
Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis

 , , ,
, , ,  and
and 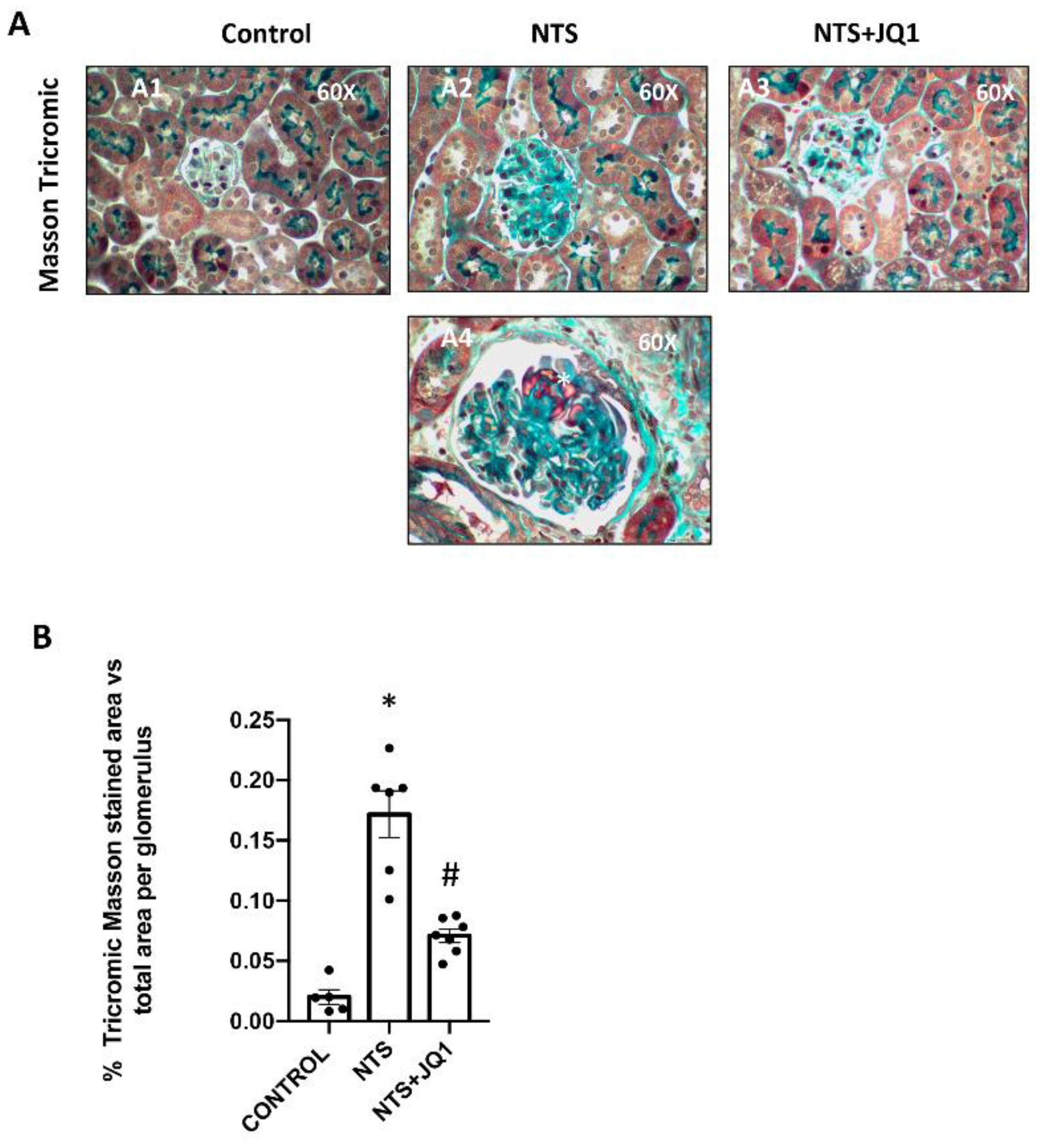{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. BET Inhibition Reduced Glomerulosclerosis in Experimental Nephrotoxic Nephritis
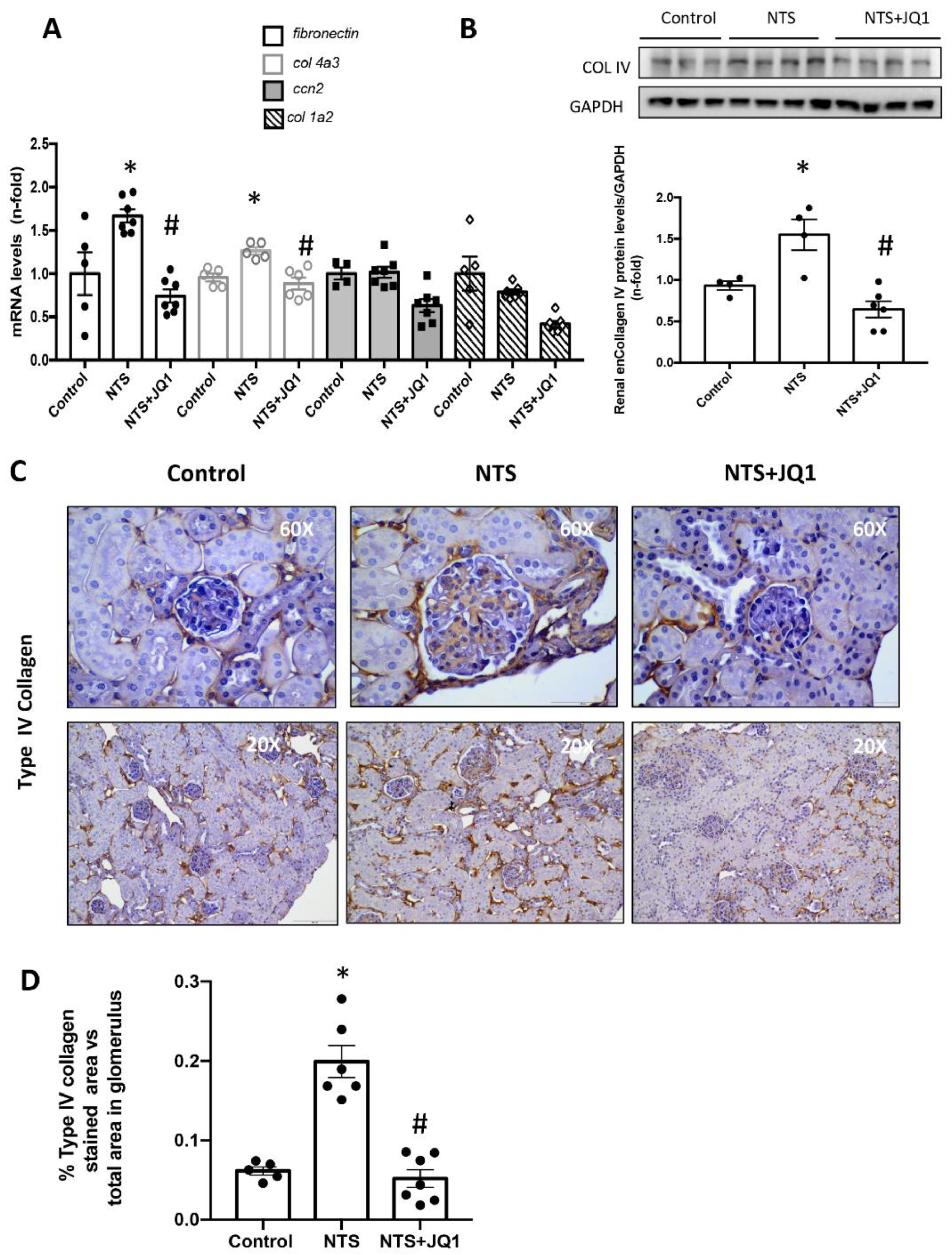
2.2. BET Inhibition Reduced Glomerular Type IV Collagen in Experimental Glomerulonephritis
2.3. Type IV Collagen Is One of the Specific Targets of BET Inhibition
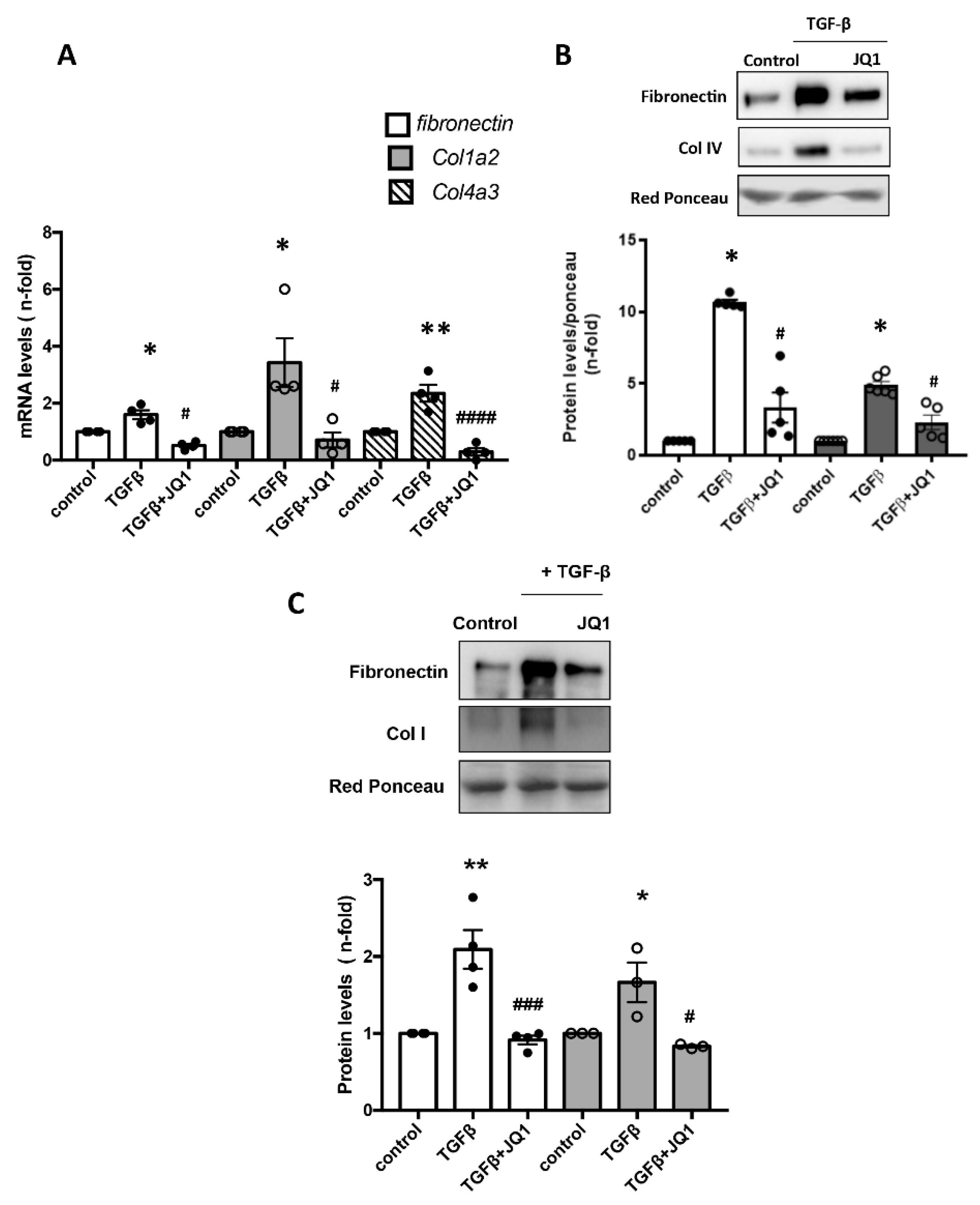
2.4. BET Inhibition Reduced ECM Production in Activated Mesangial Cells In Vitro
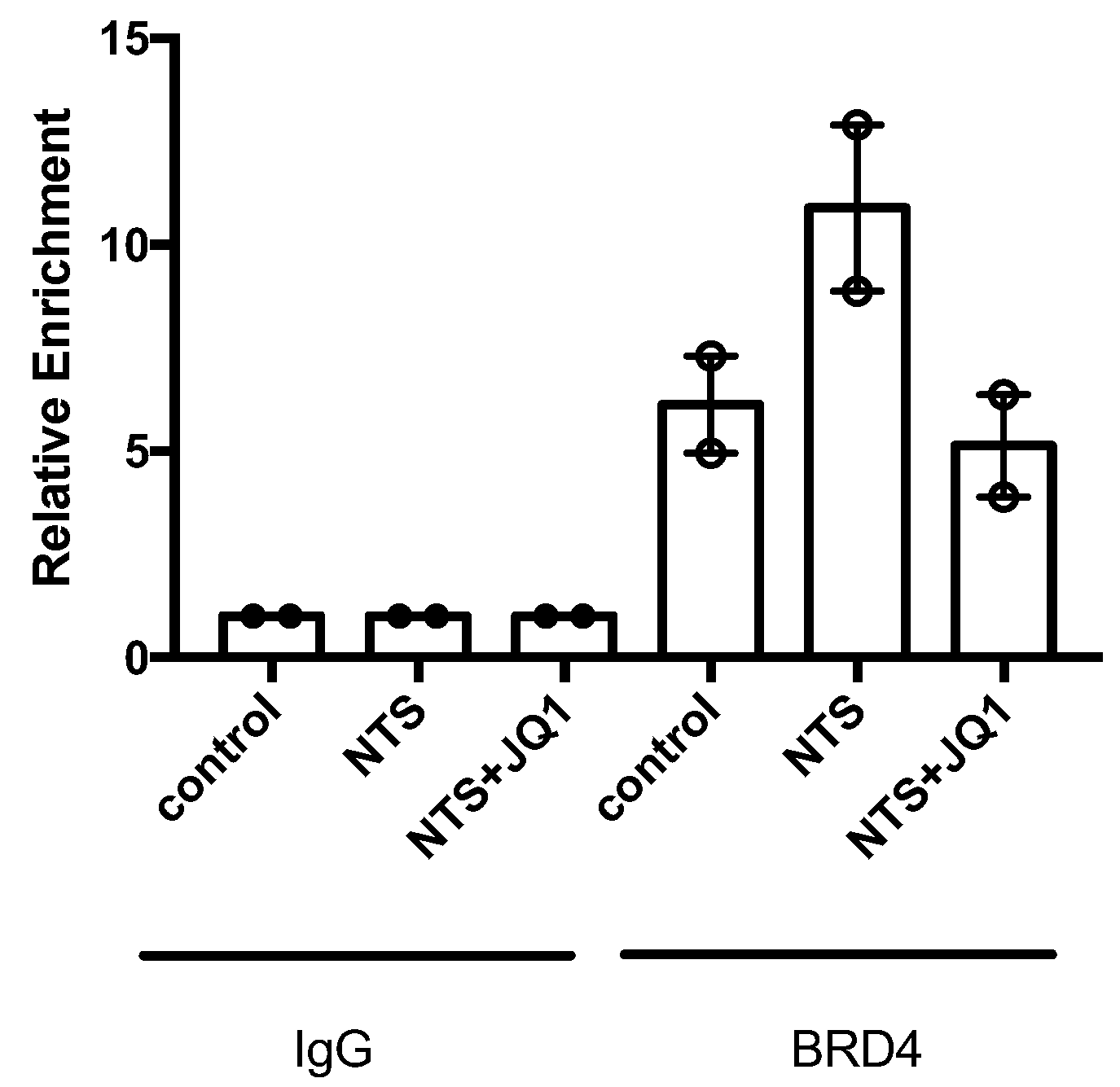
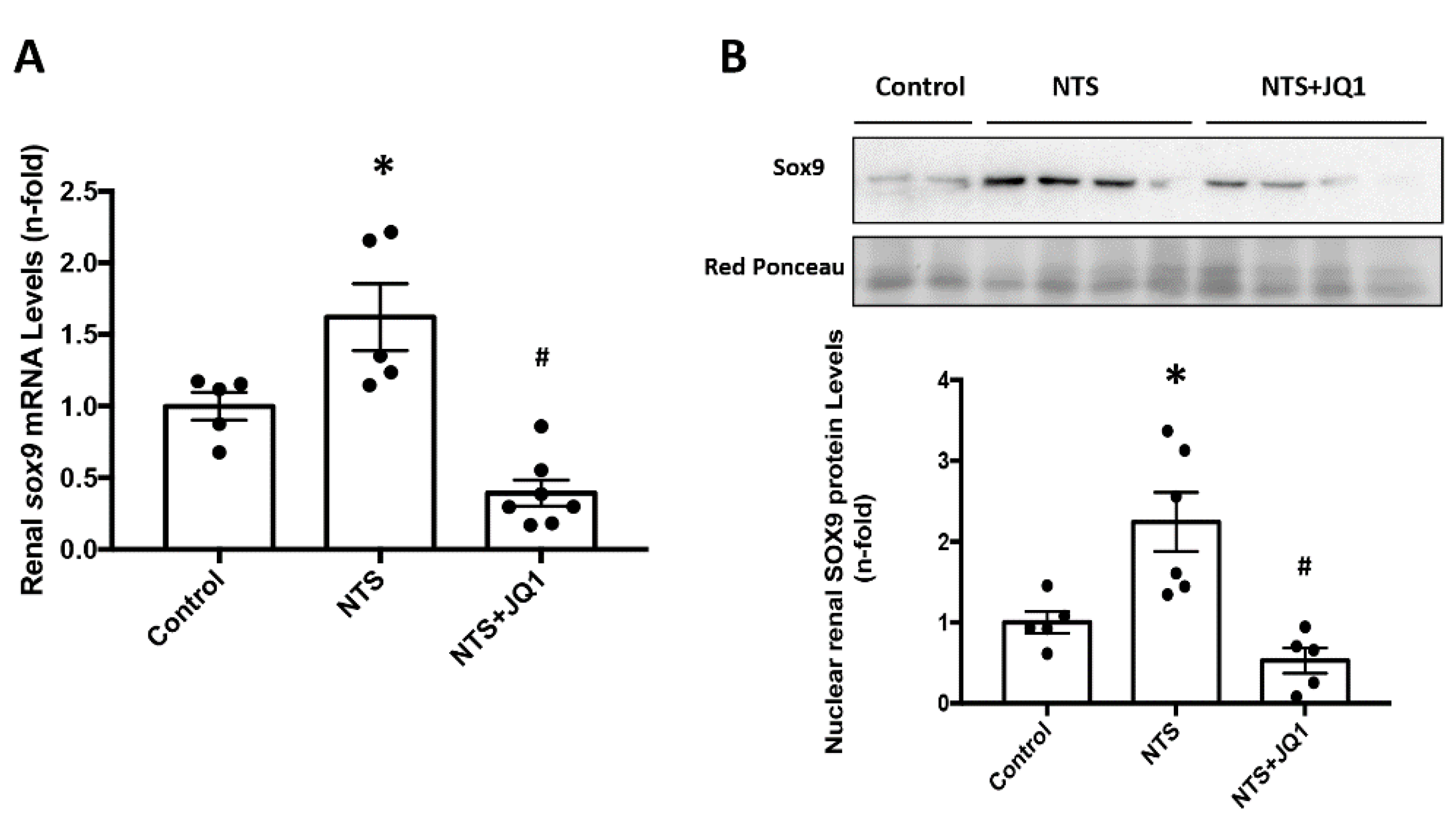
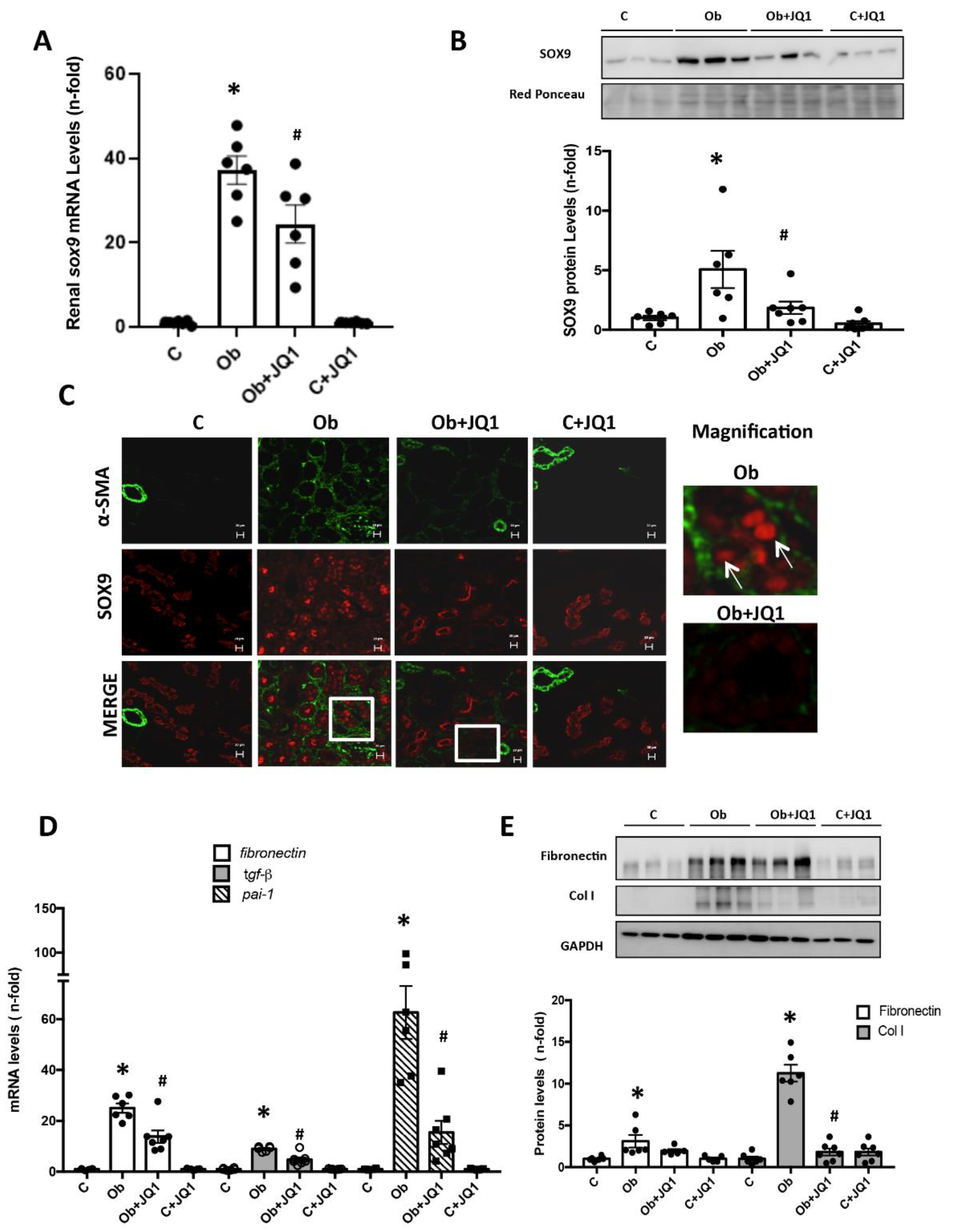
2.5. JQ1 Blocks SOX9 Activation in Experimental Nephrotoxic Nephritis
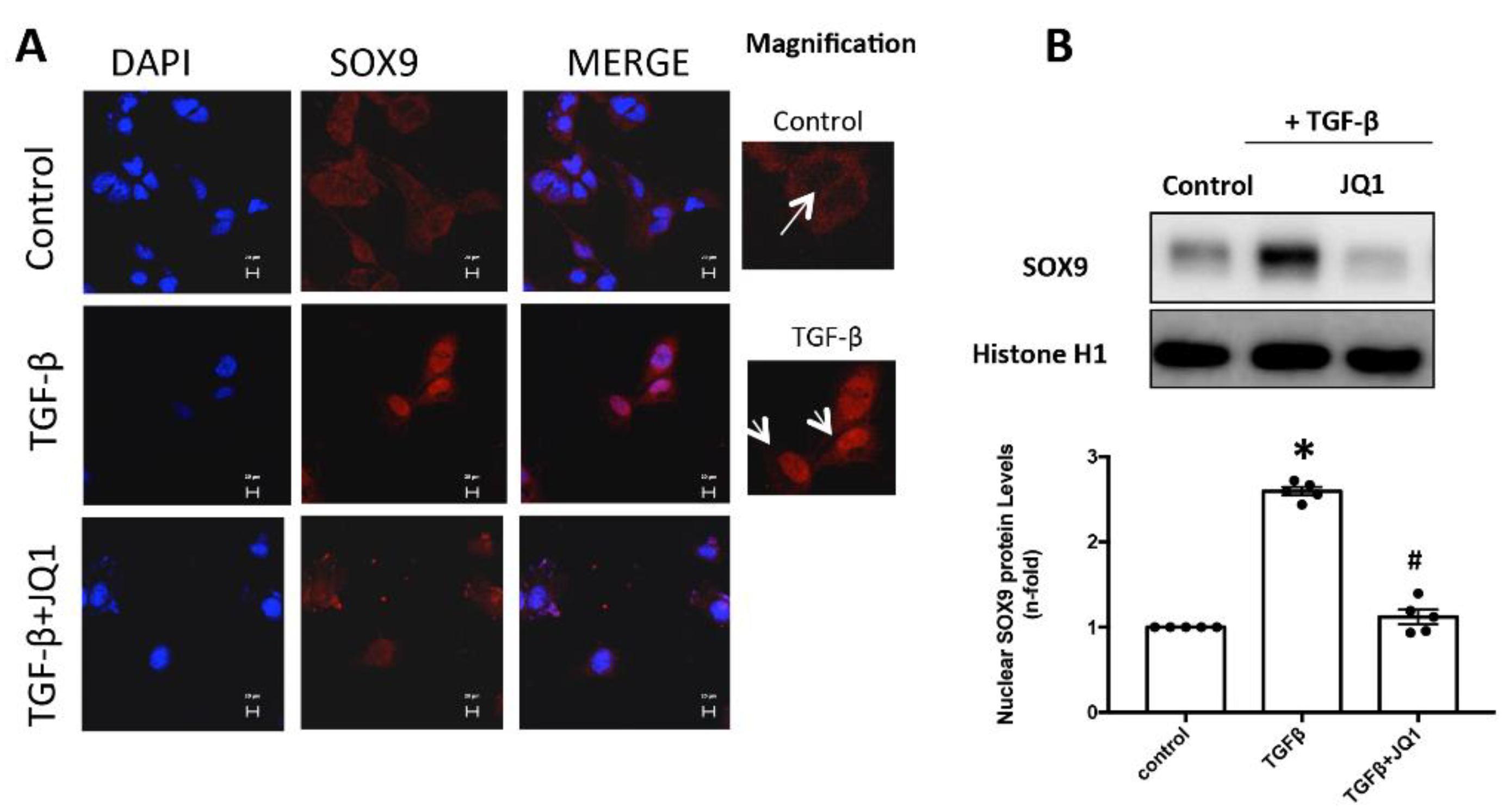
2.6. JQ1 Blocks SOX9 Activation in Cultured Mesangial Cells
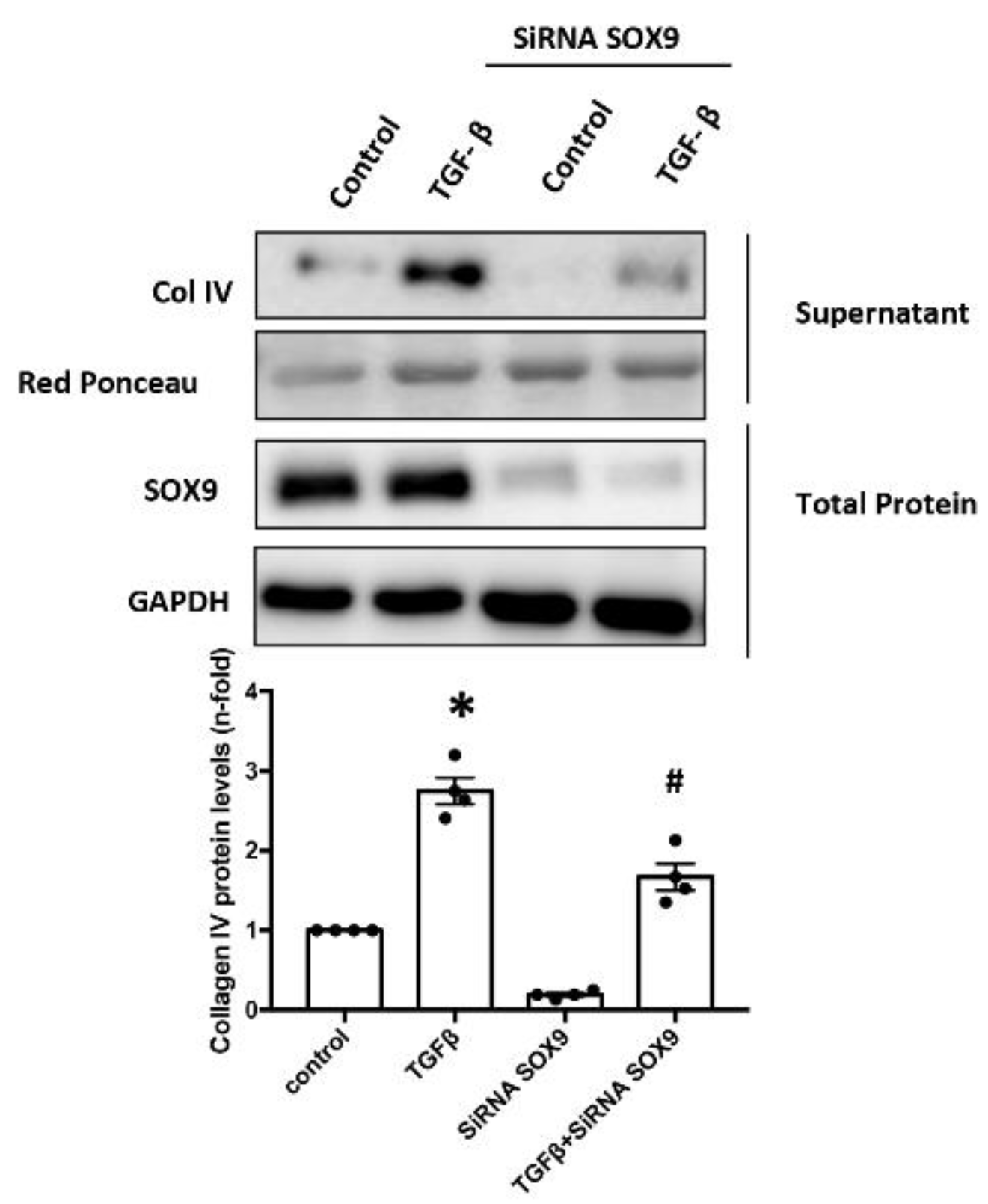
2.7. SOX9 Regulates Type IV Collagen Production in Cultured Mesangial
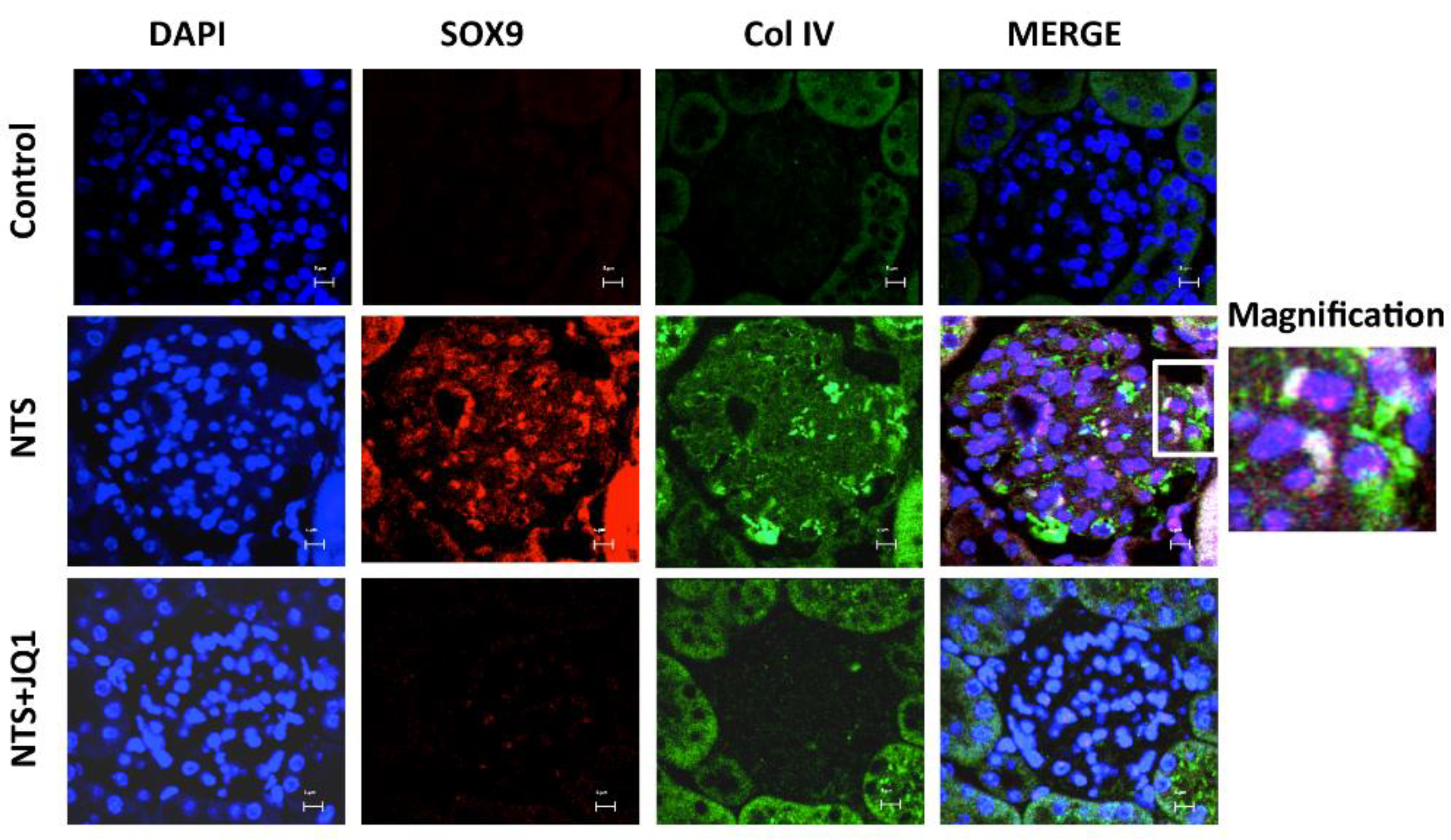
2.8. Activation of SOX9 and Increased Collagen IV in Experimental Nephrotoxic Nephritis
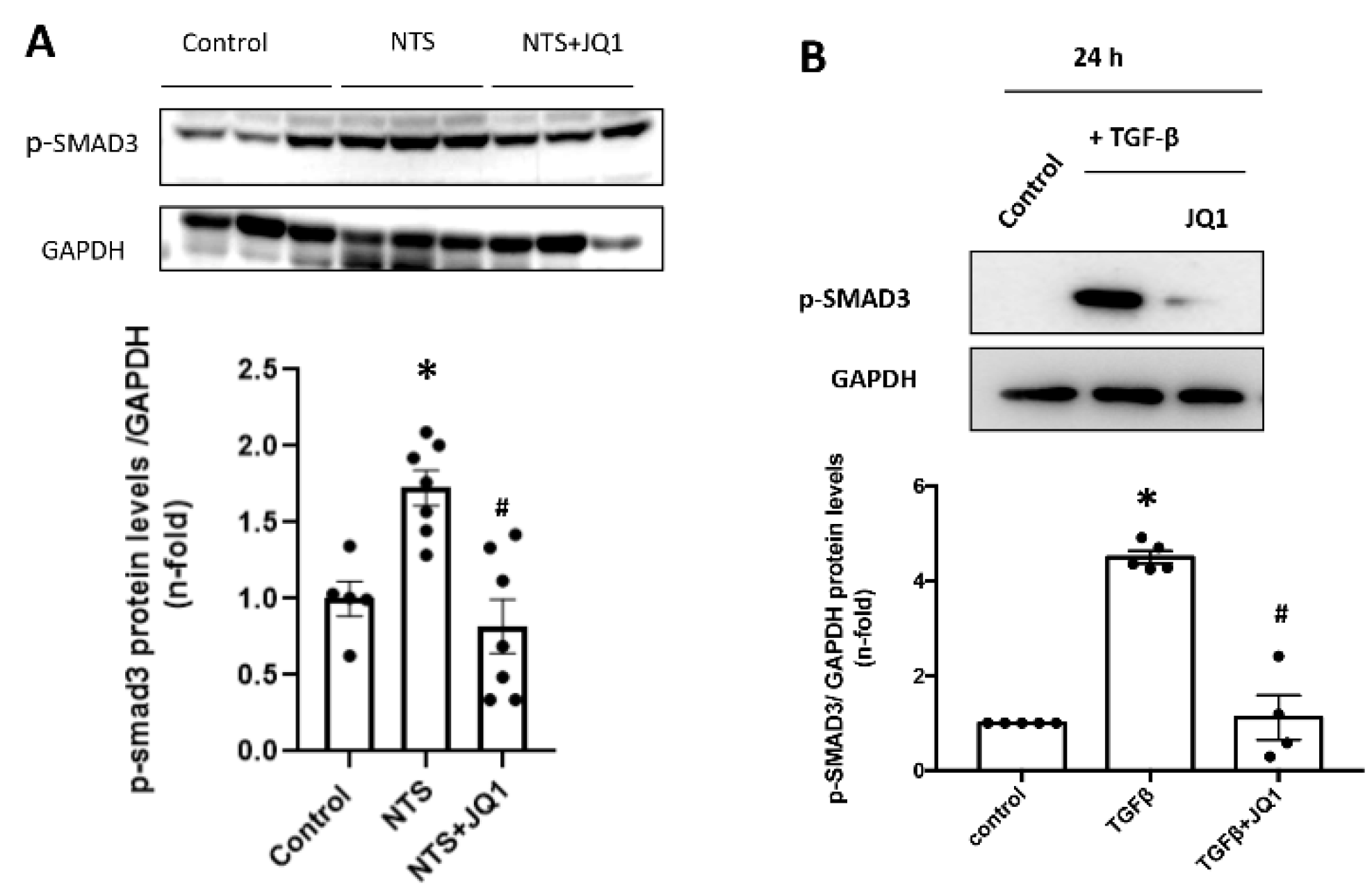
2.9. JQ1 Inhibits the SMAD Pathway in Renal Cells and in Experimental Kidney Fibrosis
2.10. SOX9 Nuclear Translocation and Interstitial Fibrosis Are Inhibited by JQ1 in Experimental Renal Fibrosis Induced by Unilateral Ureteral Obstruction (UUO)
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Models
4.3. In Vitro Studies
4.4. Histology and Immunohistochemistry
4.5. Gene Expression Studies
4.6. Protein Studies
4.7. Immunofluorescence Staining of Cells
4.8. Immunofluorescence Staining of Tissue Samples
4.9. Gene Silencing
4.10. Chromatin Immunoprecipitation
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.W.; et al. Forecasting Life Expectancy, Years of Life Lost, and All-Cause and Cause-Specific Mortality for 250 Causes of Death: Reference and Alternative Scenarios for 2016-40 for 195 Countries and Territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [PubMed]
- Healy, E.; Brady, H.R. Role of Tubule Epithelial Cells in the Pathogenesis of Tubulointerstitial Fibrosis Induced by Glomerular Disease. Curr. Opin. Nephrol. Hypertens 1998, 7, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.K.; Soo, H.I.; Hwang, C.-I.; Lee, Y.J.; Kim, J.; Yang, S.H.; Yon, S.K.; Yun, A.C.; Choi, S.; Park, W.Y. Gene Expression Profiling of Anti-GBM Glomerulonephritis Model: The Role of NF-KappaB in Immune Complex Kidney Disease. Kidney Int. 2004, 66, 1826–1837. [Google Scholar] [CrossRef]
- Crescentic Glomerulonephritis—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/28613478/ (accessed on 25 August 2022).
- Leinonen, A.; Netzer, K.O.; Boutaud, A.; Gunwar, S.; Hudson, B.G. Goodpasture Antigen: Expression of the Full-Length Alpha3(IV) Chain of Collagen IV and Localization of Epitopes Exclusively to the Noncollagenous Domain. Kidney Int. 1999, 55, 926–935. [Google Scholar] [CrossRef]
- Hellmark, T.; Segelmark, M.; Unger, C.; Burkhardt, H.; Saus, J.; Wieslander, J. Identification of a Clinically Relevant Immunodominant Region of Collagen IV in Goodpasture Disease. Kidney Int. 1999, 55, 936–944. [Google Scholar] [CrossRef][Green Version]
- [Collagen Type IV Nephropathy: From Thin Basement Membrane Nephropathy to Alport Syndrome]—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/16468607/ (accessed on 25 August 2022).
- Cosgrove, D.; Dufek, B.; Meehan, D.T.; Delimont, D.; Hartnett, M.; Samuelson, G.; Gratton, M.A.; Phillips, G.; MacKenna, D.A.; Bain, G. Lysyl Oxidase Like-2 Contributes to Renal Fibrosis in Col4α3/Alport Mice. Kidney Int. 2018, 94, 303–314. [Google Scholar] [CrossRef]
- Rubel, D.; Stock, J.; Ciner, A.; Hiller, H.; Girgert, R.; Müller, G.A.; Gross, O. Antifibrotic, Nephroprotective Effects of Paricalcitol versus Calcitriol on Top of ACE-Inhibitor Therapy in the COL4A3 Knockout Mouse Model for Progressive Renal Fibrosis. Nephrol. Dial. Transpl. 2014, 29, 1012–1019. [Google Scholar] [CrossRef][Green Version]
- Borza, D.B. Autoepitopes and Alloepitopes of Type IV Collagen: Role in the Molecular Pathogenesis of Anti-GBM Antibody Glomerulonephritis. Nephron. Exp. Nephrol. 2007, 106, e37–e43. [Google Scholar] [CrossRef]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s Syndrome, Goodpasture’s Syndrome, and Type IV Collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef]
- Endreffy, E.; Ondrik, Z.; Iványi, B.; Maróti, Z.; Bereczki, C.; Haszon, I.; Györke, Z.; Worum, E.; Németh, K.; Rikker, C.; et al. Collagen Type IV Nephropathy: Genetic Heterogeneity Examinations in Affected Hungarian Families. Mol. Cell. Probes 2011, 25, 28–34. [Google Scholar] [CrossRef]
- Smyth, L.J.; McKay, G.J.; Maxwell, A.P.; McKnight, A.J. DNA Hypermethylation and DNA Hypomethylation Is Present at Different Loci in Chronic Kidney Disease. Epigenetics 2014, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediat. Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef] [PubMed]
- The Role of Human Bromodomains in Chromatin Biology and Gene Transcription—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19736624/ (accessed on 25 August 2022).
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.F. Brd4 Maintains Constitutively Active NF-ΚB in Cancer Cells by Binding to Acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Domínguez-Andrés, J.; Ferreira, A.V.; Jansen, T.; Smithers, N.; Prinjha, R.K.; Furze, R.C.; Netea, M.G. Bromodomain Inhibitor I-BET151 Suppresses Immune Responses during Fungal-Immune Interaction. Eur. J. Immunol. 2019, 49, 2044–2050. [Google Scholar] [CrossRef]
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain Proteins Regulate Human Cytomegalovirus Latency and Reactivation Allowing Epigenetic Therapeutic Intervention. Proc. Natl. Acad. Sci. USA 2021, 118, e2023025118. [Google Scholar] [CrossRef]
- Kaneshita, S.; Kida, T.; Yoshioka, M.; Nishioka, K.; Raje, M.; Sakashita, A.; Hirano, A.; Sagawa, T.; Kasahara, A.; Inoue, T.; et al. CG223, a Novel BET Inhibitor, Exerts TGF-Β1-Mediated Antifibrotic Effects in a Murine Model of Bleomycin-Induced Pulmonary Fibrosis. Pulm. Pharmacol. Ther. 2021, 70, 102057. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Xu, Y.; Vakoc, C.R. Brd4 Is on the Move during Inflammation. Trends Cell Biol. 2014, 24, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET Bromodomain-Targeting Compounds Reactivate HIV from Latency via a Tat-Independent Mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Spriano, F.; Stathis, A.; Bertoni, F. Targeting BET Bromodomain Proteins in Cancer: The Example of Lymphomas. Pharmacol. Ther. 2020, 215, 107631. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Wu, P.; Liu, Z.; Xia, F.; Zhu, L.; Zheng, Z.; Yang, T.; Qi, J. BET Bromodomain Inhibition Suppresses Adipogenesis in Mice. Endocrine 2020, 67, 264–267. [Google Scholar] [CrossRef]
- Tang, X.; Peng, R.; Phillips, J.E.; Deguzman, J.; Ren, Y.; Apparsundaram, S.; Luo, Q.; Bauer, C.M.; Fuentes, M.E.; Demartino, J.A.; et al. Assessment of Brd4 Inhibition in Idiopathic Pulmonary Fibrosis Lung Fibroblasts and in Vivo Models of Lung Fibrosis. Am. J. Pathol. 2013, 183, 470–479. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, F.; Li, Z.; Mei, H.; Wang, Y.; Ma, H.; Shi, L.; Huang, A.; Zhang, T.; Lin, Z.; et al. Pharmacological Targeting of BET Proteins Attenuates Radiation-Induced Lung Fibrosis. Sci. Rep. 2018, 8, 998. [Google Scholar] [CrossRef]
- Middleton, S.A.; Rajpal, N.; Cutler, L.; Mander, P.; Rioja, I.; Prinjha, R.K.; Rajpal, D.; Agarwal, P.; Kumar, V. BET Inhibition Improves NASH and Liver Fibrosis. Sci. Rep. 2018, 8, 17257. [Google Scholar] [CrossRef]
- Xiong, C.; Masucci, M.V.; Zhou, X.; Liu, N.; Zang, X.; Tolbert, E.; Zhao, T.C.; Zhuang, S. Pharmacological Targeting of BET Proteins Inhibits Renal Fibroblast Activation and Alleviates Renal Fibrosis. Oncotarget 2016, 7, 69291–69308. [Google Scholar] [CrossRef]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 Is a Novel Therapeutic Target for Liver Fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Peng, Y.; Huang, T.; Xia, F.; Yang, T.; Duan, Q.; Zhang, W. Bromodomain-Containing Protein 4 Contributes to Renal Fibrosis through the Induction of Epithelial-Mesenchymal Transition. Exp. Cell Res. 2019, 383, 111507. [Google Scholar] [CrossRef]
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the SOX Family of Developmental Transcription Factors Based on Sequence and Structural Indicators. Dev. Biol. 2000, 227, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Chaboissier, M.C.; Kobayashi, A.; Vidal, V.I.P.; Lützkendorf, S.; van de Kant, H.J.G.; Wegner, M.; de Rooij, D.G.; Behringer, R.R.; Schedl, A. Functional Analysis of Sox8 and Sox9 during Sex Determination in the Mouse. Development 2004, 131, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Stolt, C.C.; Lommes, P.; Sock, E.; Chaboissier, M.C.; Schedl, A.; Wegner, M. The Sox9 Transcription Factor Determines Glial Fate Choice in the Developing Spinal Cord. Genes Dev. 2003, 17, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Huang, S.; Reidy, K.; Han, S.H.; Chinga, F.; Susztak, K. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep. 2016, 14, 861–871. [Google Scholar] [CrossRef]
- Kumar, S.; Liu, J.; Pang, P.; Krautzberger, A.M.; Reginensi, A.; Akiyama, H.; Schedl, A.; Humphreys, B.D.; McMahon, A.P. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep. 2015, 12, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Potential Use of Sox9 Gene Therapy for Intervertebral Degenerative Disc Disease—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/12698117/ (accessed on 25 August 2022).
- Gajjala, P.R.; Kasam, R.K.; Soundararajan, D.; Sinner, D.; Huang, S.K.; Jegga, A.G.; Madala, S.K. Dysregulated Overexpression of Sox9 Induces Fibroblast Activation in Pulmonary Fibrosis. JCI Insight 2021, 6, e152503. [Google Scholar] [CrossRef]
- Kishi, S.; Abe, H.; Akiyama, H.; Tominaga, T.; Murakami, T.; Mima, A.; Nagai, K.; Kishi, F.; Matsuura, M.; Matsubara, T.; et al. SOX9 Protein Induces a Chondrogenic Phenotype of Mesangial Cells and Contributes to Advanced Diabetic Nephropathy. J. Biol. Chem. 2011, 286, 32162–32169. [Google Scholar] [CrossRef]
- Wang, Y.F.; Dang, H.F.; Luo, X.; Wang, Q.Q.; Gao, C.; Tian, Y.X. Downregulation of SOX9 Suppresses Breast Cancer Cell Proliferation and Migration by Regulating Apoptosis and Cell Cycle Arrest. Oncol. Lett. 2021, 22, 517. [Google Scholar] [CrossRef]
- Wang, X.; Ju, Y.; Zhou, M.; Liu, X.; Zhou, C. Upregulation of SOX9 Promotes Cell Proliferation, Migration and Invasion in Lung Adenocarcinoma. Oncol. Lett. 2015, 10, 990–994. [Google Scholar] [CrossRef]
- Zhou, T.; Wu, L.; Ma, N.; Tang, F.; Yu, Z.; Jiang, Z.; Li, Y.; Zong, Z.; Hu, K. SOX9-Activated FARSA-AS1 Predetermines Cell Growth, Stemness, and Metastasis in Colorectal Cancer through Upregulating FARSA and SOX9. Cell Death Dis. 2020, 11, 1071. [Google Scholar] [CrossRef]
- Athwal, V.S.; Pritchett, J.; Martin, K.; Llewellyn, J.; Scott, J.; Harvey, E.; Zaitoun, A.M.; Mullan, A.F.; Zeef, L.A.H.; Friedman, S.L.; et al. SOX9 Regulated Matrix Proteins Are Increased in Patients Serum and Correlate with Severity of Liver Fibrosis. Sci. Rep. 2018, 8, 17905. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, J.; Harvey, E.; Athwal, V.; Berry, A.; Rowe, C.; Oakley, F.; Moles, A.; Mann, D.A.; Bobola, N.; Sharrocks, A.D.; et al. Osteopontin Is a Novel Downstream Target of SOX9 with Diagnostic Implications for Progression of Liver Fibrosis in Humans. Hepatology 2012, 56, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Qing, M.A.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of Neutrophil Gelatinase-Associated Lipocalin as a Novel Early Urinary Biomarker for Ischemic Renal Injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Bollée, G.; Flamant, M.; Schordan, S.; Fligny, C.; Rumpel, E.; Milon, M.; Schordan, E.; Sabaa, N.; Vandermeersch, S.; Galaup, A.; et al. Epidermal Growth Factor Receptor Promotes Glomerular Injury and Renal Failure in Rapidly Progressive Crescentic Glomerulonephritis. Nat. Med. 2011, 17, 1242–1250. [Google Scholar] [CrossRef]
- Lazareth, H.; Henique, C.; Lenoir, O.; Puelles, V.G.; Flamant, M.; Bollée, G.; Fligny, C.; Camus, M.; Guyonnet, L.; Millien, C.; et al. The Tetraspanin CD9 Controls Migration and Proliferation of Parietal Epithelial Cells and Glomerular Disease Progression. Nat. Commun. 2019, 10, 3303. [Google Scholar] [CrossRef]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2017, 28, 504–519. [Google Scholar] [CrossRef]
- Tejedor-Santamaria, L.; Morgado-Pascual, J.L.; Marquez-Exposito, L.; Suarez-Alvarez, B.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marchant, V.; Mezzano, S.; Lopez-Larrea, C.; Sola, A.; et al. Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis. Pharmaceuticals 2022, 15, 121. [Google Scholar] [CrossRef]
- Zhao, J.H. Mesangial Cells and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 165–194. [Google Scholar] [CrossRef]
- Bennett, M.R.; Czech, K.A.; Arend, L.J.; Witte, D.P.; Devarajan, P.; Potter, S.S. Laser Capture Microdissection-Microarray Analysis of Focal Segmental Glomerulosclerosis Glomeruli. Nephron. Exp. Nephrol. 2007, 107, e30–e40. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse Roles of TGF-β/Smads in Renal Fibrosis and Inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef]
- Bedore, J.; Leask, A.; Séguin, C.A. Targeting the Extracellular Matrix: Matricellular Proteins Regulate Cell-Extracellular Matrix Communication within Distinct Niches of the Intervertebral Disc. Matrix Biol. 2014, 37, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Vizjak, A.; Ferluga, D. Spectrum of Collagen Type IV Nephropathies: From Thin Basement Membrane Nephropathy to Alport Syndrome. Srp. Arh. Celok. Lek. 2008, 136 (Suppl. S4), 323–326. [Google Scholar] [CrossRef] [PubMed]
- Alexakis, C.; Maxwell, P.; Bou-Gharios, G. Organ-Specific Collagen Expression: Implications for Renal Disease. Nephron. Exp. Nephrol. 2006, 102, e71–e75. [Google Scholar] [CrossRef] [PubMed]
- Glomerular Cells, Extracellular Matrix Accumulation, and the Development of Glomerulosclerosis in the Remnant Kidney Model—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/1583888/ (accessed on 18 September 2022).
- Shiiki, H.; Nishino, T.; Uyama, H.; Kimura, T.; Nishimoto, K.; Hashimoto, T.; Fujii, Y.; Dohi, K. Alterations in Extracellular Matrix Components and Integrins in Patients with Preeclamptic Nephropathy. Virchows Arch. 1996, 427, 567–573. [Google Scholar] [CrossRef]
- Collagen Distribution in Focal and Segmental Glomerulosclerosis: An Immunofluorescence and Ultrastructural Immunogold Study—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/8758212/ (accessed on 18 September 2022).
- Zhou, B.; Mu, J.; Gong, Y.; Lu, C.; Zhao, Y.; He, T.; Qin, Z. Brd4 Inhibition Attenuates Unilateral Ureteral Obstruction-Induced Fibrosis by Blocking TGF-β-Mediated Nox4 Expression. Redox Biol. 2017, 11, 390–402. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Lyv, X.; Jennifer Zhou, Q.; Xiang, Z.; Stanford, D.; Bodduluri, S.; Rowe, S.M.; Thannickal, V.J. Brd4-P300 Inhibition Downregulates Nox4 and Accelerates Lung Fibrosis Resolution in Aged Mice. JCI Insight 2020, 5, e137127. [Google Scholar] [CrossRef]
- Kumar, K.; DeCant, B.T.; Grippo, P.J.; Hwang, R.F.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET Inhibitors Block Pancreatic Stellate Cell Collagen I Production and Attenuate Fibrosis in Vivo. JCI Insight 2017, 2, e88032. [Google Scholar] [CrossRef]
- Xiong, C.; Deng, J.; Wang, X.; Shao, X.; Zhou, Q.; Zou, H.; Zhuang, S. Pharmacologic Targeting of BET Proteins Attenuates Hyperuricemic Nephropathy in Rats. Front. Pharmacol. 2021, 12, 636154. [Google Scholar] [CrossRef]
- Ortiz, A.; Ucero, A.C.; Egido, J. Unravelling Fibrosis: Two Newcomers and an Old Foe. Nephrol. Dial. Transpl. 2010, 25, 3492–3495. [Google Scholar] [CrossRef][Green Version]
- Mele, D.A.; Salmeron, A.; Ghosh, S.; Huang, H.R.; Bryant, B.M.; Lora, J.M. BET Bromodomain Inhibition Suppresses TH17-Mediated Pathology. J. Exp. Med. 2013, 210, 2181–2190. [Google Scholar] [CrossRef]
- Bandukwala, H.S.; Gagnon, J.; Togher, S.; Greenbaum, J.A.; Lamperti, E.D.; Parr, N.J.; Molesworth, A.M.H.; Smithers, N.; Lee, K.; Witherington, J.; et al. Selective Inhibition of CD4+ T-Cell Cytokine Production and Autoimmunity by BET Protein and c-Myc Inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 14532–14537. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET Bromodomain Inhibition Suppresses Innate Inflammatory and Profibrotic Transcriptional Networks in Heart Failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef] [PubMed]
- Shakya, R.; Watanabe, T.; Costantini, F. The Role of GDNF/Ret Signaling in Ureteric Bud Cell Fate and Branching Morphogenesis. Dev. Cell 2005, 8, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Reginensi, A.; Clarkson, M.; Neirijnck, Y.; Lu, B.; Ohyama, T.; Groves, A.K.; Sock, E.; Wegner, M.; Costantini, F.; Chaboissier, M.C.; et al. SOX9 Controls Epithelial Branching by Activating RET Effector Genes during Kidney Development. Hum. Mol. Genet. 2011, 20, 1143–1153. [Google Scholar] [CrossRef]
- Ling, S.; Chang, X.; Schultz, L.; Lee, T.K.; Chaux, A.; Marchionni, L.; Netto, G.J.; Sidransky, D.; Berman, D.M. An EGFR-ERK-SOX9 Signaling Cascade Links Urothelial Development and Regeneration to Cancer. Cancer Res. 2011, 71, 3812–3821. [Google Scholar] [CrossRef]
- Lacraz, G.P.A.; Junker, J.P.; Gladka, M.M.; Molenaar, B.; Scholman, K.T.; Vigil-Garcia, M.; Versteeg, D.; De Ruiter, H.; Vermunt, M.W.; Creyghton, M.P.; et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis During Ischemic Injury. Circulation 2017, 136, 1396–1409. [Google Scholar] [CrossRef]
- Möbius, P.; Preidl, R.H.M.; Weber, M.; Amann, K.; Neukam, F.W.; Wehrhan, F. Re-Expression of pro-Fibrotic, Embryonic Preserved Mediators in Irradiated Arterial Vessels of the Head and Neck Region. Strahlenther. Onkol. 2017, 193, 951–960. [Google Scholar] [CrossRef]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.H.W.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq Identifies Novel Myocardial Gene Expression Signatures of Heart Failure. Genomics 2015, 105, 83–89. [Google Scholar] [CrossRef]
- Matsushita, K.; Toyoda, T.; Yamada, T.; Morikawa, T.; Ogawa, K. Specific Expression of Survivin, SOX9, and CD44 in Renal Tubules in Adaptive and Maladaptive Repair Processes after Acute Kidney Injury in Rats. J. Appl. Toxicol. 2021, 41, 607–617. [Google Scholar] [CrossRef]
- ZHANG, Z.; WU, W.; FANG, X.; LU, M.; WU, H.; GAO, C.; XIA, Z. Sox9 Promotes Renal Tubular Epithelial-mesenchymal Transition and Extracellular Matrix Aggregation via the PI3K/AKT Signaling Pathway. Mol. Med. Rep. 2020, 22, 4017–4030. [Google Scholar] [CrossRef]
- Sumi, E.; Iehara, N.; Akiyama, H.; Matsubara, T.; Mima, A.; Kanamori, H.; Fukatsu, A.; Salant, D.J.; Kita, T.; Arai, H.; et al. SRY-Related HMG Box 9 Regulates the Expression of Col4a2 through Transactivating Its Enhancer Element in Mesangial Cells. Am. J. Pathol. 2007, 170, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; You, J.S. SOX9 Is Controlled by the BRD4 Inhibitor JQ1 via Multiple Regulation Mechanisms. Biochem. Biophys. Res. Commun. 2019, 511, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Mu, W.; Tomita, N.; Huang, X.R.; Li, J.H.; Zhu, H.J.; Morishita, R.; Johnson, R.J. Inhibition of Renal Fibrosis by Gene Transfer of Inducible Smad7 Using Ultrasound-Microbubble System in Rat UUO Model. J. Am. Soc. Nephrol. 2003, 14, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-Beta Signaling in Vascular Fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.C.; Wang, W.; Huang, X.R.; Fu, P.; Chen, T.H.; Sheikh-Hamad, D.; Lan, H.Y. Ultrasound-Microbubble-Mediated Gene Transfer of Inducible Smad7 Blocks Transforming Growth Factor-Beta Signaling and Fibrosis in Rat Remnant Kidney. Am. J. Pathol. 2005, 166, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Zu, S.J.; Liu, Y.J.; Li, J.C.; Dang, W.Z.; Liao, L.P.; Liu, L.P.; Chen, P.Y.; Huang, H.M.; Wu, K.H.; et al. Selective Bromodomain and Extra-Terminal Bromodomain Inhibitor Inactivates Macrophages and Hepatic Stellate Cells to Inhibit Liver Inflammation and Fibrosis. Bioengineered 2022, 13, 10914–10930. [Google Scholar] [CrossRef]
- Song, S.; Liu, L.; Yu, Y.; Zhang, R.; Li, Y.; Cao, W.; Xiao, Y.; Fang, G.; Li, Z.; Wang, X.; et al. Inhibition of BRD4 Attenuates Transverse Aortic Constriction- and TGF-β-Induced Endothelial-Mesenchymal Transition and Cardiac Fibrosis. J. Mol. Cell. Cardiol. 2019, 127, 83–96. [Google Scholar] [CrossRef]
- Simonsson, M.; Kanduri, M.; Grönroos, E.; Heldin, C.H.; Ericsson, J. The DNA Binding Activities of Smad2 and Smad3 Are Regulated by Coactivator-Mediated Acetylation. J. Biol. Chem. 2006, 281, 39870–39880. [Google Scholar] [CrossRef]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads Orchestrate Specific Histone Modifications and Chromatin Remodeling to Activate Transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef]
- Lee, D.U.; Katavolos, P.; Palanisamy, G.; Katewa, A.; Sioson, C.; Corpuz, J.; Pang, J.; DeMent, K.; Choo, E.; Ghilardi, N.; et al. Nonselective Inhibition of the Epigenetic Transcriptional Regulator BET Induces Marked Lymphoid and Hematopoietic Toxicity in Mice. Toxicol. Appl. Pharmacol. 2016, 300, 47–54. [Google Scholar] [CrossRef]
- Ucero, A.C.; Benito-Martin, A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Sanz, A.B.; Ramos, A.M.; Berzal, S.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Unilateral Ureteral Obstruction: Beyond Obstruction. Int. Urol. Nephrol. 2014, 46, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Díez, R.; Rodrigues-Díez, R.R.; Rayego-Mateos, S.; Suarez-Alvarez, B.; Lavoz, C.; Stark Aroeira, L.; Sánchez-López, E.; Orejudo, M.; Alique, M.; Lopez-Larrea, C.; et al. The C-Terminal Module IV of Connective Tissue Growth Factor Is a Novel Immune Modulator of the Th17 Response. Lab. Investig. 2013, 93, 812–824. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgado-Pascual, J.L.; Suarez-Alvarez, B.; Marchant, V.; Basantes, P.; Tharaux, P.-L.; Ortiz, A.; Lopez-Larrea, C.; Ruiz-Ortega, M.; Rayego-Mateos, S. Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis. Int. J. Mol. Sci. 2023, 24, 486. https://doi.org/10.3390/ijms24010486
Morgado-Pascual JL, Suarez-Alvarez B, Marchant V, Basantes P, Tharaux P-L, Ortiz A, Lopez-Larrea C, Ruiz-Ortega M, Rayego-Mateos S. Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis. International Journal of Molecular Sciences. 2023; 24(1):486. https://doi.org/10.3390/ijms24010486
Chicago/Turabian StyleMorgado-Pascual, José Luis, Beatriz Suarez-Alvarez, Vanessa Marchant, Pamela Basantes, Pierre-Louis Tharaux, Alberto Ortiz, Carlos Lopez-Larrea, Marta Ruiz-Ortega, and Sandra Rayego-Mateos. 2023. "Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis" International Journal of Molecular Sciences 24, no. 1: 486. https://doi.org/10.3390/ijms24010486
APA StyleMorgado-Pascual, J. L., Suarez-Alvarez, B., Marchant, V., Basantes, P., Tharaux, P.-L., Ortiz, A., Lopez-Larrea, C., Ruiz-Ortega, M., & Rayego-Mateos, S. (2023). Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis. International Journal of Molecular Sciences, 24(1), 486. https://doi.org/10.3390/ijms24010486