
What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances


Abstract
1. Introduction
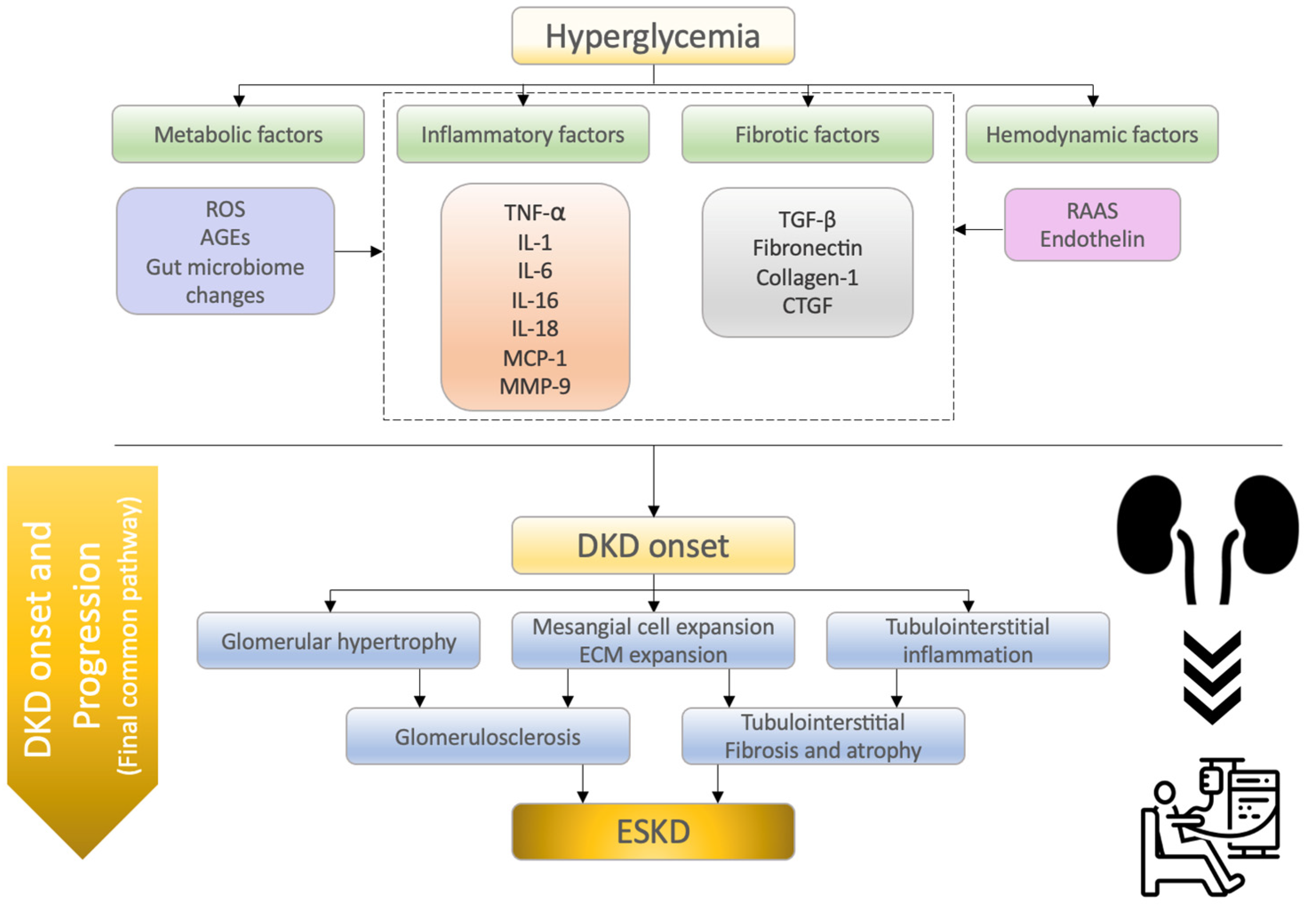
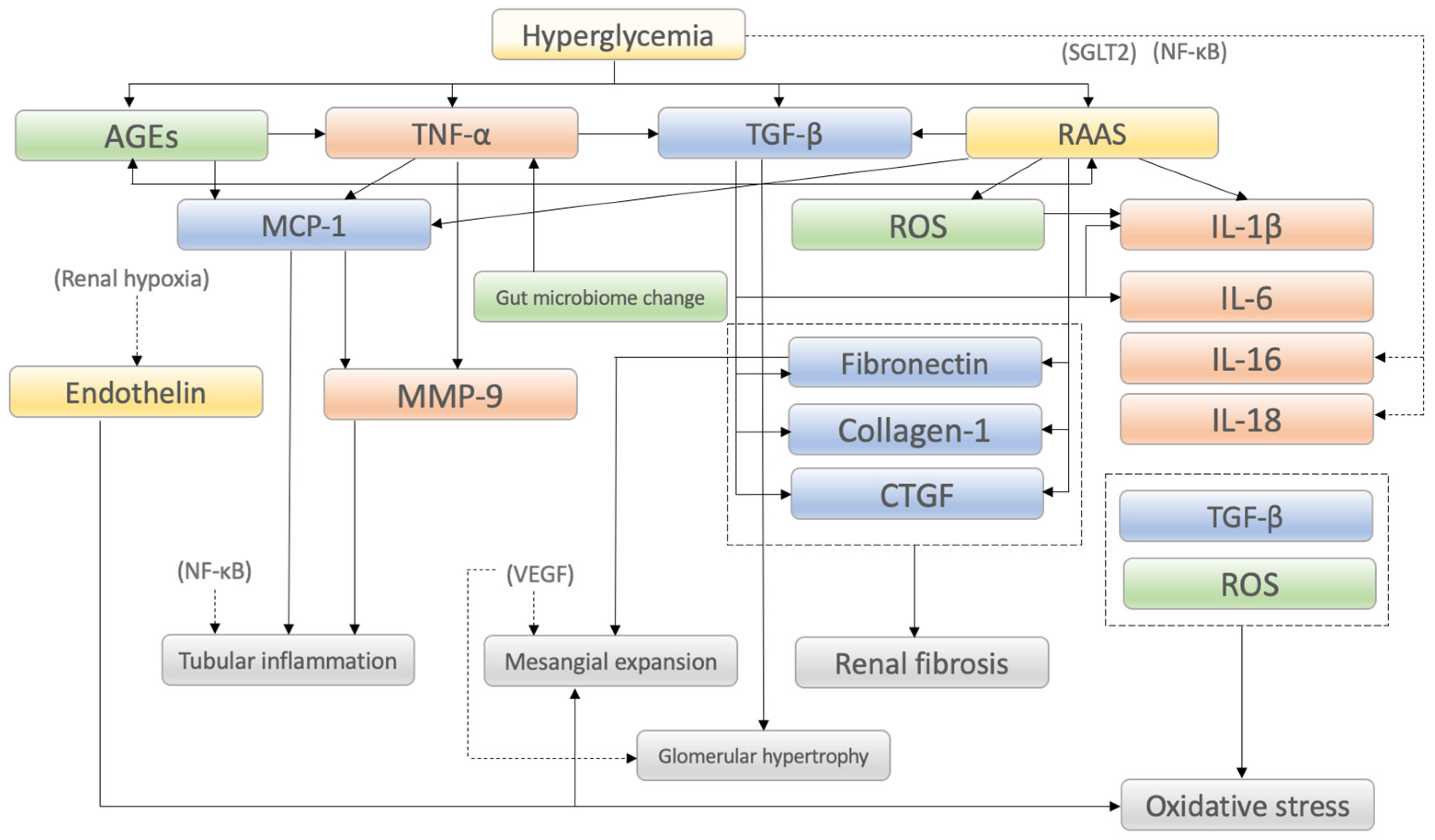
2. Molecular Mechanisms of Diabetic Kidney Disease
2.1. Inflammatory Factors
2.1.1. Tumor Necrosis Factor-α (TNF-α)
2.1.2. Interleukin-1 (IL-1)
2.1.3. Interleukin-6 (IL-6)
2.1.4. Interleukin-16 (IL-16)
2.1.5. Interleukin-18 (IL-18)
2.1.6. Monocyte Chemoattractant Protein-1 (MCP-1)
2.1.7. Matrix Metalloproteinase-9 (MMP-9)
2.2. Fibrotic Factors
2.2.1. Transforming Growth Factor-β (TGF-β)
2.2.2. Fibronectin
2.2.3. Collagen-1
2.2.4. Connective Tissue Growth Factor (CTGF)
2.3. Metabolic Factors
2.3.1. Reactive Oxygen Species (ROS)
2.3.2. Advanced Glycation End Products (AGEs)
2.3.3. Gut Microbiome Changes
2.4. Hemodynamic Factors
2.4.1. Renin–Angiotensin–Aldosterone System (RAAS)
2.4.2. Endothelin (ET)
2.5. Recent Advances in the Treatment of DKD
2.5.1. SGLT-2 Inhibitors
2.5.2. Nonsteroidal Mineralocorticoid Receptor Antagonists (MRAs)
2.5.3. Glucagon-like Peptide-1 Receptor (GLP-1R) Agonists
2.5.4. Endothelin Receptor Antagonists (ERAs)
2.5.5. Other Agents
2.6. Significance of Molecular Mechanisms as Biomarkers and Therapeutic Targets
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Doshi, S.M.; Friedman, A.N. Diagnosis and Management of Type 2 Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, L.; House, A.; Gorini, A.; Santoboni, A.; Russo, D.; Ronco, C. Chronic kidney disease and cardiovascular complications. Heart Fail. Rev. 2015, 20, 259–272. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Lima-Posada, I.; Bakris, G.L.; Jaisser, F. Mineralocorticoid receptor antagonists in diabetic kidney disease—Mechanistic and therapeutic effects. Nat. Rev. Nephrol. 2022, 18, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Langkilde, A.M.; Wheeler, D.C. Dapagliflozin in Patients with Chronic Kidney Disease. Reply. N. Engl. J. Med. 2021, 384, 389–390. [Google Scholar] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Marx, N.; Lam, C.S.P.; Schnaidt, S.; Ofstad, A.P.; Brueckmann, M.; Jamal, W.; et al. Effect of Empagliflozin on Cardiovascular and Renal Outcomes in Patients With Heart Failure by Baseline Diabetes Status: Results From the EMPEROR-Reduced Trial. Circulation 2021, 143, 337–349. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef]
- Tang, S.C.; Chan, L.Y.; Leung, J.C.; Cheng, A.S.; Chan, K.W.; Lan, H.Y.; Lai, K.N. Bradykinin and high glucose promote renal tubular inflammation. Nephrol. Dial. Transplant. 2010, 25, 698–710. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Han, Q.; Zhu, H.; Chen, X.; Liu, Z. Non-genetic mechanisms of diabetic nephropathy. Front. Med. 2017, 11, 319–332. [Google Scholar] [CrossRef]
- Klemis, V.; Ghura, H.; Federico, G.; Wurfel, C.; Bentmann, A.; Gretz, N.; Miyazaki, T.; Grone, H.J.; Nakchbandi, I.A. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int. 2017, 91, 1374–1385. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Draves, S.O.; Rosca, M. Mitochondria in Diabetic Kidney Disease. Cells 2021, 10, 2945. [Google Scholar] [CrossRef]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; lglesias-de la Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef]
- Xiang, E.; Han, B.; Zhang, Q.; Rao, W.; Wang, Z.F.; Chang, C.; Zhang, Y.Q.; Tu, C.S.; Li, C.Y.; Wu, D.C. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res. Ther. 2020, 11, 336. [Google Scholar] [CrossRef]
- Yang, H.; Xie, T.; Li, D.; Du, X.; Wang, T.; Li, C.; Song, X.; Xu, L.; Yi, F.; Liang, X.; et al. Tim-3 aggravates podocyte injury in diabetic nephropathy by promoting macrophage activation via the NF-kappaB/TNF-alpha pathway. Mol. Metab. 2019, 23, 24–36. [Google Scholar] [CrossRef]
- Caamano, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Md Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Interleukin-18 and diabetic nephropathy: A review. J. Cell. Physiol. 2019, 234, 5674–5682. [Google Scholar] [CrossRef]
- Han, Y.C.; Tang, S.Q.; Liu, Y.T.; Li, A.M.; Zhan, M.; Yang, M.; Song, N.; Zhang, W.; Wu, X.Q.; Peng, C.H.; et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021, 12, 925. [Google Scholar] [CrossRef]
- Kume, S.; Koya, D. Autophagy: A Novel Therapeutic Target for Diabetic Nephropathy. Diabetes Metab. J. 2015, 39, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Dusabimana, T.; Park, E.J.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. Geniposide Improves Diabetic Nephropathy by Enhancing ULK1-Mediated Autophagy and Reducing Oxidative Stress through AMPK Activation. Int. J. Mol. Sci. 2021, 22, 1651. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Hu, Q.S.; Li, C.L.; Liang, K.; Xiang, Y.; Hsiao, H.D.; Nguyen, T.K.; Park, P.K.; Egranov, S.D.; Ambati, C.R.; et al. PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J. Clin. Investig. 2019, 129, 1129–1151. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Liu, B.; Kanasaki, K.; Koya, D.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat. Commun. 2021, 12, 2368. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Liao, G.; Chen, Y.; Luo, A.; Liu, J.; Yuan, Y.; Li, L.; Yang, L.; Wang, H.; Liu, F.; et al. Intervention for early diabetic nephropathy by mesenchymal stem cells in a preclinical nonhuman primate model. Stem Cell Res. Ther. 2019, 10, 363. [Google Scholar] [CrossRef]
- Chen, X.; Han, Y.; Gao, P.; Yang, M.; Xiao, L.; Xiong, X.; Zhao, H.; Tang, C.; Chen, G.; Zhu, X.; et al. Disulfide-bond A oxidoreductase-like protein protects against ectopic fat deposition and lipid-related kidney damage in diabetic nephropathy. Kidney Int. 2019, 95, 880–895. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Tang, P.M.; Tang, P.C.; Xiao, J.; Huang, X.R.; Yu, C.; Ma, R.C.W.; Lan, H.Y. LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1-Dependent Renal Inflammation. Diabetes 2019, 68, 1485–1498. [Google Scholar] [CrossRef]
- Sugahara, M.; Tanaka, S.; Tanaka, T.; Saito, H.; Ishimoto, Y.; Wakashima, T.; Ueda, M.; Fukui, K.; Shimizu, A.; Inagi, R.; et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. J. Am. Soc. Nephrol. 2020, 31, 560–577. [Google Scholar] [CrossRef]
- Hasegawa, S.; Tanaka, T.; Saito, T.; Fukui, K.; Wakashima, T.; Susaki, E.A.; Ueda, H.R.; Nangaku, M. The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney Int. 2020, 97, 934–950. [Google Scholar] [CrossRef]
- Tian, S.M.; Vieira Jr, J.M.; Dominguez, W.V.; Moreira, S.R.S.; Pereira, A.B.; Barros, R.T.; Zatz, R. Urinary MCP-1 and RBP: Independent predictors of renal outcome in macroalbuminuric diabetic nephropathy. J. Diabetes Complicat. 2012, 26, 546–553. [Google Scholar]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson, A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; et al. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J. Am. Soc. Nephrol. 2021, 32, 115–126. [Google Scholar] [CrossRef]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eur. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef]
- Yang, H.; Chen, H.; Liu, F.; Ma, Q. Up-regulation of matrix metalloproteinases-9 in the kidneys of diabetic rats and the association with neutrophil gelatinase-associated lipocalin. BMC Nephrol. 2021, 22, 211. [Google Scholar] [CrossRef]
- Yue, Y.; Yeh, J.N.; Chiang, J.Y.; Sung, P.H.; Chen, Y.L.; Liu, F.; Yip, H.K. Intrarenal arterial administration of human umbilical cord-derived mesenchymal stem cells effectively preserved the residual renal function of diabetic kidney disease in rat. Stem Cell Res. Ther. 2022, 13, 186. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. [Google Scholar] [CrossRef]
- Yang, C.; Chen, X.C.; Li, Z.H.; Wu, H.L.; Jing, K.P.; Huang, X.R.; Ye, L.; Wei, B.; Lan, H.Y.; Liu, H.F. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy 2021, 17, 2325–2344. [Google Scholar] [CrossRef]
- Hong, Q.; Zhang, L.; Fu, J.; Verghese, D.A.; Chauhan, K.; Nadkarni, G.N.; Li, Z.; Ju, W.; Kretzler, M.; Cai, G.Y.; et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J. Am. Soc. Nephrol. 2019, 30, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Cai, H.; Zhang, L.; Li, Z.; Zhong, F.; Ni, Z.; Cai, G.; Chen, X.M.; He, J.C.; Lee, K. Modulation of transforming growth factor-beta-induced kidney fibrosis by leucine-rich-2 glycoprotein-1. Kidney Int. 2022, 101, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.S.; Li, H.Y.; Yang, Q.; Chen, G.Y.; Lin, S.J.; Liao, C.L.; Zhou, T.B. Administration of mesenchymal stem cells in diabetic kidney disease: A systematic review and meta-analysis. Stem Cell Res. Ther. 2021, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Hu, F.; Xue, M.; Jia, Y.J.; Zheng, Z.J.; Li, Y.; Xue, Y.M. Early growth response protein-1 upregulates long noncoding RNA Arid2-IR to promote extracellular matrix production in diabetic kidney disease. Am. J. Physiol. Cell Physiol. 2019, 316, C340–C352. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm. Regen. 2018, 38, 14. [Google Scholar] [CrossRef]
- Looker, H.C.; Lin, C.; Nair, V.; Kretzler, M.; Mauer, M.; Najafian, B.; Nelson, R.G. Serum Level of Polyubiquitinated PTEN and Loss of Kidney Function in American Indians With Type 2 Diabetes. Am. J. Kidney Dis. 2022, 79, 497–506. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; He, Q.; Bulus, N.; Fogo, A.B.; Zhang, M.Z.; Harris, R.C. YAP Activation in Renal Proximal Tubule Cells Drives Diabetic Renal Interstitial Fibrogenesis. Diabetes 2020, 69, 2446–2457. [Google Scholar] [CrossRef]
- Lee, E.; Choi, J.; Lee, H.S. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J. Cell. Physiol. 2017, 232, 3209–3217. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef]
- Song, S.; Qiu, D.; Shi, Y.; Wang, S.; Zhou, X.; Chen, N.; Wei, J.; Wu, M.; Wu, H.; Duan, H. Thioredoxin-interacting protein deficiency alleviates phenotypic alterations of podocytes via inhibition of mTOR activation in diabetic nephropathy. J. Cell. Physiol. 2019, 234, 16485–16502. [Google Scholar] [CrossRef]
- Wu, M.; Yang, Z.; Zhang, C.; Shi, Y.; Han, W.; Song, S.; Mu, L.; Du, C.; Shi, Y. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism 2021, 118, 154748. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Q.; Han, B.; Chen, Y.; Qiao, X.; Wang, L. CD36 promotes NLRP3 inflammasome activation via the mtROS pathway in renal tubular epithelial cells of diabetic kidneys. Cell Death Dis. 2021, 12, 523. [Google Scholar] [CrossRef]
- Yang, X.C.; Okamura, D.M.; Lu, X.F.; Chen, Y.X.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Shi, Q.; Lee, D.Y.; Feliers, D.; Abboud, H.E.; Bhat, M.A.; Gorin, Y. Interplay between RNA-binding protein HuR and Nox4 as a novel therapeutic target in diabetic kidney disease. Mol. Metab. 2020, 36, 100968. [Google Scholar] [CrossRef]
- Jha, J.C.; Dai, A.; Garzarella, J.; Charlton, A.; Urner, S.; Ostergaard, J.A.; Okabe, J.; Holterman, C.E.; Skene, A.; Power, D.A.; et al. Independent of Renox, NOX5 Promotes Renal Inflammation and Fibrosis in Diabetes by Activating ROS-Sensitive Pathways. Diabetes 2022, 71, 1282–1298. [Google Scholar] [CrossRef]
- Tanji, N.; Markowitz, G.S.; Fu, C.F.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef]
- Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Tan, Y.Q.; Yu, X.Y.; Zhao, Y.Y. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic. Biol. Med. 2021, 171, 260–271. [Google Scholar] [CrossRef]
- Hagiwara, S.; Sourris, K.; Ziemann, M.; Tieqiao, W.; Mohan, M.; McClelland, A.D.; Brennan, E.; Forbes, J.; Coughlan, M.; Harcourt, B.; et al. RAGE Deletion Confers Renoprotection by Reducing Responsiveness to Transforming Growth Factor-beta and Increasing Resistance to Apoptosis. Diabetes 2018, 67, 960–973. [Google Scholar] [CrossRef]
- Hou, B.Y.; Qiang, G.F.; Zhao, Y.R.; Yang, X.Y.; Chen, X.; Yan, Y.; Wang, X.B.; Liu, C.; Zhang, L.; Du, G.H. Salvianolic Acid A Protects Against Diabetic Nephropathy through Ameliorating Glomerular Endothelial Dysfunction via Inhibiting AGE-RAGE Signaling. Cell. Physiol. Biochem. 2017, 44, 2378–2394. [Google Scholar] [CrossRef]
- Watson, A.M.D.; Gray, S.P.; Jiaze, L.; Soro-Paavonen, A.; Wong, B.; Cooper, M.E.; Bierhaus, A.; Pickering, R.; Tikellis, C.; Tsorotes, D.; et al. Alagebrium Reduces Glomerular Fibrogenesis and Inflammation Beyond Preventing RAGE Activation in Diabetic Apolipoprotein E Knockout Mice. Diabetes 2012, 61, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Azegami, T.; Nakayama, T.; Hayashi, K.; Hishikawa, A.; Yoshimoto, N.; Nakamichi, R.; Itoh, H. Vaccination Against Receptor for Advanced Glycation End Products Attenuates the Progression of Diabetic Kidney Disease. Diabetes 2021, 70, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Alam, S.S.; Riaz, S.; Larkin, J.R.; Akhtar, M.W.; Shafi, T.; Thornalley, P.J. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: A randomised, double-blind placebo-controlled pilot study. Diabetologia 2009, 52, 208–212. [Google Scholar] [CrossRef]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef]
- Alkhalaf, A.; Klooster, A.; van Oeveren, W.; Achenbach, U.; Kleefstra, N.; Slingerland, R.J.; Mijnhout, G.S.; Bilo, H.J.G.; Gans, R.O.B.; Navis, G.J.; et al. A double-blind, randomized, placebo-controlled clinical trial on benfotiamine treatment in patients with diabetic nephropathy. Diabetes Care 2010, 33, 1598–1601. [Google Scholar] [CrossRef]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Beeri, M.S.; de la Maza, M.P.; Rojas, A.; Salazar-Villanea, S.; Uribarri, J. The potential role of dietary advanced glycation endproducts in the development of chronic non-infectious diseases: A narrative review. Nutr. Res. Rev. 2020, 33, 298–311. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The Gut Microbiome, Kidney Disease, and Targeted Interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef]
- Ma, J.; Chadban, S.J.; Zhao, C.Y.; Chen, X.C.; Kwan, T.; Panchapakesan, U.; Pollock, C.A.; Wu, H.L. TLR4 Activation Promotes Podocyte Injury and Interstitial Fibrosis in Diabetic Nephropathy. PLoS ONE 2014, 9, e97985. [Google Scholar] [CrossRef]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Linh, H.T.; Iwata, Y.; Senda, Y.; Sakai-Takemori, Y.; Nakade, Y.; Oshima, M.; Nakagawa-Yoneda, S.; Ogura, H.; Sato, K.; Minami, T.; et al. Intestinal Bacterial Translocation Contributes to Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1105–1119. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.C.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.Z.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid?Mediated Activation of G Protein? Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef]
- Premaratne, E.; Verma, S.; Ekinci, E.I.; Theverkalam, G.; Jerums, G.; MacIsaac, R.J. The impact of hyperfiltration on the diabetic kidney. Diabetes Metab. 2015, 41, 5–17. [Google Scholar] [CrossRef]
- Anders, H.J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Tuttle, K.R. Back to the Future: Glomerular Hyperfiltration and the Diabetic Kidney. Diabetes 2017, 66, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Wu, H.; Gonzalez Villalobos, R.; Yao, X.; Reilly, D.; Chen, T.; Rankin, M.; Myshkin, E.; Breyer, M.D.; Humphreys, B.D. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metab. 2022, 34, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, K.; Cherney, D.Z.I.; Bozovic, A.; Nagasu, H.; Satoh, M.; Kanda, E.; Sasaki, T.; Kashihara, N. Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using In Vivo Imaging. Circulation 2019, 140, 303–315. [Google Scholar] [CrossRef]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Renal Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef]
- Ravindran, S.; Munusamy, S. Renoprotective mechanisms of sodium-glucose co-transporter 2 (SGLT2) inhibitors against the progression of diabetic kidney disease. J. Cell. Physiol. 2022, 237, 1182–1205. [Google Scholar] [CrossRef]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Hinden, L.; Riahi, Y.; Israeli, T.; Tirosh, B.; Cerasi, E.; Mizrachi, E.B.; Tam, J.; Mosenzon, O.; Leibowitz, G. Proximal Tubule mTORC1 Is a Central Player in the Pathophysiology of Diabetic Nephropathy and Its Correction by SGLT2 Inhibitors. Cell Rep. 2020, 32, 107954. [Google Scholar] [CrossRef]
- Gurley, S.B.; Ghosh, S.; Johnson, S.A.; Azushima, K.; Sakban, R.B.; George, S.E.; Maeda, M.; Meyer, T.W.; Coffman, T.M. Inflammation and Immunity Pathways Regulate Genetic Susceptibility to Diabetic Nephropathy. Diabetes 2018, 67, 2096–2106. [Google Scholar] [CrossRef]
- Singh, R.; Singh, A.K.; Leehey, D.J. A novel mechanism for angiotensin II formation in streptozotocin-diabetic rat glomeruli. Am. J. Physiol. Renal Physiol. 2005, 288, F1183–F1190. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; Habibi, J.; Nistala, R.; Cooper, S.A.; Karuparthi, P.R.; Hayden, M.R.; Rehmer, N.; DeMarco, V.G.; Andresen, B.T.; Wei, Y.Z.; et al. Attenuation of NADPH oxidase activation and glomerular filtration barrier remodeling with statin treatment. Hypertension 2008, 51, 474–480. [Google Scholar] [CrossRef][Green Version]
- Ilatovskaya, D.V.; Blass, G.; Palygin, O.; Levchenko, V.; Pavlov, T.S.; Grzybowski, M.N.; Winsor, K.; Shuyskiy, L.S.; Geurts, A.M.; Cowley, A.W.; et al. A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1917–1927. [Google Scholar] [CrossRef]
- Haruhara, K.; Suzuki, T.; Wakui, H.; Azushima, K.; Kurotaki, D.; Kawase, W.; Uneda, K.; Kobayashi, R.; Ohki, K.; Kinguchi, S.; et al. Deficiency of the kidney tubular angiotensin II type1 receptor-associated protein ATRAP exacerbates streptozotocin-induced diabetic glomerular injury via reducing protective macrophage polarization. Kidney Int. 2022, 101, 912–928. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef]
- Ritz, E.; Tomaschitz, A. Aldosterone, a vasculotoxic agent--novel functions for an old hormone. Nephrol. Dial. Transplant. 2009, 24, 2302–2305. [Google Scholar] [CrossRef][Green Version]
- Guan, Z.; VanBeusecum, J.P.; Inscho, E.W. Endothelin and the renal microcirculation. Semin. Nephrol. 2015, 35, 145–155. [Google Scholar] [CrossRef]
- Lytvyn, Y.; Bjornstad, P.; van Raalte, D.H.; Heerspink, H.L.; Cherney, D.Z.I. The New Biology of Diabetic Kidney Disease-Mechanisms and Therapeutic Implications. Endocr. Rev. 2020, 41, 202–231. [Google Scholar] [CrossRef]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef]
- Boels, M.G.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; van der Vlag, J.; Koster, A.J.; van Zonneveld, A.J.; van Faassen, E.; Grone, H.J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef]
- Zhang, L.; Xue, S.; Hou, J.; Chen, G.; Xu, Z.G. Endothelin receptor antagonists for the treatment of diabetic nephropathy: A meta-analysis and systematic review. World J. Diabetes 2020, 11, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K.R. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Li, J.; Neuen, B.L.; Neal, B.; Arnott, C.; Parikh, C.R.; Coca, S.G.; Perkovic, V.; Mahaffey, K.W.; Yavin, Y.; et al. Effects of the SGLT2 inhibitor canagliflozin on plasma biomarkers TNFR-1, TNFR-2 and KIM-1 in the CANVAS trial. Diabetologia 2021, 64, 2147–2158. [Google Scholar] [CrossRef]
- Wanner, C.; Marx, N. SGLT2 inhibitors: The future for treatment of type 2 diabetes mellitus and other chronic diseases. Diabetologia 2018, 61, 2134–2139. [Google Scholar] [CrossRef]
- American Diabetes Association. 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2020. Diabetes Care. 2020, 43 (Suppl. 1), S135–S151. [Google Scholar] [CrossRef]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Salah, H.M.; Al’Aref, S.J.; Khan, M.S.; Al-Hawwas, M.; Vallurupalli, S.; Mehta, J.L.; Mounsey, J.P.; Greene, S.J.; McGuire, D.K.; Lopes, R.D.; et al. Effect of sodium-glucose cotransporter 2 inhibitors on cardiovascular and kidney outcomes-Systematic review and meta-analysis of randomized placebo-controlled trials. Am. Heart J. 2021, 232, 10–22. [Google Scholar] [CrossRef]
- Group, E.-K.C.; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2022. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, Inflammation, Immune System, and Hypertension. Am. J. Hypertens. 2021, 34, 15–27. [Google Scholar] [CrossRef]
- De Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes management in chronic kidney disease: A consensus report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2022, 102, 974–989. [Google Scholar] [CrossRef]
- Rossing, P.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: An update based on rapidly emerging new evidence. Kidney Int. 2022, 102, 990–999. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef]
- Kolkhof, P.; Hartmann, E.; Freyberger, A.; Pavkovic, M.; Mathar, I.; Sandner, P.; Droebner, K.; Joseph, A.; Huser, J.; Eitner, F. Effects of Finerenone Combined with Empagliflozin in a Model of Hypertension-Induced End-Organ Damage. Am. J. Nephrol. 2021, 52, 642–652. [Google Scholar] [CrossRef]
- Shen, L.; Kristensen, S.L.; Bengtsson, O.; Bohm, M.; de Boer, R.A.; Docherty, K.F.; Inzucchi, S.E.; Katova, T.; Kober, L.; Kosiborod, M.N.; et al. Dapagliflozin in HFrEF Patients Treated With Mineralocorticoid Receptor Antagonists: An Analysis of DAPA-HF. JACC Heart Fail. 2021, 9, 254–264. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Zannad, F.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Jamal, W.; Steubl, D.; Schueler, E.; et al. Interplay of Mineralocorticoid Receptor Antagonists and Empagliflozin in Heart Failure: EMPEROR-Reduced. J. Am. Coll. Cardiol. 2021, 77, 1397–1407. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Cox, E.J.; Neumiller, J.J.; Tuttle, K.R. Incretin drugs in diabetic kidney disease: Biological mechanisms and clinical evidence. Nat. Rev. Nephrol. 2021, 17, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.K.; Carr, M.J.; Kontopantelis, E.; Leelarathna, L.; Thabit, H.; Emsley, R.; Buchan, I.; Mamas, M.A.; van Staa, T.P.; Sattar, N.; et al. Primary Prevention of Cardiovascular and Heart Failure Events With SGLT2 Inhibitors, GLP-1 Receptor Agonists, and Their Combination in Type 2 Diabetes. Diabetes Care 2022, 45, 909–918. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef]
- Nangaku, M.; Takama, H.; Ichikawa, T.; Mukai, K.; Kojima, M.; Suzuki, Y.; Watada, H.; Wada, T.; Ueki, K.; Narita, I.; et al. Randomized, double-blind, placebo-controlled phase 3 study of bardoxolone methyl in patients with diabetic kidney disease: Design and baseline characteristics of AYAME study. Nephrol. Dial. Transplant. 2022, gfac242. [Google Scholar] [CrossRef]
- Chertow, G.M.; Pergola, P.E.; Chen, F.; Kirby, B.J.; Sundy, J.S.; Patel, U.D.; GS-US-223-1015 Investigators. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 1980–1990. [Google Scholar] [CrossRef]
- Gilead Sciences. Study Evaluating the Efficacy and Safety of Selonsertib in Participants with Moderate to Advanced Diabetic Kidney Disease (MOSAIC). Available online: https://clinicaltrials.gov/ct2/show/NCT04026165 (accessed on 15 November 2022).
- Rayego-Mateos, S.; Rodrigues-Diez, R.; Fernandez-Fernandez, B.; Mora-Fernandez, C.; Marchant, V.; Donate-Correa, J.; Navarro-Gonzalez, J.F.; Ortiz, A.; Ruiz-Ortega, M. Targeting inflammation to treat diabetic kidney disease: The road to 2030. Kidney Int. 2022. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category/ Factors | Mechanisms of Action in DKD Onset and Progression and/or Characteristic Findings | Reference |
|---|---|---|
| Inflammatory factors | ||
| TNF-α | Produced by activated macrophages and induces other cytokines, chemokines, apoptosis and cytotoxic effects | [7,15,16,17,18] |
| IL-1 | IL-1β plays a significant role in the association between hyperglycemia and macrophage infiltration | [19,20] |
| IL-6 | Induces neutrophil infiltration of the tubulointerstitium and is associated with thickening of the GBM and podocyte hypertrophy | [7,23,24] |
| IL-16 | Immunomodulatory cytokine that correlates with the severity of DKD, although concise mechanisms have not been clarified | [7,25] |
| IL-18 | Correlates most strongly with DKD severity among cytokines and is induced by inflammasome (NLRP3) | [19,26,27] |
| MCP-1 | Enhances the expression of other inflammatory factors such as inflammatory cytokines and inflammatory cells such as monocytes/macrophages | [28,29,30] |
| MMP-9 | Regulates extracellular matrix degradation during fibrosis in the proximal renal tubular epithelial cells | [34,35,36] |
| Fibrotic factors | ||
| TGF-β | Master regulator of inflammation and fibrosis. TGF-β1 and Smad3 (downstream of TGF-β) are particularly pathogenic | [39,40,41,42] |
| Fibronectin | Accumulation in glomerular mesangial lesions is associated with deterioration of kidney function | [12,43] |
| Collagen-1 | Deeply involved in the pathogenesis of renal fibrosis and excessive accumulation of extracellular matrix | [20,44] |
| CTGF | Its expression level correlates with the degree of glomerulosclerosis and tubulointerstitial fibrosis | [23,45,46] |
| Metabolic factors | ||
| ROS | Excess in podocytes promotes DKD. Induces epithelial-to-mesenchymal transition and apoptosis | [48,49,50,51,52,53,54,55,56,57] |
| AGEs | Alters extracellular matrix architecture. Modulation of cellular functions through interaction with RAGE | [58,59,60,61,62,63,64] |
| Gut microbiome changes | Dietary AGEs interact with colonic microbiota and trigger local inflammation and release of inflammatory mediators | [71,72,73,74,75,76,77] |
| Hemodynamic factors | ||
| RAAS | Induces ROS production and podocyte damage via angiotensin-II mediated calcium influx into podocytes | [81,91,92,93,94] |
| Endothelin | ET-1 activates proinflammatory and profibrotic pathways and induces endothelial dysfunction and oxidative stress | [7,97,98,99,100,101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, K.; Sato, E.; Mishima, E.; Miyazaki, M.; Tanaka, T. What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances. Int. J. Mol. Sci. 2023, 24, 570. https://doi.org/10.3390/ijms24010570
Watanabe K, Sato E, Mishima E, Miyazaki M, Tanaka T. What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances. International Journal of Molecular Sciences. 2023; 24(1):570. https://doi.org/10.3390/ijms24010570
Chicago/Turabian StyleWatanabe, Kimio, Emiko Sato, Eikan Mishima, Mariko Miyazaki, and Tetsuhiro Tanaka. 2023. "What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances" International Journal of Molecular Sciences 24, no. 1: 570. https://doi.org/10.3390/ijms24010570
APA StyleWatanabe, K., Sato, E., Mishima, E., Miyazaki, M., & Tanaka, T. (2023). What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances. International Journal of Molecular Sciences, 24(1), 570. https://doi.org/10.3390/ijms24010570