa-Pereira, A.P. A novel requirement for Janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS ONE 2011, 6, e19861. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Durie, D.; Li, H.; Liu, B.-Q.; Skehel, J.M.; Mauri, F.; Cuorvo, L.V.; Barbareschi, M.; Guo, L.; Holcik, M.; et al. hnRNPA1 couples nuclear export and translation of specific mRNAs downstream of FGF-2/S6K2 signalling. Nucleic Acids Res. 2014, 42, 12483–12497. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.; Bossert, M.; Schwander, A.; Akgün, E.; Fackelmayer, F.O. Arginine Methylation of Scaffold Attachment Factor A by Heterogeneous Nuclear Ribonucleoprotein Particle-associated PRMT1. J. Biol. Chem. 2004, 279, 48774–48779. [Google Scholar] [CrossRef]
- Lorton, B.M.; Shechter, D. Cellular consequences of arginine methylation. Cell Mol. Life Sci. 2019, 76, 2933–2956. [Google Scholar] [CrossRef]
- Singh, V.; Miranda, T.B.; Jiang, W.; Frankel, A.; Roemer, M.E.; Robb, V.A.; Gutmann, D.H.; Herschman, H.R.; Clarke, S.; Newsham, I.F. DAL-1/4.1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene 2004, 23, 7761–7771. [Google Scholar] [CrossRef]
- Liang, Z.; Wen, C.; Jiang, H.; Ma, S.; Liu, X. Protein Arginine Methyltransferase 5 Functions via Interacting Proteins. Front. Cell Dev. Biol. 2021, 9, 725301. [Google Scholar] [CrossRef]
- Lin, W.-J.; Gary, J.D.; Yang, M.C.; Clarke, S.; Herschman, H.R. The Mammalian Immediate-early TIS21 Protein and the Leukemia-associated BTG1 Protein Interact with a Protein-arginine N-Methyltransferase. J. Biol. Chem. 1996, 271, 15034–15044. [Google Scholar] [CrossRef]
- Berthet, C.; Guéhenneux, F.; Revol, V.; Samarut, C.; Lukaszewicz, A.; Dehay, C.; Dumontet, C.; Magaud, J.-P.; Rouault, J.-P. Interaction of PRMT1 with BTG/TOB proteins in cell signalling: Molecular analysis and functional aspects. Genes Cells 2002, 7, 29–39. [Google Scholar] [CrossRef]
- Nichols, R.C.; Wang, X.W.; Tang, J.; Hamilton, B.J.; High, F.A.; Herschman, H.R.; Rigby, W.F.C. The RGG Domain in hnRNP A2 Affects Subcellular Localization. Exp. Cell Res. 2000, 256, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 Is a Histone Arginine Demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.-H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Henikoff, S. The Histone Variant H3.3 Marks Active Chromatin by Replication-Independent Nucleosome Assembly. Mol. Cell. 2002, 9, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Ostareck-Lederer, A.; Ostareck, D.H.; Cans, C.; Neubauer, G.; Bomsztyk, K.; Superti-Furga, G.; Hentze, M.W. c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol. Cell Biol. 2002, 22, 4535–4543. [Google Scholar] [CrossRef]
- Bedford, M.T.; Frankel, A.; Yaffe, M.B.; Clarke, S.; Leder, P.; Richard, S. Arginine Methylation Inhibits the Binding of Proline-rich Ligands to Src Homology 3, but Not WW, Domains. J. Biol. Chem. 2000, 275, 16030–16036. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, H.; Panasyuk, G.; Fenton, T.; Nemazanyy, I.; Valovka, T.; Flajolet, M.; Ronnstrand, L.; Stephens, L.; West, A.; Gout, I.T. Receptor association and tyrosine phosphorylation of S6 kinases. FEBS J. 2006, 273, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; Chénard, C.A.; Richard, S. Protein Interfaces in Signaling Regulated by Arginine Methylation. Sci. STKE 2005, 2005, re2. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Lee, J.; Tessari, M.A.; Altamura, S.; Spolaore, B.; Giancotti, V.; Bedford, M.T.; Manfioletti, G. The AT-hook of the Chromatin Architectural Transcription Factor High Mobility Group A1a Is Arginine-methylated by Protein Arginine Methyltransferase 6. J. Biol. Chem. 2006, 281, 3764–3772. [Google Scholar] [CrossRef]
- Zou, Y.; Webb, K.; Perna, A.D.; Zhang, Q.; Clarke, S.; Wang, Y. A Mass Spectrometric Study on the in vitro Methylation of HMGA1a and HMGA1b Proteins by PRMTs: Methylation Specificity, the Effect of Binding to AT-Rich Duplex DNA, and the Effect of C-Terminal Phosphorylation. Biochemistry 2007, 46, 7896–7906. [Google Scholar] [CrossRef]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Hassa, P.O.; Gehrig, P.; Covic, M.; Hübscher, U.; Hottiger, M.O. Methylation of DNA polymerase ß by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. FASEB J. 2007, 21, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Belyanskaya, L.L.; Gehrig, P.M.; Gehring, H. Exposure on Cell Surface and Extensive Arginine Methylation of Ewing Sarcoma (EWS) Protein. J. Biol. Chem. 2001, 276, 18681–18687. [Google Scholar] [CrossRef] [PubMed]
- Rajpurohit, R.; Paik, W.K.; Kim, S. Effect of enzymic methylation of heterogeneous ribonucleoprotein particle A1 on its nucleic-acid binding and controlled proteolysis. Biochem. J. 1994, 304 Pt 3, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Mowen, K.A.; Tang, J.; Zhu, W.; Schurter, B.T.; Shuai, K.; Herschman, H.R.; David, M. Arginine Methylation of STAT1 Modulates IFNα/β-Induced Transcription. Cell 2001, 104, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.L.; Gabor, P.; Nagy, L. Genome-wide localization of histone 4 arginine 3 methylation in a differentiation primed myeloid leukemia cell line. Immunobiology 2005, 210, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Côté, J.; Boisvert, F.-M.; Boulanger, M.-C.; Bedford, M.T.; Richard, S. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell 2003, 14, 274–287. [Google Scholar] [CrossRef]
- Chen, W.; Daines, M.O.; Hershey, G.K.K. Methylation of STAT6 Modulates STAT6 Phosphorylation, Nuclear Translocation, and DNA-Binding Activity. J. Immunol. 2004, 172, 6744–6750. [Google Scholar] [CrossRef]
- Zhu, W.; Mustelin, T.; David, M. Arginine Methylation of STAT1 Regulates Its Dephosphorylation by T Cell Protein Tyrosine Phosphatase. J. Biol. Chem. 2002, 277, 35787–35790. [Google Scholar] [CrossRef]
- Yun, C.Y.; Fu, X.D. Conserved SR protein kinase functions in nuclear import and its action is counteracted by arginine methylation in Saccharomyces cerevisiae. J. Cell Biol. 2000, 150, 707–718. [Google Scholar] [CrossRef]
- Petritsch, C.; Beug, H.; Balmain, A.; Oft, M. TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G1 arrest. Genes Dev. 2000, 14, 3093–3101. [Google Scholar] [CrossRef]
- Duong, F.H.T.; Christen, V.; Berke, J.M.; Penna, S.H.; Moradpour, D.; Heim, M.H. Upregulation of protein phosphatase 2Ac by hepatitis C virus modulates NS3 helicase activity through inhibition of protein arginine methyltransferase 1. J. Virol. 2005, 79, 15342–15350. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Ho, P.-C.; Huq, M.D.M.; Khan, A.A.; Tsai, N.-P.; Wei, L.-N. PKCepsilon stimulated arginine methylation of RIP140 for its nuclear-cytoplasmic export in adipocyte differentiation. PLoS ONE 2008, 3, e2658. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Arcaro, A.; Salerno, G.; Tetley, T.D.; Valovka, T.; Gout, I.; Seckl, M.J. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene 2001, 20, 7658–7667. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Daitoku, H.; Takahashi, Y.; Namiki, K.; Hisatake, K.; Kako, K.; Mukai, H.; Kasuya, Y.; Fukamizu, A. Arginine Methylation of FOXO Transcription Factors Inhibits Their Phosphorylation by Akt. Mol. Cell. 2008, 32, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-L.; Panasyuk, G.; Gwalter, J.; Nemazanyy, I.; Fenton, T.; Filonenko, V.; Gout, I. Regulation of ribosomal protein S6 kinases by ubiquitination. Biochem. Biophys. Res. Commun. 2008, 369, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Gordon, J.; Rogg, H. N4-Acetylcytidine. A previously unidentified labile component of the small subunit of eukaryotic ribosomes. J. Biol. Chem. 1978, 253, 1101–1105. [Google Scholar] [CrossRef]
- Ismail, H.M.S.; Hurd, P.J.; Khalil, M.I.M.; Kouzarides, T.; Bannister, A.; Gout, I. S6 Kinase 2 Is Bound to Chromatin-Nuclear Matrix Cellular Fractions and Is Able to Phosphorylate Histone H3 at Threonine 45 In vitro and In vivo. J. Cell Biochem. 2014, 115, 1048–1062. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, M.I.; Ismail, H.M.; Panasyuk, G.; Bdzhola, A.; Filonenko, V.; Gout, I.; Pardo, O.E. Asymmetric Dimethylation of Ribosomal S6 Kinase 2 Regulates Its Cellular Localisation and Pro-Survival Function. Int. J. Mol. Sci. 2023, 24, 8806. https://doi.org/10.3390/ijms24108806
Khalil MI, Ismail HM, Panasyuk G, Bdzhola A, Filonenko V, Gout I, Pardo OE. Asymmetric Dimethylation of Ribosomal S6 Kinase 2 Regulates Its Cellular Localisation and Pro-Survival Function. International Journal of Molecular Sciences. 2023; 24(10):8806. https://doi.org/10.3390/ijms24108806
Chicago/Turabian StyleKhalil, Mahmoud I., Heba M. Ismail, Ganna Panasyuk, Anna Bdzhola, Valeriy Filonenko, Ivan Gout, and Olivier E. Pardo. 2023. "Asymmetric Dimethylation of Ribosomal S6 Kinase 2 Regulates Its Cellular Localisation and Pro-Survival Function" International Journal of Molecular Sciences 24, no. 10: 8806. https://doi.org/10.3390/ijms24108806
APA StyleKhalil, M. I., Ismail, H. M., Panasyuk, G., Bdzhola, A., Filonenko, V., Gout, I., & Pardo, O. E. (2023). Asymmetric Dimethylation of Ribosomal S6 Kinase 2 Regulates Its Cellular Localisation and Pro-Survival Function. International Journal of Molecular Sciences, 24(10), 8806. https://doi.org/10.3390/ijms24108806