

Drug Potency Prediction of SARS-CoV-2 Main Protease Inhibitors Based on a Graph Generative Model

 , , ,
, , ,  and
and
Abstract
1. Introduction
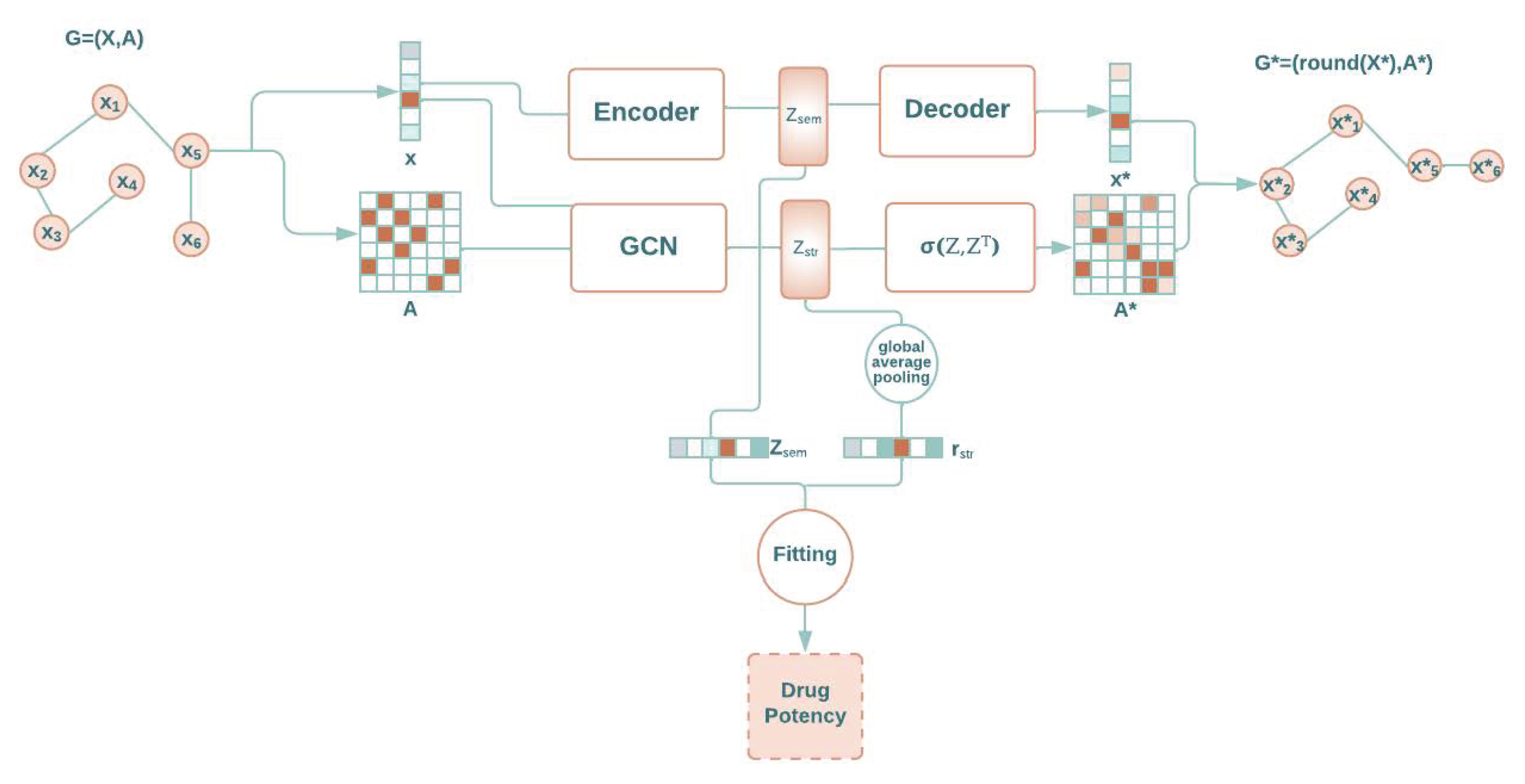
1.1. Autoencoders
1.2. Graph Autoencoders
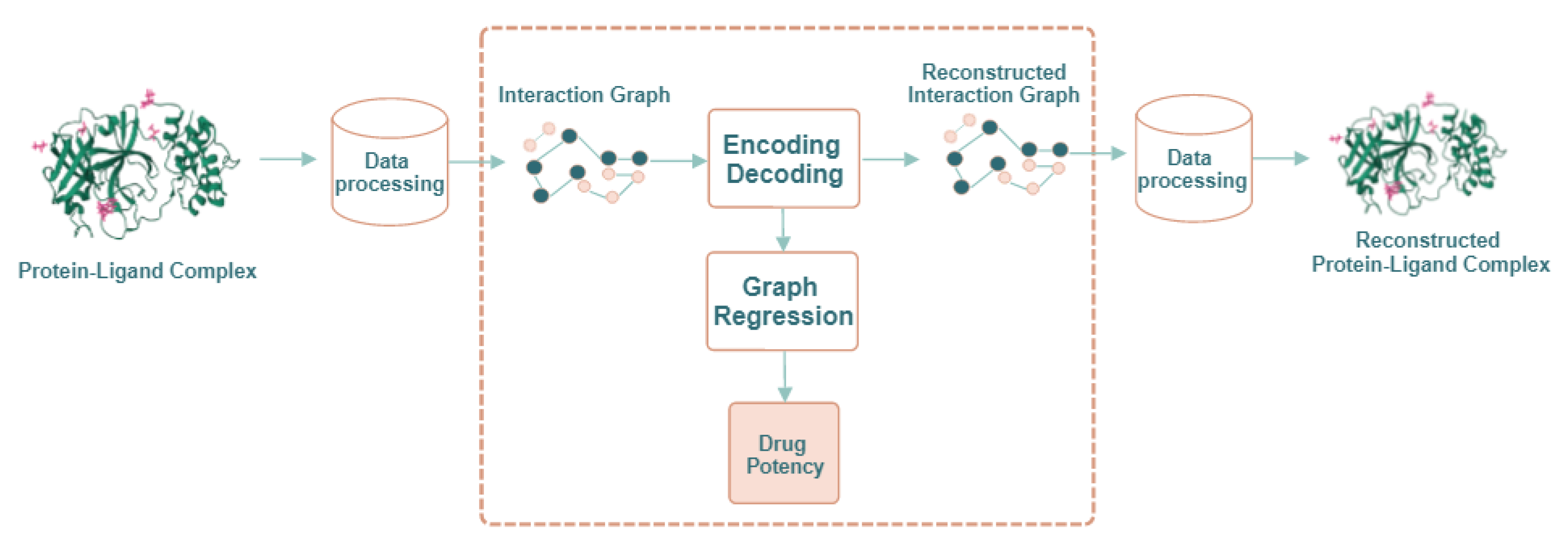
2. Results and Discussion
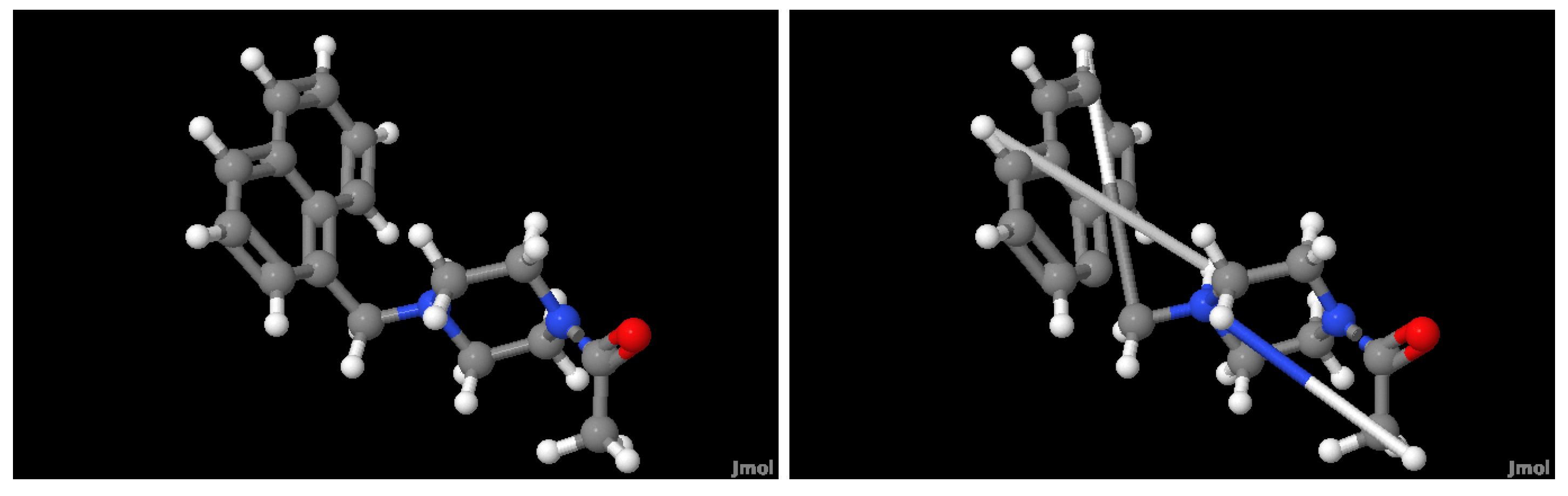
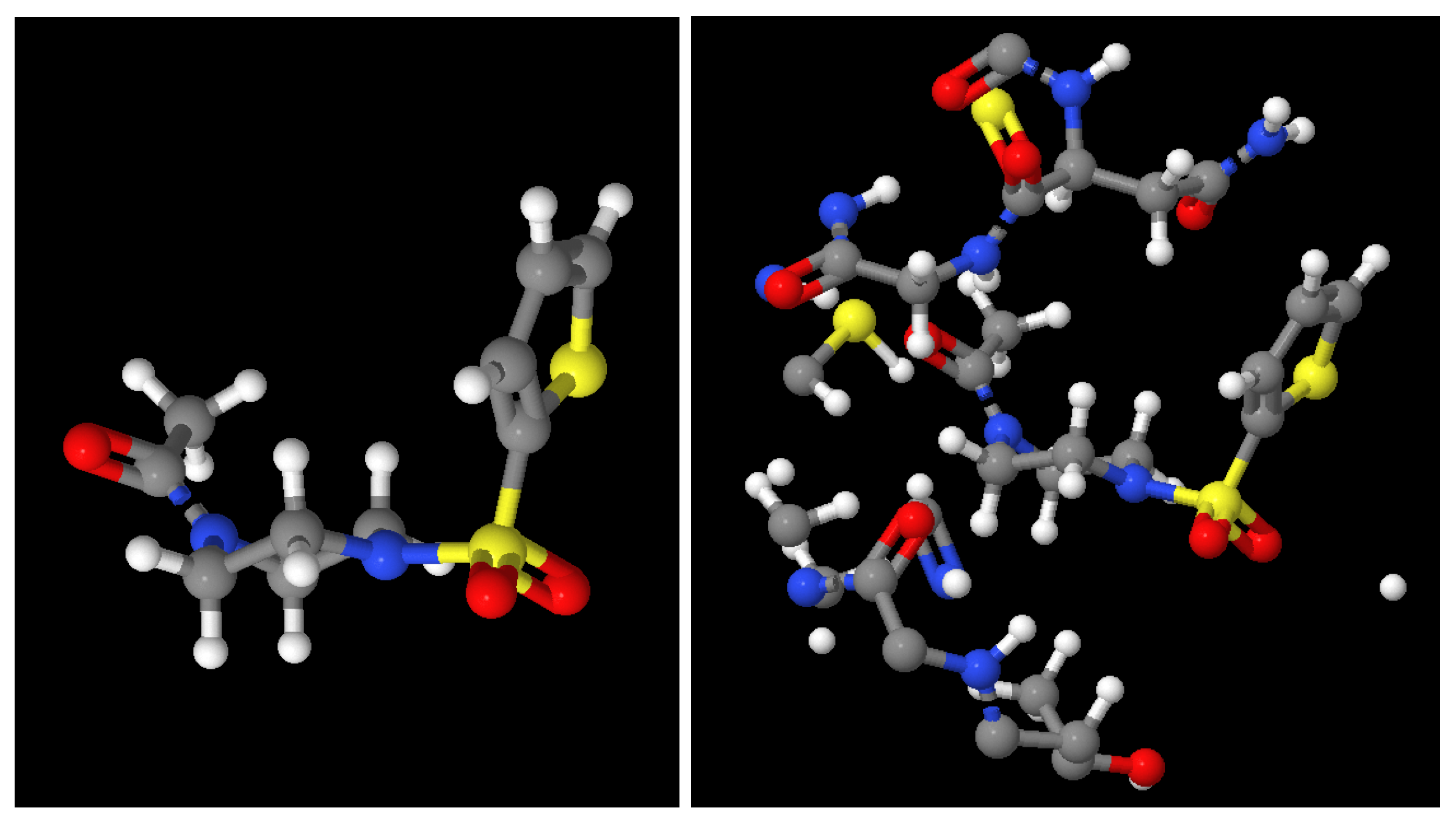
2.1. Molecule Reconstruction
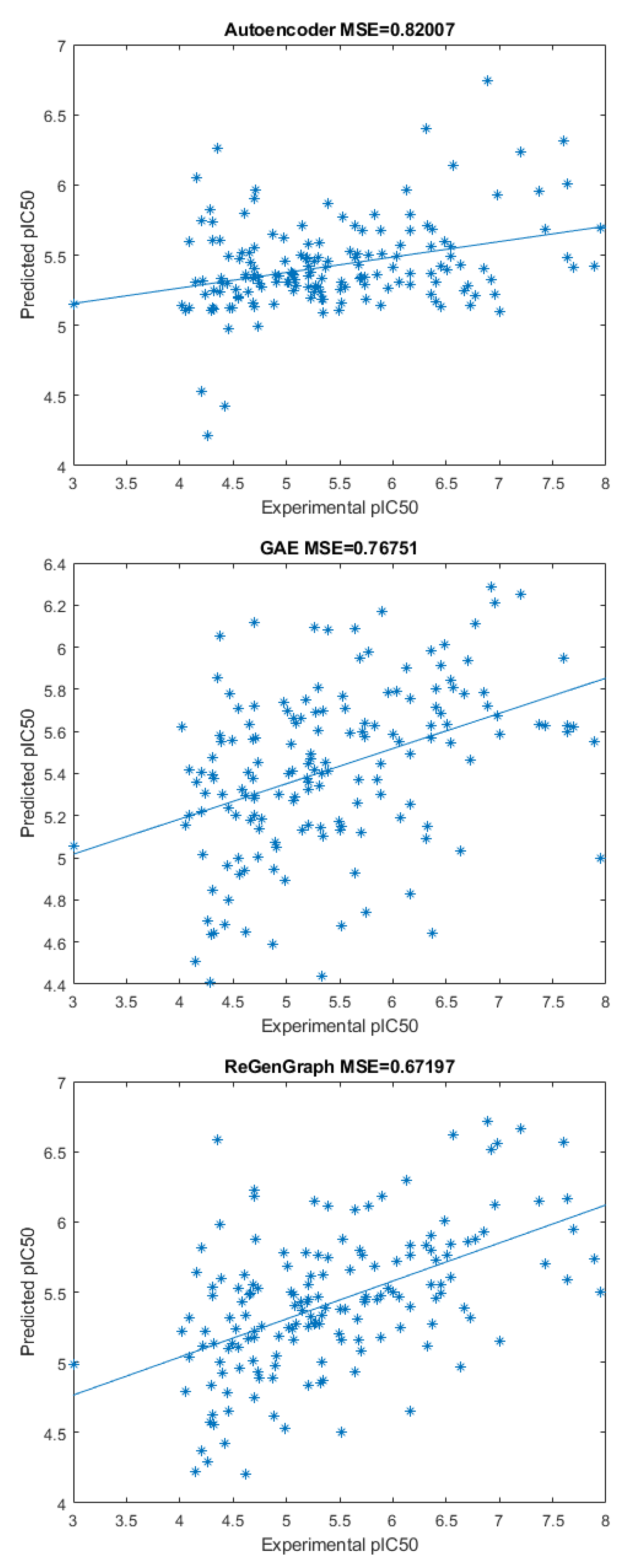
2.2. Drug Potency Prediction
2.3. Runtime Analysis
3. Materials and Methods
3.1. SARS-CoV-2 M-pro Database
3.2. Architecture Configuration
3.3. The Learning Process
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GAE | Graph Autoencoder |
| GCN | Graph Convolutional Network |
| MSE | Mean Squared Error |
| VS | Virtual Screening |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Ligand Code |
|---|---|
| FRAGALYSIS | Mpro-x0689, Mpro-x0691, Mpro-x0755, Mpro-x0770, Mpro-x0830, Mpro-x10236, Mpro-x10322, Mpro-x10338, Mpro-x10371, Mpro-x10387, Mpro-x10417, Mpro-x10422, Mpro-x10423, Mpro-x10466, Mpro-x10535, Mpro-x10565, Mpro-x10638, Mpro-x10679, Mpro-x10789, Mpro-x10820, Mpro-x10870, Mpro-x10871, Mpro-x10876, Mpro-x10942, Mpro-x10959, Mpro-x11011, Mpro-x11271, Mpro-x11276, Mpro-x11294, Mpro-x11313, Mpro-x11317, Mpro-x11318, Mpro-x11366, Mpro-x11368, Mpro-x11454, Mpro-x11458, Mpro-x11488, Mpro-x11498, Mpro-x11499, Mpro-x11501, Mpro-x11507, Mpro-x11508, Mpro-x11530, Mpro-x11541, Mpro-x11542, Mpro-x11543, Mpro-x11548, Mpro-x11562, Mpro-x11564, Mpro-x11609, Mpro-x11612, Mpro-x11616, Mpro-x11641, Mpro-x11642, Mpro-x11723, Mpro-x11742, Mpro-x11743, Mpro-x11757, Mpro-x11764, Mpro-x11789, Mpro-x11790, Mpro-x11797, Mpro-x11798, Mpro-x11801, Mpro-x11810, Mpro-x11812, Mpro-x11813, Mpro-x11831, Mpro-x12000, Mpro-x12073, Mpro-x12143, Mpro-x12171, Mpro-x12177, Mpro-x12202, Mpro-x12207, Mpro-x12300, Mpro-x12321, Mpro-x12419, Mpro-x12423, Mpro-x12582, Mpro-x12587, Mpro-x12659, Mpro-x12661, Mpro-x12674, Mpro-x12679, Mpro-x12686, Mpro-x12692, Mpro-x12695, Mpro-x12696, Mpro-x12698, Mpro-x12699, Mpro-x12710, Mpro-x12715, Mpro-x12716, Mpro-x12731, Mpro-x12740, Mpro-x1336, Mpro-x1386, Mpro-x1418, Mpro-x2563, Mpro-x2572, Mpro-x2646, Mpro-x2649, Mpro-x2908, Mpro-x2910, Mpro-x2912, Mpro-x3303 |
| PDB | 6M2N, 6W63, 7AU4, 7B2J, 7B2U, 7B5Z, 7B77, 7E18, 7E19, 7KX5, 7L0D, 7L10, 7L11, 7L12, 7L14, 7LCT, 7LMD, 7LME, 7LMF, 7M8M, 7M8N, 7M8O, 7M8P, 7M8X, 7M8Y, 7M8Z, 7M90, 7M91, 7N44, 7N8C, 7NT3, 7O46, 7P2G, 7QBB, 7RLS, 7RM2, 7RMB, 7RME, 7RMT, 7RMZ, 7RN4, 7RNH, 7RNK, 7S3K, 7S3S, 7S4B, 7TVX, 7VIC, 7VLP, 7VLQ, 7VTH, 7VU6, 7VVP, 7VVT, 7X6K, 8ACD |
References
- Achdout, H.; Aimon, A.; Bar-David, E.; Morris, G.M. COVID Moonshot: Open Science Discovery of SARS-CoV-2 Main Protease Inhibitors by Combining Crowdsourcing, High-Throughput Experiments, Computational Simulations, and Machine Learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration to Treat COVID-19. Available online: https://fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs (accessed on 20 March 2023).
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Pujadas, G.; Garcia-Vallvé, S. A review of the current landscape of SARS-CoV-2 main protease inhibitors: Have we hit the Bullseye yet? Int. J. Mol. Sci. 2021, 23, 259. [Google Scholar] [CrossRef]
- Siddiqui, S.; Upadhyay, S.; Ahmad, R.; Gupta, A.; Srivastava, A.; Trivedi, A.; Husain, I.; Ahmad, B.; Ahamed, M.; Khan, M.A. Virtual screening of phytoconstituents from miracle herb nigella sativa targeting nucleocapsid protein and papain-like protease of SARS-CoV-2 for COVID-19 treatment. J. Biomol. Struct. Dyn. 2022, 40, 3928–3948. [Google Scholar] [CrossRef]
- Chan, W.K.; Olson, K.M.; Wotring, J.W.; Sexton, J.Z.; Carlson, H.A.; Traynor, J.R. In silico analysis of SARS-CoV-2 proteins as targets for clinically available drugs. Sci. Rep. 2022, 12, 5320. [Google Scholar] [CrossRef]
- Gogoi, M.; Borkotoky, M.; Borchetia, S.; Chowdhury, P.; Mahanta, S.; Barooah, A.K. Black tea bioactives as inhibitors of multiple targets of SARS-CoV-2 (3CLpro, PLpro and RdRp): A virtual screening and molecular dynamic simulation study. J. Biomol. Struct. Dyn. 2022, 40, 7143–7166. [Google Scholar] [CrossRef]
- Deshmukh, M.G.; Ippolito, J.A.; Zhang, C.H.; Stone, E.A.; Reilly, R.A.; Miller, S.J.; Jorgensen, W.L.; Anderson, K.S. Structure-guided design of a perampanel-derived pharmacophore targeting the SARS-CoV-2 main protease. Structure 2021, 29, 823–833.e5. [Google Scholar] [CrossRef]
- Glaser, J.; Sedova, A.; Galanie, S.; Kneller, D.W.; Davidson, R.B.; Maradzike, E.; Del Galdo, S.; Labbé, A.; Hsu, D.J.; Agarwal, R.; et al. Hit expansion of a noncovalent SARS-CoV-2 main protease inhibitor. ACS Pharmacol. Transl. Sci. 2022, 5, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Sylvester, K.; Song, L.; Claff, T.; Jing, L.; Woodson, M.; Weiße, R.H.; Cheng, Y.; Schäkel, L.; Petry, M.; et al. Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high target specificity and low toxicity. J. Med. Chem. 2022, 65, 13343–13364. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef]
- Li, Z.; Wu, C.; Li, Y.; Liu, R.; Lu, K.; Wang, R.; Liu, J.; Gong, C.; Yang, C.; Wang, X.; et al. Free energy perturbation–based large-scale virtual screening for effective drug discovery against COVID-19. Int. J. High Perform. Comput. Appl. 2023, 37, 45–57. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs-19. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-Supervised Classification with Graph Convolutional Networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Garcia-Hernandez, C.; Fernández, A.; Serratosa, F. Ligand-based virtual screening using graph edit distance as molecular similarity measure. J. Chem. Inf. Model. 2019, 59, 1410–1421. [Google Scholar] [CrossRef]
- Rica, E.; Álvarez, S.; Serratosa, F. Ligand-based virtual screening based on the graph edit distance. Int. J. Mol. Sci. 2021, 22, 12751. [Google Scholar] [CrossRef]
- Garcia-Hernandez, C.; Fernandez, A.; Serratosa, F. Learning the Edit Costs of Graph Edit Distance Applied to Ligand-Based Virtual Screening. Curr. Top. Med. Chem. 2020, 20, 1582–1592. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph Neural Networks: A Review of Methods and Applications. arXiv 2018, arXiv:1812.08434. [Google Scholar] [CrossRef]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32, 4–24. [Google Scholar] [CrossRef]
- Le, T.; Le, N.; Le, B. Knowledge graph embedding by relational rotation and complex convolution for link prediction. Expert Syst. Appl. 2023, 214, 119122. [Google Scholar] [CrossRef]
- Kramer, M.A. Nonlinear principal component analysis using autoassociative neural networks. Aiche J. 1991, 37, 233–243. [Google Scholar] [CrossRef]
- Fadlallah, S.; Julià, C.; Serratosa, F. Graph Regression Based on Graph Autoencoders. In Proceedings of the Structural, Syntactic, and Statistical Pattern Recognition; Krzyzak, A., Suen, C.Y., Torsello, A., Nobile, N., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 142–151. [Google Scholar]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph Attention Networks. arXiv 2017, arXiv:1710.10903. [Google Scholar] [CrossRef]
- Mpro: Fragalysis. Available online: https://fragalysis.diamond.ac.uk/viewer/react/preview/target/Mpro (accessed on 20 March 2023).
- Majumdar, A. Graph structured autoencoder. Neural Netw. 2018, 106, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Kipf, T.N. Deep Learning with Graph-Structured Representations. Ph.D. Thesis, University of Amsterdam, Amsterdam, The Netherlands, 2020. [Google Scholar]





| Autoencoder | GAE | ReGenGraph | |
|---|---|---|---|
| Mean | 0.83576 | 0.7456 | 0.6717 |
| Std. Dev. | 0.3188 | 0.9382 | 0.1796 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadlallah, S.; Julià, C.; García-Vallvé, S.; Pujadas, G.; Serratosa, F. Drug Potency Prediction of SARS-CoV-2 Main Protease Inhibitors Based on a Graph Generative Model. Int. J. Mol. Sci. 2023, 24, 8779. https://doi.org/10.3390/ijms24108779
Fadlallah S, Julià C, García-Vallvé S, Pujadas G, Serratosa F. Drug Potency Prediction of SARS-CoV-2 Main Protease Inhibitors Based on a Graph Generative Model. International Journal of Molecular Sciences. 2023; 24(10):8779. https://doi.org/10.3390/ijms24108779
Chicago/Turabian StyleFadlallah, Sarah, Carme Julià, Santiago García-Vallvé, Gerard Pujadas, and Francesc Serratosa. 2023. "Drug Potency Prediction of SARS-CoV-2 Main Protease Inhibitors Based on a Graph Generative Model" International Journal of Molecular Sciences 24, no. 10: 8779. https://doi.org/10.3390/ijms24108779
APA StyleFadlallah, S., Julià, C., García-Vallvé, S., Pujadas, G., & Serratosa, F. (2023). Drug Potency Prediction of SARS-CoV-2 Main Protease Inhibitors Based on a Graph Generative Model. International Journal of Molecular Sciences, 24(10), 8779. https://doi.org/10.3390/ijms24108779