

The Mutational Landscape of SARS-CoV-2

 , , , ,
, , , ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
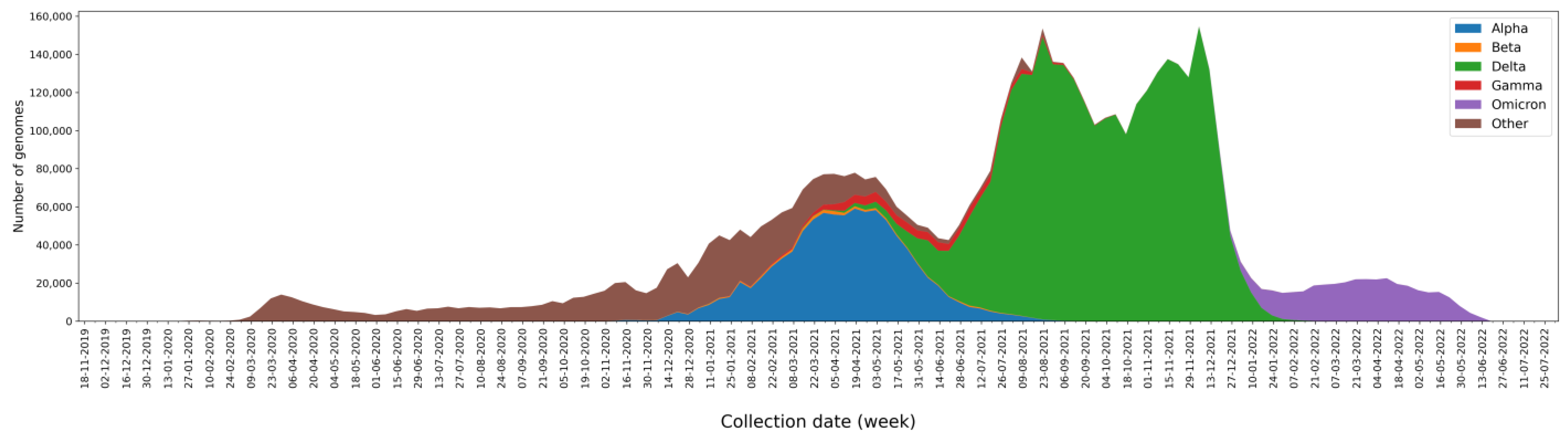
2.1. SARS-CoV-2 Genomes Analyzed
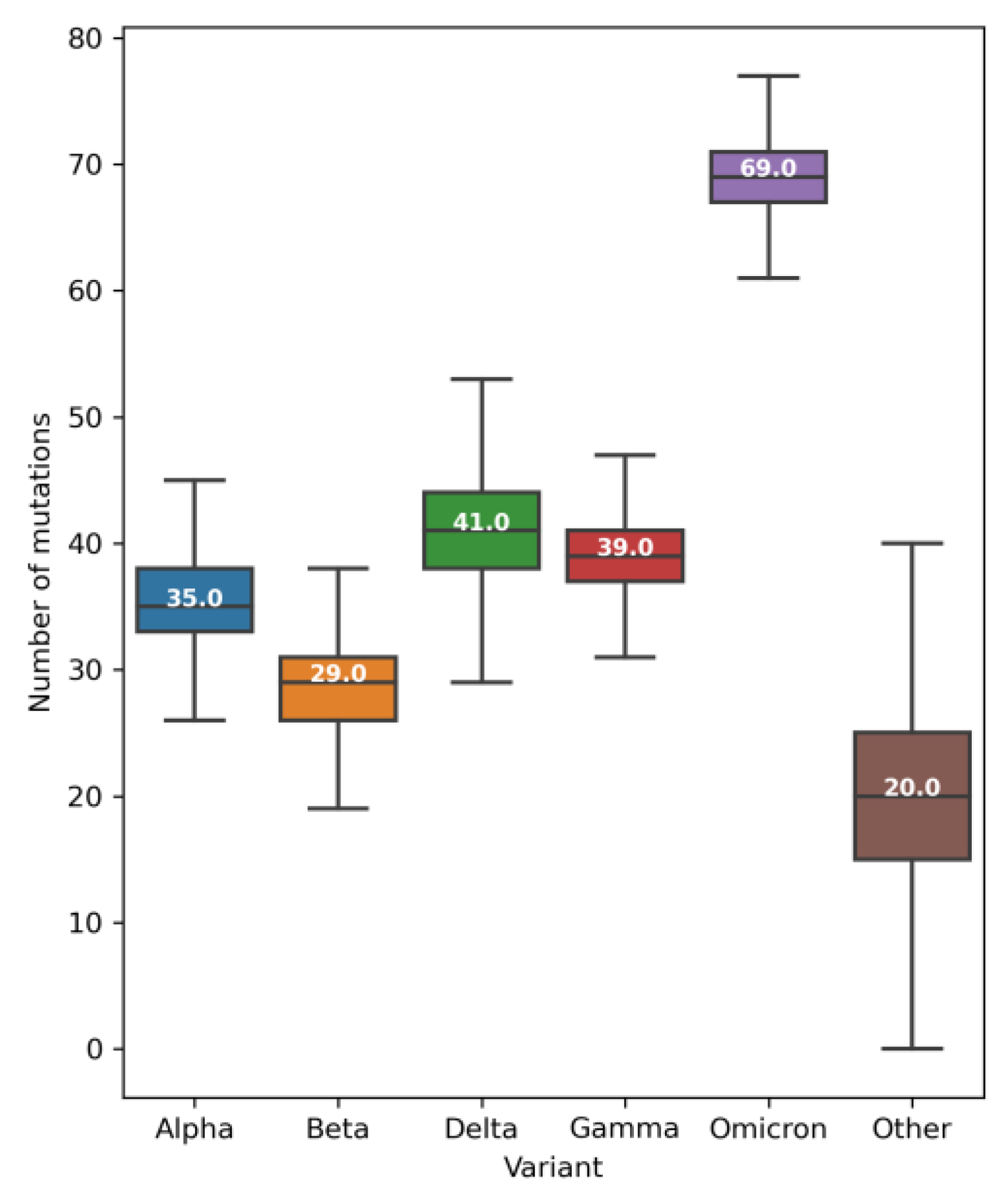
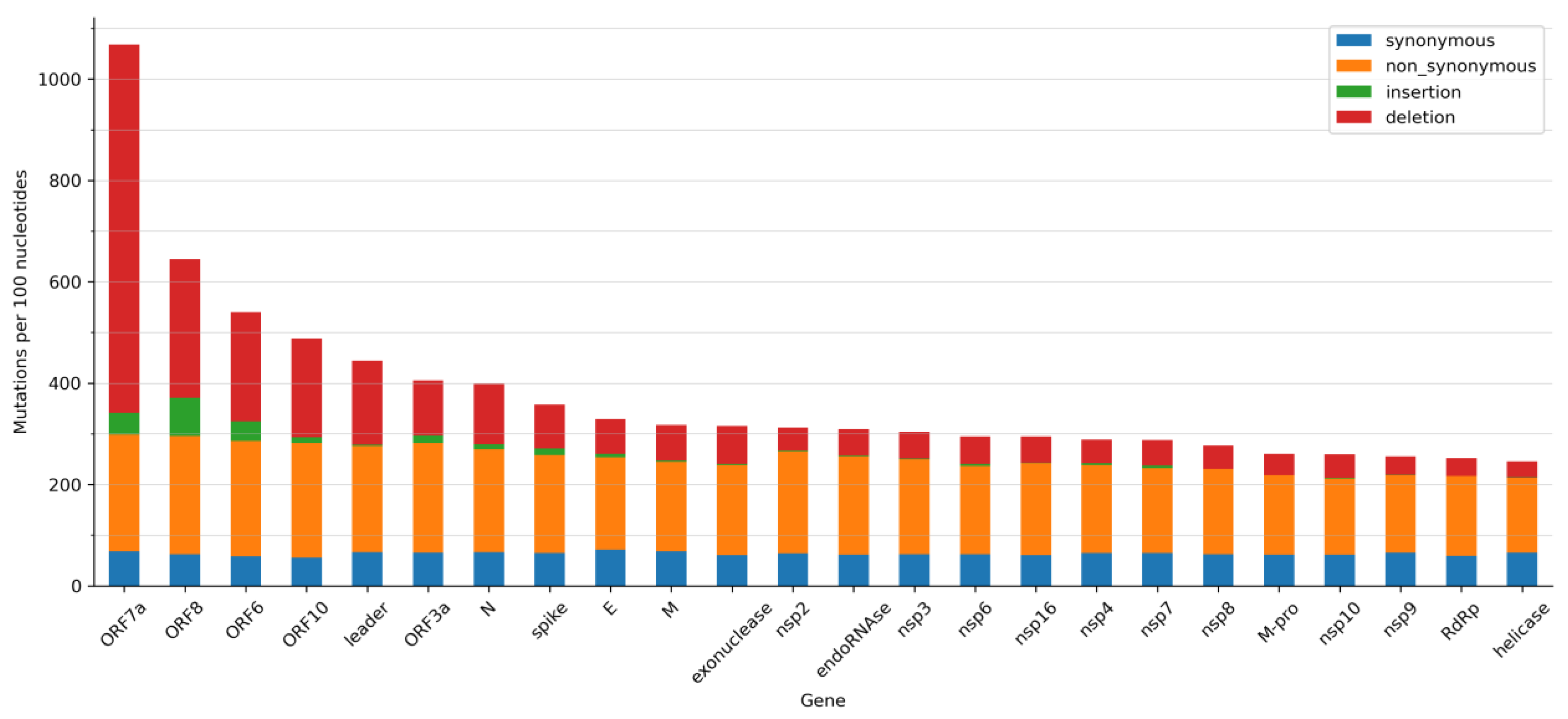
2.2. Mutations, Deletions, and Insertions per Genome per Week
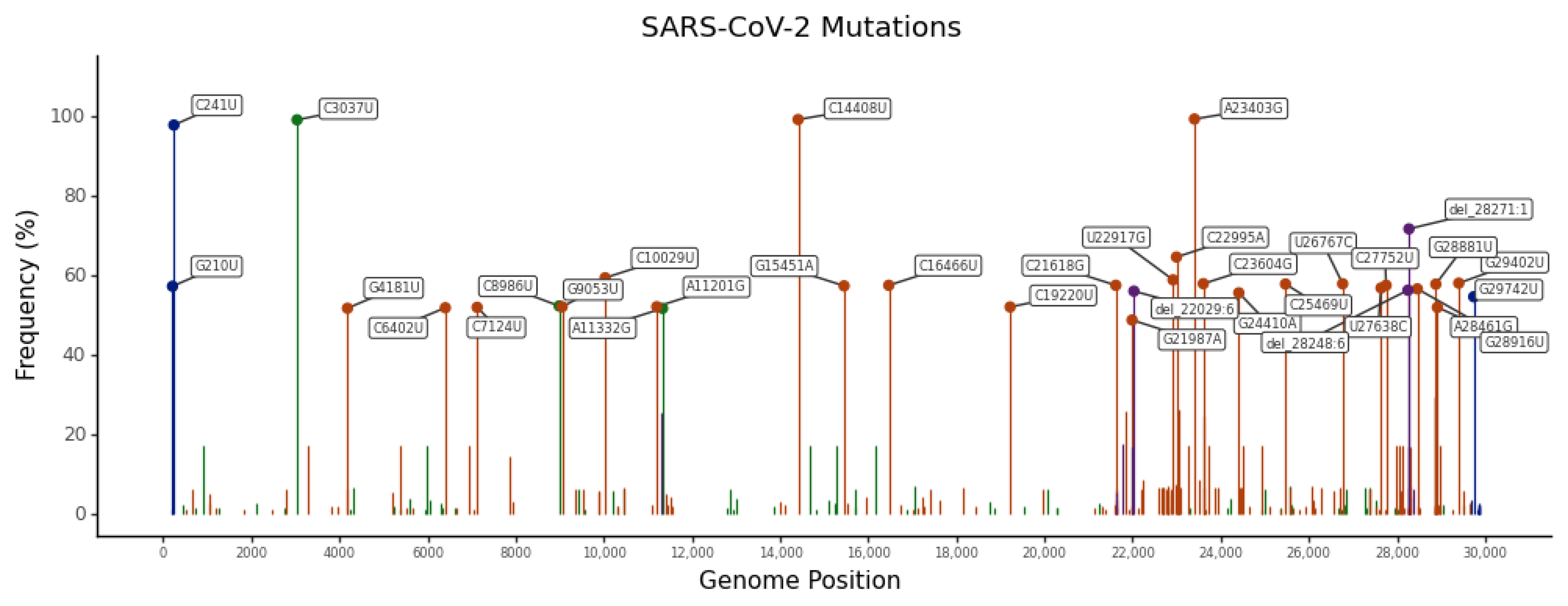
2.3. Most Frequent SNVs
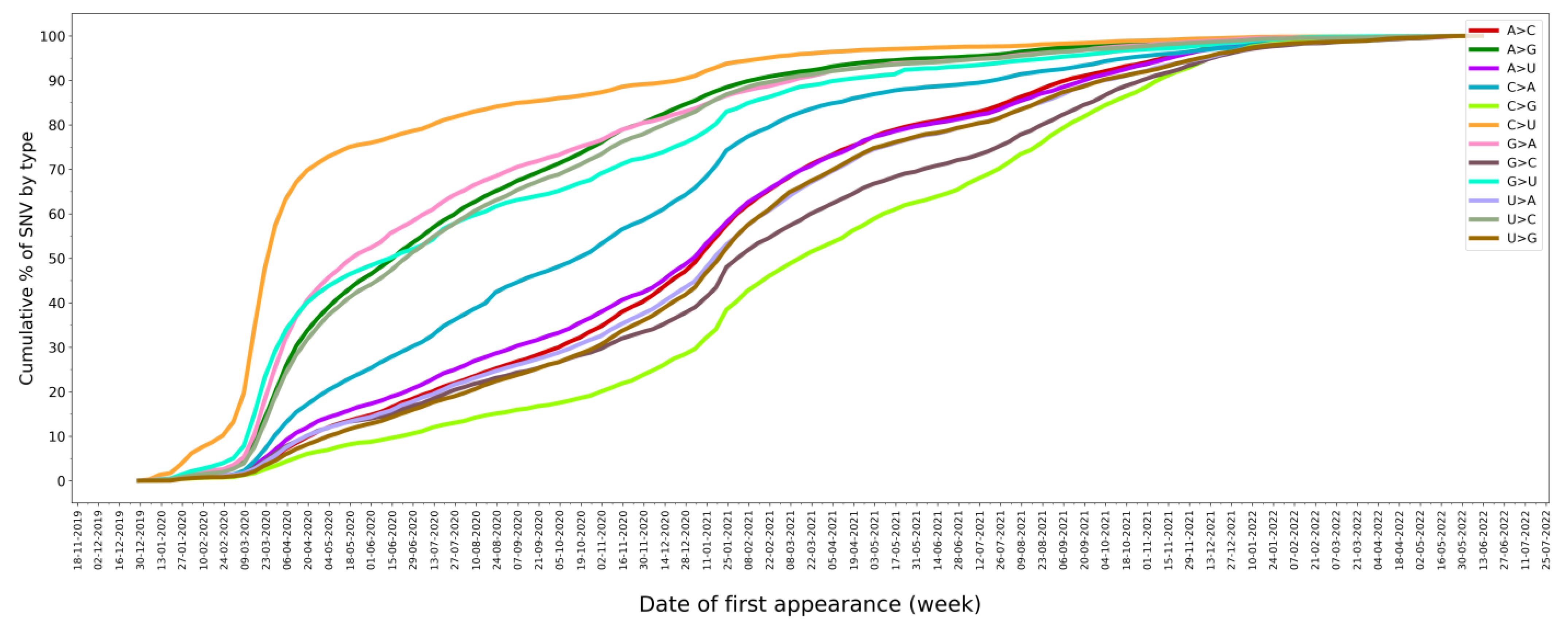
2.4. SNV Signature Analysis
2.5. Mutations in the Target Regions of the COVID-19 Diagnostic RT-qPCR Tests
2.6. SARS-CoV-2 Mutation Portal
3. Materials and Methods
Origin and Characterization of the SARS-CoV-2 Genomes Analyzed
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Belém, M.O.; Pinto, Y.O.; Moreira, H.H.T.; et al. The Spike Glycoprotein of SARS-CoV-2: A Review of How Mutations of Spike Glycoproteins Have Driven the Emergence of Variants with High Transmissibility and Immune Escape. Int. J. Biol. Macromol. 2022, 208, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Alkhatib, M.; Svicher, V.; Salpini, R.; Ambrosio, F.A.; Bellocchi, M.C.; Carioti, L.; Piermatteo, L.; Scutari, R.; Costa, G.; Artese, A.; et al. SARS-CoV-2 Variants and Their Relevant Mutational Profiles: Update Summer 2021. Microbiol. Spectr. 2021, 9, e0109621. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Agoramoorthy, G.; Lee, S.-S. Evolution, Mode of Transmission, and Mutational Landscape of Newly Emerging SARS-CoV-2 Variants. mBio 2021, 12, e0114021. [Google Scholar] [CrossRef]
- Callaway, E. Beyond Omicron: What’s next for COVID’s Viral Evolution. Nature 2021, 600, 204–207. [Google Scholar] [CrossRef]
- Kannan, S.; Shaik Syed Ali, P.; Sheeza, A. Omicron (B.1.1.529)—Variant of Concern—Molecular Profile and Epidemiology: A Mini Review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8019–8022. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5, e00408-20. [Google Scholar] [CrossRef]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef]
- Ratcliff, J.; Simmonds, P. Potential APOBEC-Mediated RNA Editing of the Genomes of SARS-CoV-2 and Other Coronaviruses and Its Impact on Their Longer Term Evolution. Virology 2021, 556, 62–72. [Google Scholar] [CrossRef]
- Simmonds, P.; Ansari, M.A. Extensive C->U Transition Biases in the Genomes of a Wide Range of Mammalian RNA Viruses; Potential Associations with Transcriptional Mutations, Damage- or Host-Mediated Editing of Viral RNA. PLoS Pathog. 2021, 17, e1009596. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The Emergence, Genomic Diversity and Global Spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Wang, J.; Wu, L.; Pu, X.; Liu, B.; Cao, M. Evidence Supporting That C-to-U RNA Editing Is the Major Force That Drives SARS-CoV-2 Evolution. J. Mol. Evol. 2023, 91, 214–224. [Google Scholar] [CrossRef]
- Kim, K.; Calabrese, P.; Wang, S.; Qin, C.; Rao, Y.; Feng, P.; Chen, X.S. The Roles of APOBEC-Mediated RNA Editing in SARS-CoV-2 Mutations, Replication and Fitness. Sci. Rep. 2022, 12, 14972. [Google Scholar] [CrossRef]
- Song, Y.; He, X.; Yang, W.; Wu, Y.; Cui, J.; Tang, T.; Zhang, R. Virus-Specific Editing Identification Approach Reveals the Landscape of A-to-I Editing and Its Impacts on SARS-CoV-2 Characteristics and Evolution. Nucleic Acids Res. 2022, 50, 2509–2521. [Google Scholar] [CrossRef]
- Nakata, Y.; Ode, H.; Kubota, M.; Kasahara, T.; Matsuoka, K.; Sugimoto, A.; Imahashi, M.; Yokomaku, Y.; Iwatani, Y. Cellular APOBEC3A Deaminase Drives Mutations in the SARS-CoV-2 Genome. Nucleic Acids Res. 2023, 51, 783–795. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. Prediction of Recurrent Mutations in SARS-CoV-2 Using Artificial Neural Networks. Int. J. Mol. Sci. 2022, 23, 14683. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and Virus Restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Graudenzi, A.; Maspero, D.; Angaroni, F.; Piazza, R.; Ramazzotti, D. Mutational Signatures and Heterogeneous Host Response Revealed via Large-Scale Characterization of SARS-CoV-2 Genomic Diversity. iScience 2021, 24, 102116. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA Editing—Immune Protector and Transcriptome Diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Verrou, K.-M.; Stellos, K.; Sfikakis, P.P.; Paraskevis, D. The Role of A-to-I RNA Editing in Infections by RNA Viruses: Possible Implications for SARS-CoV-2 Infection. Clin. Immunol. 2021, 226, 108699. [Google Scholar] [CrossRef]
- Zimmermann, F.; Urban, M.; Krüger, C.; Walter, M.; Wölfel, R.; Zwirglmaier, K. In Vitro Evaluation of the Effect of Mutations in Primer Binding Sites on Detection of SARS-CoV-2 by RT-QPCR. J. Virol. Methods 2022, 299, 114352. [Google Scholar] [CrossRef]
- Jian, M.-J.; Chung, H.-Y.; Chang, C.-K.; Lin, J.-C.; Yeh, K.-M.; Chen, C.-W.; Lin, D.-Y.; Chang, F.-Y.; Hung, K.-S.; Perng, C.-L.; et al. SARS-CoV-2 Variants with T135I Nucleocapsid Mutations May Affect Antigen Test Performance. Int. J. Infect. Dis. 2022, 114, 112–114. [Google Scholar] [CrossRef]
- Mentes, A.; Papp, K.; Visontai, D.; Stéger, J.; VEO Technical Working Group; Csabai, I.; Medgyes-Horváth, A.; Pipek, O.A. Identification of Mutations in SARS-CoV-2 PCR Primer Regions. Sci. Rep. 2022, 12, 18651. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef]
- Dong, H.; Wang, S.; Zhang, J.; Zhang, K.; Zhang, F.; Wang, H.; Xie, S.; Hu, W.; Gu, L. Structure-Based Primer Design Minimizes the Risk of PCR Failure Caused by SARS-CoV-2 Mutations. Front. Cell. Infect. Microbiol. 2021, 11, 741147. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes de Jesus, J.; et al. Multiplex QPCR Discriminates Variants of Concern to Enhance Global Surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.-W. Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine. J. Chem. Inf. Model. 2020, 60, 5853–5865. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Zheng, Y.-H.; Yin, C.; Wei, G.-W. Host Immune Response Driving SARS-CoV-2 Evolution. Viruses 2020, 12, 1095. [Google Scholar] [CrossRef]
- Jaroszewski, L.; Iyer, M.; Alisoltani, A.; Sedova, M.; Godzik, A. The Interplay of SARS-CoV-2 Evolution and Constraints Imposed by the Structure and Functionality of Its Proteins. PLoS Comput. Biol. 2021, 17, e1009147. [Google Scholar] [CrossRef]
- Abbasian, M.H.; Mahmanzar, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Moradi, B.; Sisakht, M.M.; Deng, Y. Global Landscape of SARS-CoV-2 Mutations and Conserved Regions. J. Transl. Med. 2023, 21, 152. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The Emergence and Epidemic Characteristics of the Highly Mutated SARS-CoV-2 Omicron Variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef]
- Focosi, D.; Quiroga, R.; McConnell, S.; Johnson, M.C.; Casadevall, A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. Int. J. Mol. Sci. 2023, 24, 2264. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Weng, S.; Zhou, H.; Ji, C.; Li, L.; Han, N.; Yang, R.; Shang, J.; Wu, A. Conserved Pattern and Potential Role of Recurrent Deletions in SARS-CoV-2 Evolution. Microbiol. Spectr. 2022, 10, e0219121. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T.; Fujiwara, K. Insertion and Deletion Mutations Preserved in SARS-CoV-2 Variants. Arch. Microbiol. 2023, 205, 154. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Saura, A.; Bykova, A.; Brover, V.; Yurchenko, V. Deletions across the SARS-CoV-2 Genome: Molecular Mechanisms and Putative Functional Consequences of Deletions in Accessory Genes. Microorganisms 2023, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Anand, P.; Lenehan, P.J.; Ghosh, P.; Suratekar, R.; Silvert, E.; Pawlowski, C.; Siroha, A.; Chowdhury, D.R.; O’Horo, J.C.; et al. Expanding Repertoire of SARS-CoV-2 Deletion Mutations Contributes to Evolution of Highly Transmissible Variants. Sci. Rep. 2023, 13, 257. [Google Scholar] [CrossRef] [PubMed]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Template Switching and Duplications in SARS-CoV-2 Genomes Give Rise to Insertion Variants That Merit Monitoring. Commun. Biol. 2021, 4, 1343. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Bai, H.; Ata, G.; Sun, Q.; Rahman, S.U.; Tao, S. Natural Selection Pressure Exerted on “Silent” Mutations during the Evolution of SARS-CoV-2: Evidence from Codon Usage and RNA Structure. Virus Res. 2022, 323, 198966. [Google Scholar] [CrossRef]
- Martínez-González, B.; Soria, M.E.; Vázquez-Sirvent, L.; Ferrer-Orta, C.; Lobo-Vega, R.; Mínguez, P.; de la Fuente, L.; Llorens, C.; Soriano, B.; Ramos-Ruíz, R.; et al. SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations. Pathogens 2022, 11, 662. [Google Scholar] [CrossRef]
- Yang, H.-C.; Chen, C.-H.; Wang, J.-H.; Liao, H.-C.; Yang, C.-T.; Chen, C.-W.; Lin, Y.-C.; Kao, C.-H.; Lu, M.-Y.J.; Liao, J.C. Analysis of Genomic Distributions of SARS-CoV-2 Reveals a Dominant Strain Type with Strong Allelic Associations. Proc. Natl. Acad. Sci. USA 2020, 117, 30679–30686. [Google Scholar] [CrossRef]
- Goldswain, H.; Dong, X.; Penrice-Randal, R.; Alruwaili, M.; Shawli, G.T.; Prince, T.; Williamson, M.K.; Raghwani, J.; Randle, N.; Jones, B.; et al. The P323L Substitution in the SARS-CoV-2 Polymerase (NSP12) Confers a Selective Advantage during Infection. Genome Biol. 2023, 24, 47. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 Gene Content and COVID-19 Mutation Impact by Comparing 44 Sarbecovirus Genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef]
- Berno, G.; Fabeni, L.; Matusali, G.; Gruber, C.E.M.; Rueca, M.; Giombini, E.; Garbuglia, A.R. SARS-CoV-2 Variants Identification: Overview of Molecular Existing Methods. Pathogens 2022, 11, 1058. [Google Scholar] [CrossRef]
- Specchiarello, E.; Matusali, G.; Carletti, F.; Gruber, C.E.M.; Fabeni, L.; Minosse, C.; Giombini, E.; Rueca, M.; Maggi, F.; Amendola, A.; et al. Detection of SARS-CoV-2 Variants via Different Diagnostics Assays Based on Single-Nucleotide Polymorphism Analysis. Diagnostics 2023, 13, 1573. [Google Scholar] [CrossRef]
- Cassari, L.; Pavan, A.; Zoia, G.; Chinellato, M.; Zeni, E.; Grinzato, A.; Rothenberger, S.; Cendron, L.; Dettin, M.; Pasquato, A. SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works. Int. J. Mol. Sci. 2023, 24, 4791. [Google Scholar] [CrossRef]
- He, X.; He, C.; Hong, W.; Yang, J.; Wei, X. Research Progress in Spike Mutations of SARS-CoV-2 Variants and Vaccine Development. Med. Res. Rev. 2023, in press. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Zhou, J.; Dong, Y.; Jiang, W.; Jiang, W. Rampant C-to-U Deamination Accounts for the Intrinsically High Mutation Rate in SARS-CoV-2 Spike Gene. RNA 2022, 28, 917–926. [Google Scholar] [CrossRef]
- Ravi, V.; Swaminathan, A.; Yadav, S.; Arya, H.; Pandey, R. SARS-CoV-2 Variants of Concern and Variations within Their Genome Architecture: Does Nucleotide Distribution and Mutation Rate Alter the Functionality and Evolution of the Virus? Viruses 2022, 14, 2499. [Google Scholar] [CrossRef]
- Oronsky, B.; Larson, C.; Caroen, S.; Hedjran, F.; Sanchez, A.; Prokopenko, E.; Reid, T. Nucleocapsid as a Next-Generation COVID-19 Vaccine Candidate. Int. J. Infect. Dis. 2022, 122, 529–530. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Macip, G.; Pujadas, G.; Garcia-Vallve, S. Could Nucleocapsid Be a Next-Generation COVID-19 Vaccine Candidate? Int. J. Infect. Dis. 2022, 125, 231–232. [Google Scholar] [CrossRef]
- Zhao, H.; Nguyen, A.; Wu, D.; Li, Y.; Hassan, S.A.; Chen, J.; Shroff, H.; Piszczek, G.; Schuck, P. Plasticity in Structure and Assembly of SARS-CoV-2 Nucleocapsid Protein. PNAS Nexus 2022, 1, pgac049. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, D.; Hassan, S.A.; Nguyen, A.; Chen, J.; Piszczek, G.; Schuck, P. A Conserved Oligomerization Domain in the Disordered Linker of Coronavirus Nucleocapsid Proteins. Sci. Adv. 2023, 9, eadg6473. [Google Scholar] [CrossRef] [PubMed]
- De Maio, N.; Walker, C.R.; Turakhia, Y.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Mutation Rates and Selection on Synonymous Mutations in SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hou, F.; Zhou, M.; Yang, X.; Yin, B.; Jiang, W.; Xu, H. C-to-U RNA Deamination Is the Driving Force Accelerating SARS-CoV-2 Evolution. Life Sci. Alliance 2023, 6, e202201688. [Google Scholar] [CrossRef] [PubMed]
- Ringlander, J.; Fingal, J.; Kann, H.; Prakash, K.; Rydell, G.; Andersson, M.; Martner, A.; Lindh, M.; Horal, P.; Hellstrand, K.; et al. Impact of ADAR-Induced Editing of Minor Viral RNA Populations on Replication and Transmission of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2022, 119, e2112663119. [Google Scholar] [CrossRef]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 Samples May Escape Detection Because of a Single Point Mutation in the N Gene. Eurosurveillance 2020, 25, 2001650. [Google Scholar] [CrossRef]
- Vanaerschot, M.; Mann, S.A.; Webber, J.T.; Kamm, J.; Bell, S.M.; Bell, J.; Hong, S.N.; Nguyen, M.P.; Chan, L.Y.; Bhatt, K.D.; et al. Identification of a Polymorphism in the N Gene of SARS-CoV-2 That Adversely Impacts Detection by Reverse Transcription-PCR. J. Clin. Microbiol. 2020, 59, e02369-20. [Google Scholar] [CrossRef]
- Hasan, R.; Hossain, M.E.; Miah, M.; Hasan, M.M.; Rahman, M.; Rahman, M.Z. Identification of Novel Mutations in the N Gene of SARS-CoV-2 That Adversely Affect the Detection of the Virus by Reverse Transcription-Quantitative PCR. Microbiol. Spectr. 2021, 9, e0054521. [Google Scholar] [CrossRef]
- Zannoli, S.; Dirani, G.; Taddei, F.; Gatti, G.; Poggianti, I.; Denicolò, A.; Arfilli, V.; Manera, M.; Mancini, A.; Battisti, A.; et al. A Deletion in the N Gene May Cause Diagnostic Escape in SARS-CoV-2 Samples. Diagn. Microbiol. Infect. Dis. 2022, 102, 115540. [Google Scholar] [CrossRef]
- Laine, P.; Nihtilä, H.; Mustanoja, E.; Lyyski, A.; Ylinen, A.; Hurme, J.; Paulin, L.; Jokiranta, S.; Auvinen, P.; Meri, T. SARS-CoV-2 Variant with Mutations in N Gene Affecting Detection by Widely Used PCR Primers. J. Med. Virol. 2022, 94, 1227–1231. [Google Scholar] [CrossRef]
- Miller, S.; Lee, T.; Merritt, A.; Pryce, T.; Levy, A.; Speers, D. Single-Point Mutations in the N Gene of SARS-CoV-2 Adversely Impact Detection by a Commercial Dual Target Diagnostic Assay. Microbiol. Spectr. 2021, 9, e0149421. [Google Scholar] [CrossRef]
- Isabel, S.; Abdulnoor, M.; Boissinot, K.; Isabel, M.R.; de Borja, R.; Zuzarte, P.C.; Sjaarda, C.P.; Barker, R.K.; Sheth, P.M.; Matukas, L.M.; et al. Emergence of a Mutation in the Nucleocapsid Gene of SARS-CoV-2 Interferes with PCR Detection in Canada. Sci. Rep. 2022, 12, 10867. [Google Scholar] [CrossRef]
- Kami, W.; Kinjo, T.; Hashioka, H.; Arakaki, W.; Uechi, K.; Takahashi, A.; Oki, H.; Tanaka, K.; Motooka, D.; Nakamura, S.; et al. Impact of G29179T Mutation on Two Commercial PCR Assays for SARS-CoV-2 Detection. J. Virol. Methods 2023, 314, 114692. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.-W. Mutations on COVID-19 Diagnostic Targets. Genomics 2020, 112, 5204–5213. [Google Scholar] [CrossRef]
- Marchini, A.; Petrillo, M.; Parrish, A.; Buttinger, G.; Tavazzi, S.; Querci, M.; Betsou, F.; Elsinga, G.; Medema, G.; Abdelrahman, T.; et al. New RT-PCR Assay for the Detection of Current and Future SARS-CoV-2 Variants. Viruses 2023, 15, 206. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Type | Count | Median Number of Genomes (%) |
|---|---|---|
| in UTRs | 1842 | 120 (2.2 × 10−3 %) |
| non-synonymous | 51,467 | 20 (3.8 × 10−4 %) |
| synonymous | 18,413 | 135 (2.6 × 10−3 %) |
| not alone | 1742 | 1 (1.9 × 10−5 %) |
| Mutation | Gene | Mutation Type | AA Change | No. Genomes (%) |
|---|---|---|---|---|
| A23403G | S | non-synonymous | D614G | 5,312,457 (99.48%) |
| C14408U | RdRp | non-synonymous | P323L | 5,306,009 (99.35%) |
| C3037U | nsp3 | synonymous | F106F | 5,301,673 (99.27%) |
| C241U | 5’-UTR | in UTRs | - | 5,231,432 (97.96%) |
| del_28271:1 | N | deletion | - | 3,835,041 (71.81%) |
| C22995A | S | non-synonymous | T478K | 3,456,098 (64.71%) |
| C10029U | nsp4 | non-synonymous | T492I | 3,176,761 (59.48%) |
| U22917G | S | non-synonymous | L452R | 3,149,402 (58.97%) |
| G29402U | N | non-synonymous | D377Y | 3,105,411 (58.15%) |
| C23604G | S | non-synonymous | P681R | 3,097,770 (58.00%) |
| Mutation | No. Genomes (% a) | No. Genomes (% b) in Variants | |||||
|---|---|---|---|---|---|---|---|
| Alpha | Beta | Delta | Epsilon | Gamma | Omicron | ||
| Δ69/70 | 945,256 (17.70%) | 895,448 (94.73%) | 14 (0.001%) | 3850 (0.41%) | 287 (0.03%) | 189 (0.02%) | 19,260 (2.04%) |
| W152C | 45,304 (0.85%) | 9 (0.02%) | 0 (0%) | 29 (0.06%) | 45,192 (99.75%) | 2 (0.004%) | 1 (0.002%) |
| K417N | 383,722 (7.18%) | 137 (0.04%) | 24,366 (6.35%) | 5353 (1.40%) | 10 (0.003%) | 2 (0.0005%) | 352,543 (91.87%) |
| K417T | 88,480 (16.32%) | 32 (0.04%) | 0 (0%) | 59 (0.07%) | 1 (0.001%) | 86,902 (98.22%) | 1426 (1.61%) |
| L452R | 3,149,260 (58.97%) | 450 (0.01%) | 28 (0.0009%) | 3,075,838 (97.67%) | 45,457 (1.44%) | 42 (0.001%) | 1847 (0.06%) |
| E484A | 357,227 (6.69%) | 20 (0.006%) | 0 (0%) | 2595 (0.73%) | 1 (0.0003%) | 0 (0%) | 354,118 (99.13%) |
| N501Y | 1,396,003 (26.14%) | 914,121 (65.48%) | 25,377 (1.82%) | 1520 (0.11%) | 25 (0.0002%) | 89,927 (6.44%) | 348,897 (24.99%) |
| To Nucleotide | ||||||
|---|---|---|---|---|---|---|
| A | G | C | U | Total SNVs | ||
| From nucleotide | A | 0 | 8416 (94.0%) | 7008 (78.3%) | 6982 (78.0%) | 22,406 |
| G | 5420 (92.4%) | 0 | 4059 (69.2%) | 5475 (93.4%) | 14,954 | |
| C | 4780 (87.0%) | 3580 (65.2%) | 0 | 5351 (97.4%) | 13,711 | |
| U | 6791 (70.8%) | 6666 (69.5%) | 8936 (93.1%) | 0 | 22,393 | |
| Total SNVs | 16,991 | 18,662 | 20,003 | 17,808 | 73,464 | |
| Name | Gene | Region Amplified | No. Different Mutations Found in Forward and Reverse Primers and Probe | Total No. Mutations and Total Frequency (%) |
|---|---|---|---|---|
| nCoV_IP2 | RdRp | 12,690–12,797 | 46 | 68 | 64 | 178 (1.75%) |
| nCoV_IP4 | RdRp | 14,080–14,186 | 50 | 66 | 65 | 181 (3.95%) |
| Charite-E | E | 26,269–26,381 | 89 | 81 | 143 | 313 (7.15%) |
| N-Sarbeco | N | 28,706–28,833 | 68 | 93 | 116 | 277 (2.14%) |
| Charite-RdRp | RdRp | 15,431–15,528 | 67 | 52 | 64 | 183 (60.84%) |
| HKU-ORF1ab | ORF1ab | 18,778–18,909 | 60 | 73 | 60 | 193 (1.18%) |
| HKU-N | N | 29,145–29,254 | 145 | 222 | 167 | 534 (3.25%) |
| China-CDC-ORF1ab | ORF1ab | 13,342–13,460 | 58 | 59 | 103 | 220 (0.79%) |
| China-CDC-N | N | 28,881–28,979 | 156 | 118 | 86 | 360 (141.28%) |
| US-CDC-N1 | N | 28,287–28,358 | 102 | 111 | 131 | 344 (14.59%) |
| US-CDC-N2 | N | 29,164–29,230 | 154 | 184 | 189 | 527 (2.73%) |
| US-CDC-N3 | N | 28,681–28,752 | 88 | 91 | 90 | 269 (3.36%) |
| Japan-N | N | 29,125–29,282 | 116 | 234 | 211 | 561 (2.02%) |
| Thailand-N | N | 28,320–28,376 | 104 | 112 | 78 | 294 (2.46%) |
| Sigma-Aldrich | N | 28,750–28,860 | 96 | 96 | -1 | 192 (2.66%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saldivar-Espinoza, B.; Garcia-Segura, P.; Novau-Ferré, N.; Macip, G.; Martínez, R.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. The Mutational Landscape of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 9072. https://doi.org/10.3390/ijms24109072
Saldivar-Espinoza B, Garcia-Segura P, Novau-Ferré N, Macip G, Martínez R, Puigbò P, Cereto-Massagué A, Pujadas G, Garcia-Vallve S. The Mutational Landscape of SARS-CoV-2. International Journal of Molecular Sciences. 2023; 24(10):9072. https://doi.org/10.3390/ijms24109072
Chicago/Turabian StyleSaldivar-Espinoza, Bryan, Pol Garcia-Segura, Nil Novau-Ferré, Guillem Macip, Ruben Martínez, Pere Puigbò, Adrià Cereto-Massagué, Gerard Pujadas, and Santiago Garcia-Vallve. 2023. "The Mutational Landscape of SARS-CoV-2" International Journal of Molecular Sciences 24, no. 10: 9072. https://doi.org/10.3390/ijms24109072
APA StyleSaldivar-Espinoza, B., Garcia-Segura, P., Novau-Ferré, N., Macip, G., Martínez, R., Puigbò, P., Cereto-Massagué, A., Pujadas, G., & Garcia-Vallve, S. (2023). The Mutational Landscape of SARS-CoV-2. International Journal of Molecular Sciences, 24(10), 9072. https://doi.org/10.3390/ijms24109072