

Roles of NAD+ in Acute and Chronic Kidney Diseases
 ,
,
Abstract
1. Introduction
2. The Role of NAD+ and NADH Redox Cycling in Mitochondria
3. NAD+ Homeostasis
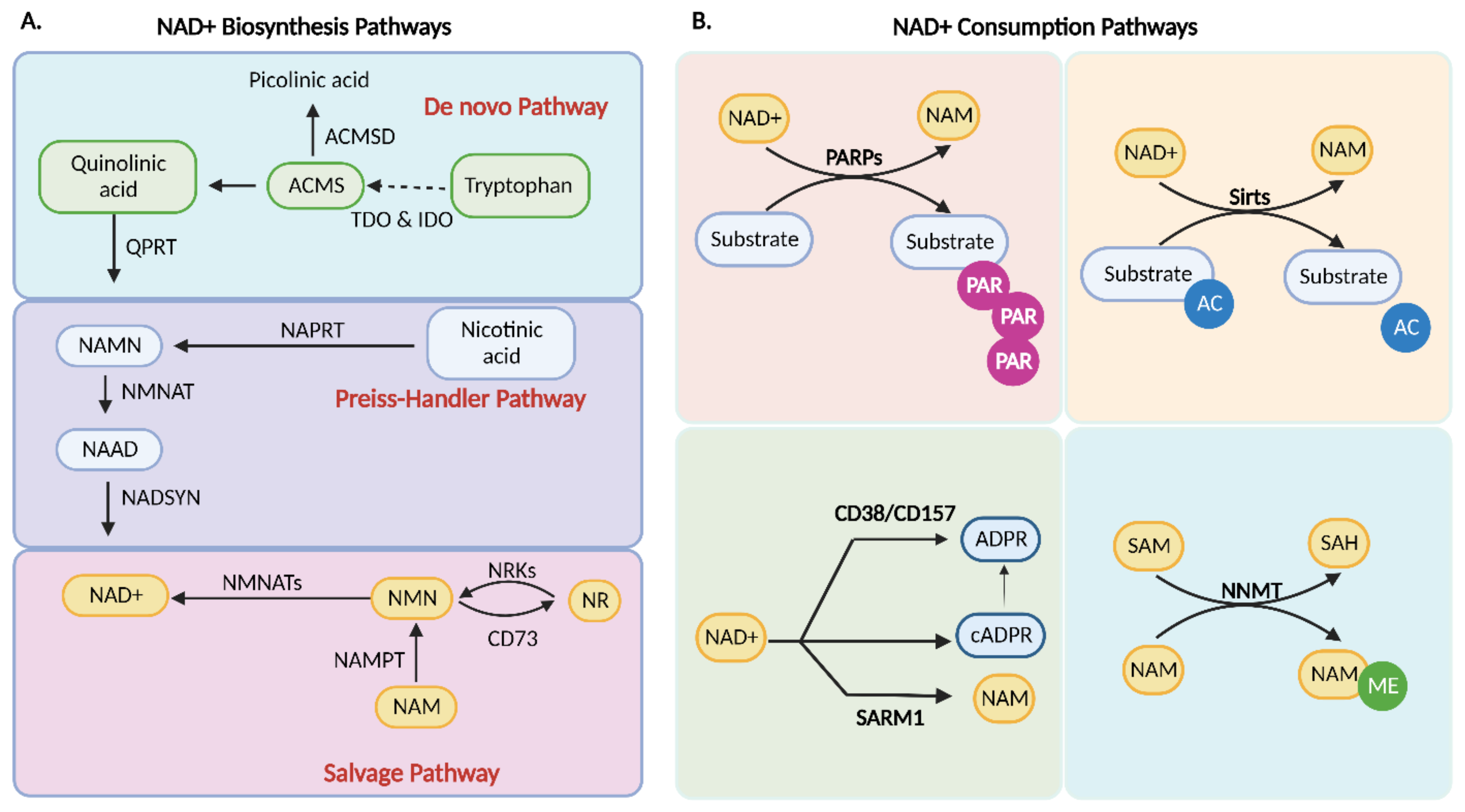
4. NAD+ Biosynthesis
4.1. De Novo Biosynthesis
4.2. Preiss–Handler Pathway
4.3. Salvage Pathway
5. Consumption of NAD+
5.1. PARP1
5.2. cADPR Synthetases
5.3. Sirtuins
5.4. Nicotinamide N-methyltransferase (NNMT)
6. Kidney Function and NAD+
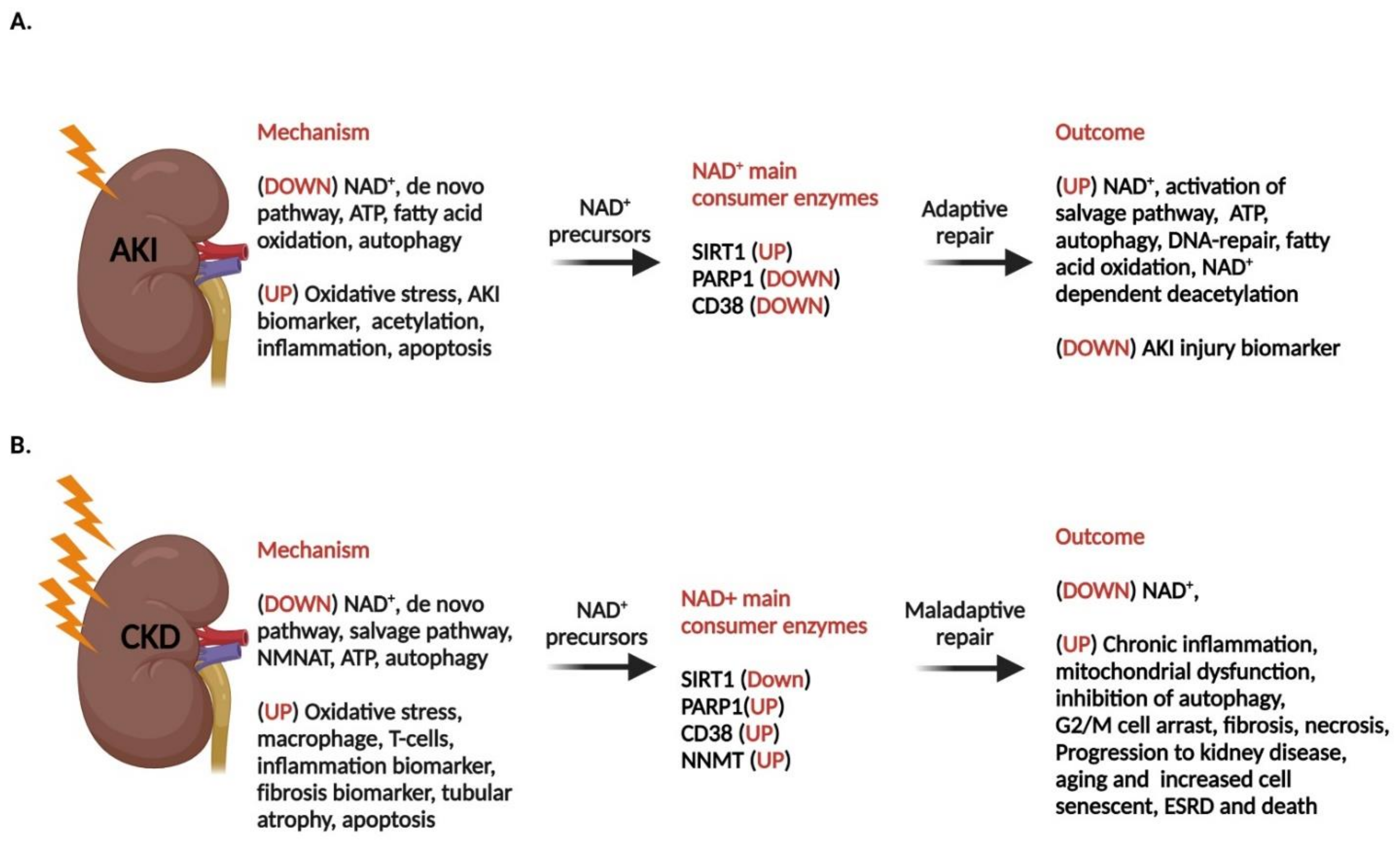
7. Acute Kidney Disease and NAD+ Level
7.1. Compromised NAD+ Homeostasis in AKI
7.2. Disrupted FAO and Level of NAD+ in AKI
7.3. Induced PARP1 in AKI and the Level of NAD+
7.4. Elevated Expression of CD38 in AKI
7.5. Decreased Level of Sirt1 in AKI
7.6. Low NAD+ and Compromised Autophagy in AKI
7.7. Klotho
8. Chronic Kidney Disease
8.1. Fibrosis in CKD
8.2. NAD+ and Fibrosis
8.3. Cellular Senescence in Kidney Dysfunction
9. Cellular Senescence in the Kidney and the Effect of NAD+
10. NAD+ as a Potential Pharmacological Option for AKI and CKD
11. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harden, A.; Young, W.J. The alcoholic ferment of yeast-juice. Part II.—The coferment of yeast-juice. Proc. R. Soc. London Ser. B Contain. Pap. A Biol. Character 1906, 78, 369–375. [Google Scholar]
- Warburg, O.; Christian, W. Pyridin, the hydrogen-transferring component of the fermentation enzymes (pyridine nucleotide). Biochem. Z 1936, 287, 1. [Google Scholar]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD(+) in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [CrossRef]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 26–33. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007, 129, 473–484. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Sauve, A.A. Vitamin B3, the nicotinamide adenine dinucleotides and aging. Mech. Ageing Dev. 2010, 131, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Hyndman, K.A.; Griffin, M.D. Could NAD(+) Precursor Supplements Induce a Legacy of Protection against Diabetic Nephropathy? J. Am. Soc. Nephrol. 2021, 32, 1270–1273. [Google Scholar] [CrossRef]
- Abebe, A.; Kumela, K.; Belay, M.; Kebede, B.; Wobie, Y. Mortality and predictors of acute kidney injury in adults: A hospital-based prospective observational study. Sci. Rep. 2021, 11, 15672. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L.; Acute Kidney Injury Advisory Group of the American Society of, N. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef]
- Ali, T.; Khan, I.; Simpson, W.; Prescott, G.; Townend, J.; Smith, W.; Macleod, A. Incidence and outcomes in acute kidney injury: A comprehensive population-based study. J. Am. Soc. Nephrol. 2007, 18, 1292–1298. [Google Scholar] [CrossRef]
- Murugan, R.; Kellum, J.A. Acute kidney injury: What's the prognosis? Nat. Rev. Nephrol. 2011, 7, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.L.; Daniels, F.; Star, R.A.; Kimmel, P.L.; Eggers, P.W.; Molitoris, B.A.; Himmelfarb, J.; Collins, A.J. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J. Am. Soc. Nephrol. 2006, 17, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Kane-Gill, S.L.; Sileanu, F.E.; Murugan, R.; Trietley, G.S.; Handler, S.M.; Kellum, J.A. Risk factors for acute kidney injury in older adults with critical illness: A retrospective cohort study. Am. J. Kidney. Dis. 2015, 65, 860–869. [Google Scholar] [CrossRef]
- Fuhrman, D.Y.; Kane-Gill, S.; Goldstein, S.L.; Priyanka, P.; Kellum, J.A. Acute kidney injury epidemiology, risk factors, and outcomes in critically ill patients 16-25 years of age treated in an adult intensive care unit. Ann. Intensive Care 2018, 8, 26. [Google Scholar] [CrossRef]
- Carlson, N.; Hommel, K.; Olesen, J.B.; Soja, A.M.; Vilsboll, T.; Kamper, A.L.; Torp-Pedersen, C.; Gislason, G. Trends in One-Year Outcomes of Dialysis-Requiring Acute Kidney Injury in Denmark 2005-2012: A Population-Based Nationwide Study. PLoS ONE 2016, 11, e0159944. [Google Scholar] [CrossRef]
- Zheng, M.; Cai, J.; Liu, Z.; Shu, S.; Wang, Y.; Tang, C.; Dong, Z. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell Mol. Med. 2019, 23, 3995–4004. [Google Scholar] [CrossRef]
- Ix, J.H.; Isakova, T.; Larive, B.; Raphael, K.L.; Raj, D.S.; Cheung, A.K.; Sprague, S.M.; Fried, L.F.; Gassman, J.J.; Middleton, J.P.; et al. Effects of Nicotinamide and Lanthanum Carbonate on Serum Phosphate and Fibroblast Growth Factor-23 in CKD: The COMBINE Trial. J. Am. Soc. Nephrol. 2019, 30, 1096–1108. [Google Scholar] [CrossRef]
- Kumakura, S.; Sato, E.; Sekimoto, A.; Hashizume, Y.; Yamakage, S.; Miyazaki, M.; Ito, S.; Harigae, H.; Takahashi, N. Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins 2021, 13, 50. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Igarashi, M.; Guarente, L. mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Ragland, T.E.; Hackett, D.P. Compartmentation of Nicotinamide Dinucleotide Dehydrogenases and Transhydrogenases in Nonphotosynthetic Plant Tissues. Arch. Biochem. Biophys. 1964, 108, 479–489. [Google Scholar] [CrossRef] [PubMed]
- White, A.T.; Schenk, S. NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E308–E321. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Hillyard, D.; Olivera, B.M. Turnover at nicotinamide adenine dinucleotide in cultures of human cells. J. Cell Physiol. 1976, 88, 207–217. [Google Scholar] [CrossRef]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD(+). Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef]
- Williams, G.T.; Lau, K.M.; Coote, J.M.; Johnstone, A.P. NAD metabolism and mitogen stimulation of human lymphocytes. Exp. Cell Res. 1985, 160, 419–426. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Youn, H.S.; Kim, T.G.; Kim, M.K.; Kang, G.B.; Kang, J.Y.; Lee, J.G.; An, J.Y.; Park, K.R.; Lee, Y.; Im, Y.J.; et al. Structural Insights into the Quaternary Catalytic Mechanism of Hexameric Human Quinolinate Phosphoribosyltransferase, a Key Enzyme in de novo NAD Biosynthesis. Sci. Rep. 2016, 6, 19681. [Google Scholar] [CrossRef]
- Li, S.; Ding, H.; Deng, Y.; Zhang, J. Knockdown of Quinolinate Phosphoribosyltransferase Results in Decreased Salicylic Acid-Mediated Pathogen Resistance in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Bignon, Y.; Rinaldi, A.; Nadour, Z.; Poindessous, V.; Nemazanyy, I.; Lenoir, O.; Fohlen, B.; Weill-Raynal, P.; Hertig, A.; Karras, A.; et al. Cell stress response impairs de novo NAD+ biosynthesis in the kidney. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Vela Parada, X.F.; Parikh, S.M.; Dellinger, R.; Guarente, L.P.; Rhee, E.P. Nicotinamide riboside with pterostilbene (NRPT) increases NAD(+) in patients with acute kidney injury (AKI): A randomized, double-blind, placebo-controlled, stepwise safety study of escalating doses of NRPT in patients with AKI. BMC Nephrol. 2020, 21, 342. [Google Scholar] [CrossRef] [PubMed]
- Marletta, A.S.; Massarotti, A.; Orsomando, G.; Magni, G.; Rizzi, M.; Garavaglia, S. Crystal structure of human nicotinic acid phosphoribosyltransferase. FEBS Open Bio. 2015, 5, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef]
- Rizzi, M.; Bolognesi, M.; Coda, A. A novel deamido-NAD+-binding site revealed by the trapped NAD-adenylate intermediate in the NAD+ synthetase structure. Structure 1998, 6, 1129–1140. [Google Scholar] [CrossRef][Green Version]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bheda, P.; Revollo, J.R.; Imai, S.; Wolberger, C. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 2006, 13, 661–662. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef]
- Zhou, T.; Kurnasov, O.; Tomchick, D.R.; Binns, D.D.; Grishin, N.V.; Marquez, V.E.; Osterman, A.L.; Zhang, H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J. Biol. Chem. 2002, 277, 13148–13154. [Google Scholar] [CrossRef]
- Werner, E.; Ziegler, M.; Lerner, F.; Schweiger, M.; Heinemann, U. Crystal structure of human nicotinamide mononucleotide adenylyltransferase in complex with NMN. FEBS Lett. 2002, 516, 239–244. [Google Scholar] [CrossRef]
- Liu, X.; Luo, D.; Huang, S.; Liu, S.; Zhang, B.; Wang, F.; Lu, J.; Chen, J.; Li, S. Impaired Nicotinamide Adenine Dinucleotide Biosynthesis in the Kidney of Chronic Kidney Disease. Front. Physiol. 2021, 12, 723690. [Google Scholar] [CrossRef]
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years and Counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef]
- Leung, A.K.L. PARPs. Curr. Biol. 2017, 27, R1256–R1258. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Lee, J.H.; Hussain, M.; Kim, E.W.; Cheng, S.J.; Leung, A.K.L.; Fakouri, N.B.; Croteau, D.L.; Bohr, V.A. Mitochondrial PARP1 regulates NAD(+)-dependent poly ADP-ribosylation of mitochondrial nucleoids. Exp. Mol. Med. 2022, 10.1038/s12276-022-00894-x. [Google Scholar] [CrossRef]
- Benjamin, R.C.; Gill, D.M. ADP-ribosylation in mammalian cell ghosts. Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J. Biol. Chem. 1980, 255, 10493–10501. [Google Scholar] [CrossRef]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef]
- van Beek, L.; McClay, E.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia de Oliveira, T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Brunyanszki, A.; Huber, A.; Szanto, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef]
- Wei, W.; Graeff, R.; Yue, J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca(2+) signaling pathway. World J. Biol. Chem. 2014, 5, 58–67. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Wang, W.; Korman, B.; Kai, L.; Wang, Q.; Wei, J.; Bale, S.; Marangoni, R.G.; Bhattacharyya, S.; Miller, S.; et al. Targeting CD38-dependent NAD(+) metabolism to mitigate multiple organ fibrosis. iScience 2021, 24, 101902. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e1335. [Google Scholar] [CrossRef]
- Mink, M.; Fogelgren, B.; Olszewski, K.; Maroy, P.; Csiszar, K. A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/beta-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics 2001, 74, 234–244. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Landry, J.; Sutton, A.; Tafrov, S.T.; Heller, R.C.; Stebbins, J.; Pillus, L.; Sternglanz, R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 5807–5811. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Vilwanathan, R. Silencing Sirtuin 6 induces cell cycle arrest and apoptosis in non-small cell lung cancer cell lines. Genomics 2020, 112, 3703–3712. [Google Scholar] [CrossRef]
- Kim, M.J.; Kang, Y.J.; Sung, B.; Jang, J.Y.; Ahn, Y.R.; Oh, H.J.; Choi, H.; Choi, I.; Im, E.; Moon, H.R.; et al. Novel SIRT Inhibitor, MHY2256, Induces Cell Cycle Arrest, Apoptosis, and Autophagic Cell Death in HCT116 Human Colorectal Cancer Cells. Biomol. Ther. (Seoul) 2020, 28, 561–568. [Google Scholar] [CrossRef]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef]
- Boily, G.; Seifert, E.L.; Bevilacqua, L.; He, X.H.; Sabourin, G.; Estey, C.; Moffat, C.; Crawford, S.; Saliba, S.; Jardine, K.; et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE 2008, 3, e1759. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Alam, F.; Syed, H.; Amjad, S.; Baig, M.; Khan, T.A.; Rehman, R. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr. Res. Physiol. 2021, 4, 119–124. [Google Scholar] [CrossRef]
- Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide riboside, an NAD(+) precursor, attenuates inflammation and oxidative stress by activating sirtuin 1 in alcohol-stimulated macrophages. Lab. Invest. 2021, 101, 1225–1237. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar] [CrossRef]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef] [PubMed]
- Vaitiekunaite, R.; Butkiewicz, D.; Krzesniak, M.; Przybylek, M.; Gryc, A.; Snietura, M.; Benedyk, M.; Harris, C.C.; Rusin, M. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech. Ageing Dev. 2007, 128, 650–661. [Google Scholar] [CrossRef]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1alpha and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Shen, H.; Holliday, M.; Sheikh-Hamad, D.; Li, Q.; Tong, Q.; Hamad, C.D.; Pan, J.S. Sirtuin-3 mediates sex differences in kidney ischemia-reperfusion injury. Transl. Res. 2021, 235, 15–31. [Google Scholar] [CrossRef]
- Raji-Amirhasani, A.; Khaksari, M.; Darvishzadeh Mahani, F.; Hajializadeh, Z. Activators of SIRT1 in the kidney and protective effects of SIRT1 during acute kidney injury (AKI) (effect of SIRT1 activators on acute kidney injury). Clin. Exp. Nephrol. 2021, 25, 807–821. [Google Scholar] [CrossRef]
- Li, W.; Yang, Y.; Li, Y.; Zhao, Y.; Jiang, H. Sirt5 Attenuates Cisplatin-Induced Acute Kidney Injury through Regulation of Nrf2/HO-1 and Bcl-2. Biomed. Res. Int. 2019, 2019, 4745132. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Navarro, A.; Martinez-Rojas, M.A.; Albarran-Godinez, A.; Perez-Villalva, R.; Auwerx, J.; de la Cruz, A.; Noriega, L.G.; Rosetti, F.; Bobadilla, N.A. Sirtuin 7 Deficiency Reduces Inflammation and Tubular Damage Induced by an Episode of Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 2573. [Google Scholar] [CrossRef]
- Miyasato, Y.; Yoshizawa, T.; Sato, Y.; Nakagawa, T.; Miyasato, Y.; Kakizoe, Y.; Kuwabara, T.; Adachi, M.; Ianni, A.; Braun, T.; et al. Sirtuin 7 Deficiency Ameliorates Cisplatin-induced Acute Kidney Injury Through Regulation of the Inflammatory Response. Sci. Rep. 2018, 8, 5927. [Google Scholar] [CrossRef]
- Kitada, M.; Kume, S.; Koya, D. Role of sirtuins in kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e1065. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef] [PubMed]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD(+) metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. Suppl. 2003, 10.1046/j.1523-1755.63.s84.36.x. S19–S21. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-methyl-2-pyridone-5-carboxamide (2PY)-Major Metabolite of Nicotinamide: An Update on an Old Uremic Toxin. Toxins 2016, 8, 339. [Google Scholar] [CrossRef]
- Slominska, E.M.; Rutkowski, P.; Smolenski, R.T.; Szutowicz, A.; Rutkowski, B.; Swierczynski, J. The age-related increase in N-methyl-2-pyridone-5-carboxamide (NAD catabolite) in human plasma. Mol. Cell Biochem. 2004, 267, 25–30. [Google Scholar] [CrossRef]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 2014, 9, e113939. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef]
- Tian, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 963. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Weidemann, M.J.; Krebs, H.A. The fuel of respiration of rat kidney cortex. Biochem. J. 1969, 112, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox. Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Estrada, J.C.; Torres, Y.; Benguria, A.; Dopazo, A.; Roche, E.; Carrera-Quintanar, L.; Perez, R.A.; Enriquez, J.A.; Torres, R.; Ramirez, J.C.; et al. Human mesenchymal stem cell-replicative senescence and oxidative stress are closely linked to aneuploidy. Cell Death Dis. 2013, 4, e691. [Google Scholar] [CrossRef] [PubMed]
- Harbo, M.; Koelvraa, S.; Serakinci, N.; Bendix, L. Telomere dynamics in human mesenchymal stem cells after exposure to acute oxidative stress. DNA Repair. 2012, 11, 774–779. [Google Scholar] [CrossRef]
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 377, 544–552. [Google Scholar] [CrossRef]
- Morevati, M.; Egstrand, S.; Nordholm, A.; Mace, M.L.; Andersen, C.B.; Salmani, R.; Olgaard, K.; Lewin, E. Effect of NAD+ boosting on kidney ischemia-reperfusion injury. PLoS ONE 2021, 16, e0252554. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef]
- Bellomo, R.; See, E.J. Novel renal biomarkers of acute kidney injury and their implications. Intern. Med. J. 2021, 51, 316–318. [Google Scholar] [CrossRef]
- Manjunath, G.; Sarnak, M.J.; Levey, A.S. Prediction equations to estimate glomerular filtration rate: An update. Curr. Opin. Nephrol. Hypertens. 2001, 10, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Bellomo, R.; Goldsmith, D.; Bates, S.; Ronco, C. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit. Care Med. 2006, 34, 1913–1917. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, M.; Chang, R. The RIFLE criteria: Are the foundations robust? Crit. Care Med. 2007, 35, 2669–2670. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.K.; Wong, H.R.; Krawczeski, C.D.; Wheeler, D.S.; Manning, P.B.; Chawla, L.S.; Devarajan, P.; Goldstein, S.L. Combining functional and tubular damage biomarkers improves diagnostic precision for acute kidney injury after cardiac surgery. J. Am. Coll. Cardiol. 2014, 64, 2753–2762. [Google Scholar] [CrossRef]
- Katagiri, D.; Doi, K.; Matsubara, T.; Negishi, K.; Hamasaki, Y.; Nakamura, K.; Ishii, T.; Yahagi, N.; Noiri, E. New biomarker panel of plasma neutrophil gelatinase-associated lipocalin and endotoxin activity assay for detecting sepsis in acute kidney injury. J. Crit. Care 2013, 28, 564–570. [Google Scholar] [CrossRef]
- Katagiri, D.; Doi, K.; Honda, K.; Negishi, K.; Fujita, T.; Hisagi, M.; Ono, M.; Matsubara, T.; Yahagi, N.; Iwagami, M.; et al. Combination of two urinary biomarkers predicts acute kidney injury after adult cardiac surgery. Ann. Thorac. Surg. 2012, 93, 577–583. [Google Scholar] [CrossRef]
- Zhou, D.; Li, Y.; Lin, L.; Zhou, L.; Igarashi, P.; Liu, Y. Tubule-specific ablation of endogenous beta-catenin aggravates acute kidney injury in mice. Kidney Int. 2012, 82, 537–547. [Google Scholar] [CrossRef]
- El-Khoury, J.M.; Hoenig, M.P.; Jones, G.R.D.; Lamb, E.J.; Parikh, C.R.; Tolan, N.V.; Wilson, F.P. AACC Guidance Document on Laboratory Investigation of Acute Kidney Injury. J. Appl. Lab. Med. 2021, 6, 1316–1337. [Google Scholar] [CrossRef]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A.; Acute Kidney Injury, N. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Zhang, J.; Cui, L.Y.; Yang, S. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp. Biol. Med. (Maywood) 2015, 240, 1590–1598. [Google Scholar] [CrossRef]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2010, 333, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol. 2013, 273, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, Y.; Karamercan, M.A.; Ye, L.; Bhatti, T.; Becker, L.B.; Baur, J.A.; Sims, C.A. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock 2015, 44, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef]
- Johnson, A.C.; Stahl, A.; Zager, R.A. Triglyceride accumulation in injured renal tubular cells: Alterations in both synthetic and catabolic pathways. Kidney Int. 2005, 67, 2196–2209. [Google Scholar] [CrossRef] [PubMed]
- Portilla, D. Role of fatty acid beta-oxidation and calcium-independent phospholipase A2 in ischemic acute renal failure. Curr. Opin. Nephrol. Hypertens. 1999, 8, 473–477. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Li, Q.; Wei, S.; Zhou, Y.; Yu, W.; Xue, L.; Zhou, L.; Shen, L.; Lu, G.; et al. STAT6 contributes to renal fibrosis by modulating PPARalpha-mediated tubular fatty acid oxidation. Cell Death Dis. 2022, 13, 66. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. (Lausanne) 2015, 2, 52. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J.; Xin, W.; Chen, L.; Zhao, X.; Lv, Z.; Liu, Y.; Wan, Q. Lipid accumulation is ahead of epithelial-to-mesenchymal transition and therapeutic intervention by acetyl-CoA carboxylase 2 silence in diabetic nephropathy. Metabolism 2014, 63, 716–726. [Google Scholar] [CrossRef]
- Martin, D.R.; Lewington, A.J.; Hammerman, M.R.; Padanilam, B.J. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1834–R1840. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Liu, J.; Liu, D.W.; Wang, X.T.; Yang, R.L. Inhibition of Poly-(ADP-Ribose) Polymerase Protects the Kidney in a Canine Model of Endotoxic Shock. Nephron 2015, 130, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Devalaraja-Narashimha, K.; Singaravelu, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal. Physiol. 2005, 288, F387–F398. [Google Scholar] [CrossRef]
- Bruin, M.A.C.; Korse, C.M.; van Wijnen, B.; de Jong, V.M.T.; Linn, S.C.; van Triest, B.; Rosing, H.; Beijnen, J.H.; van den Broek, D.; Huitema, A.D.R. A real or apparent decrease in glomerular filtration rate in patients using olaparib? Eur. J. Clin. Pharmacol. 2021, 77, 179–188. [Google Scholar] [CrossRef]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. Recent Results Cancer Res. 2018, 211, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef]
- Pletcher, J.P.; Bhattacharjee, S.; Doan, J.P.; Wynn, R.; Sindhwani, P.; Nadiminty, N.; Petros, F.G. The Emerging Role of Poly (ADP-Ribose) Polymerase Inhibitors as Effective Therapeutic Agents in Renal Cell Carcinoma. Front. Oncol. 2021, 11, 681441. [Google Scholar] [CrossRef]
- Tarrago, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095.e1010. [Google Scholar] [CrossRef]
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kappaB signaling suppression. Cell Signal. 2018, 42, 249–258. [Google Scholar] [CrossRef]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O'Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef]
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef]
- Gao, P.; Xu, T.T.; Lu, J.; Li, L.; Xu, J.; Hao, D.L.; Chen, H.Z.; Liu, D.P. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. 2014, 92, 347–357. [Google Scholar] [CrossRef]
- Bordone, L.; Cohen, D.; Robinson, A.; Motta, M.C.; van Veen, E.; Czopik, A.; Steele, A.D.; Crowe, H.; Marmor, S.; Luo, J.; et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 2007, 6, 759–767. [Google Scholar] [CrossRef]
- Chen, D.; Steele, A.D.; Lindquist, S.; Guarente, L. Increase in activity during calorie restriction requires Sirt1. Science 2005, 310, 1641. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Yoshioka, K.; Tatematsu, S.; Hara, Y.; Minakuchi, H.; Sueyasu, K.; Washida, N.; Tokuyama, H.; Tzukerman, M.; et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J. Biol. Chem. 2010, 285, 13045–13056. [Google Scholar] [CrossRef]
- Wakino, S.; Hasegawa, K.; Itoh, H. Sirtuin and metabolic kidney disease. Kidney Int. 2015, 88, 691–698. [Google Scholar] [CrossRef]
- Fan, H.; Yang, H.C.; You, L.; Wang, Y.Y.; He, W.J.; Hao, C.M. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2013, 83, 404–413. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D'Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Ralto, K.M.; Zsengeller, Z.K.; Raman, V.; Bhasin, S.S.; Sun, N.; Chen, X.; Brown, D.; Rovira, I.I.; et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1alpha-dependent renal stress resistance. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef]
- Li, P.; Shi, M.; Maique, J.; Shaffer, J.; Yan, S.; Moe, O.W.; Hu, M.C. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am. J. Physiol. Renal. Physiol. 2020, 318, F772–F792. [Google Scholar] [CrossRef]
- Minami, S.; Nakamura, S. Therapeutic potential of Beclin1 for transition from AKI to CKD: Autophagy-dependent and autophagy-independent functions. Kidney Int. 2022, 101, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Colbert, C.L.; Becker, N.; Wei, Y.; Levine, B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008, 4, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Nordholm, A.; Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Morevati, M.; Olgaard, K.; Lewin, E. Klotho and activin A in kidney injury: Plasma Klotho is maintained in unilateral obstruction despite no upregulation of Klotho biosynthesis in the contralateral kidney. Am. J. Physiol. Renal. Physiol. 2018, 314, F753–F762. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef]
- Lim, K.; Groen, A.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.L.; et al. alpha-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Kuro-o, M.; Moe, O.W. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010, 78, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Oishi, H.; Doi, S.; Nakashima, A.; Ike, T.; Maeoka, Y.; Sasaki, K.; Doi, T.; Masaki, T. Klotho overexpression protects against renal aging along with suppression of transforming growth factor-beta1 signaling pathways. Am. J. Physiol. Renal. Physiol. 2021, 321, F799–F811. [Google Scholar] [CrossRef]
- Kawano, K.; Ogata, N.; Chiano, M.; Molloy, H.; Kleyn, P.; Spector, T.D.; Uchida, M.; Hosoi, T.; Suzuki, T.; Orimo, H.; et al. Klotho gene polymorphisms associated with bone density of aged postmenopausal women. J. Bone Miner Res. 2002, 17, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; Matsumura, Y.; Shiraki, M.; Kawano, K.; Koshizuka, Y.; Hosoi, T.; Nakamura, K.; Kuro, O.M.; Kawaguchi, H. Association of klotho gene polymorphism with bone density and spondylosis of the lumbar spine in postmenopausal women. Bone 2002, 31, 37–42. [Google Scholar] [CrossRef]
- Kuro, O.M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Sugiura, H.; Yoshida, T.; Mitobe, M.; Shiohira, S.; Nitta, K.; Tsuchiya, K. Recombinant human erythropoietin mitigates reductions in renal klotho expression. Am. J. Nephrol. 2010, 32, 137–144. [Google Scholar] [CrossRef]
- Shi, M.; Maique, J.; Shaffer, J.; Davidson, T.; Sebti, S.; Fernandez, A.F.; Zou, Z.; Yan, S.; Levine, B.; Moe, O.W.; et al. The tripartite interaction of phosphate, autophagy, and alphaKlotho in health maintenance. FASEB J. 2020, 34, 3129–3150. [Google Scholar] [CrossRef]
- Hsu, S.C.; Huang, S.M.; Chen, A.; Sun, C.Y.; Lin, S.H.; Chen, J.S.; Liu, S.T.; Hsu, Y.J. Resveratrol increases anti-aging Klotho gene expression via the activating transcription factor 3/c-Jun complex-mediated signaling pathway. Int. J. Biochem. Cell Biol. 2014, 53, 361–371. [Google Scholar] [CrossRef]
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154. [Google Scholar] [CrossRef]
- Leung, K.C.; Tonelli, M.; James, M.T. Chronic kidney disease following acute kidney injury-risk and outcomes. Nat. Rev. Nephrol. 2013, 9, 77–85. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Basile, D.; Liu, K.D.; McKay, D.; Molitoris, B.A.; Nath, K.A.; Nickolas, T.L.; Okusa, M.D.; Palevsky, P.M.; Schnellmann, R.; et al. AKI: A path forward. Clin. J. Am. Soc. Nephrol. 2013, 8, 1606–1608. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O'Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.J.; Fraser, S.D.S. The ascending rank of chronic kidney disease in the global burden of disease study. Nephrol. Dial. Transplant. 2017, 32, ii121–ii128. [Google Scholar] [CrossRef] [PubMed]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Collaboration, G.B.D.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Vaidya, S.R.; Aeddula, N.R. Chronic Renal Failure. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Korstanje, R.; Deutsch, K.; Bolanos-Palmieri, P.; Hanke, N.; Schroder, P.; Staggs, L.; Brasen, J.H.; Roberts, I.S.; Sheehan, S.; Savage, H.; et al. Loss of Kynurenine 3-Mono-oxygenase Causes Proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3271–3277. [Google Scholar] [CrossRef]
- Schefold, J.C.; Zeden, J.P.; Fotopoulou, C.; von Haehling, S.; Pschowski, R.; Hasper, D.; Volk, H.D.; Schuett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543, 531p following 143. [Google Scholar] [CrossRef]
- Bulow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Baues, M.; Klinkhammer, B.M.; Ehling, J.; Gremse, F.; van Zandvoort, M.; Reutelingsperger, C.P.M.; Daniel, C.; Amann, K.; Babickova, J.; Kiessling, F.; et al. A collagen-binding protein enables molecular imaging of kidney fibrosis in vivo. Kidney Int. 2020, 97, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Genovese, F.; Manresa, A.A.; Leeming, D.J.; Karsdal, M.A.; Boor, P. The extracellular matrix in the kidney: A source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair 2014, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Sparding, N.; Rasmussen, D.G.K.; Genovese, F.; Karsdal, M.A.; Hornum, M.; Feldt-Rasmussen, B.; Packington, R.; Selby, N.M. Circulating Levels of Endotrophin Are Prognostic for Long-Term Mortality after AKI. Kidney360 2022, 3, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Sparding, N.; Genovese, F.; Rasmussen, D.G.K.; Karsdal, M.A.; Neprasova, M.; Maixnerova, D.; Satrapova, V.; Frausova, D.; Hornum, M.; Bartonova, L.; et al. Endotrophin, a collagen type VI-derived matrikine, reflects the degree of renal fibrosis in patients with IgA nephropathy and in patients with ANCA-associated vasculitis. Nephrol. Dial. Transplant. 2022, 37, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Group, A.X.W. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef]
- Clements, M.E.; Chaber, C.J.; Ledbetter, S.R.; Zuk, A. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS ONE 2013, 8, e70464. [Google Scholar] [CrossRef]
- Wang, W.J.; Chen, X.M.; Cai, G.Y. Cellular senescence and the senescence-associated secretory phenotype: Potential therapeutic targets for renal fibrosis. Exp. Gerontol. 2021, 151, 111403. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, L.; Liu, Y. Cellular Senescence in Kidney Fibrosis: Pathologic Significance and Therapeutic Strategies. Front. Pharmacol. 2020, 11, 601325. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhou, S.; Zhou, Z.; Liu, Y.; Yang, L.; Liu, J.; Zhang, Y.; Li, H.; Liu, Y.; Hou, F.F.; et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2018, 29, 1238–1256. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Renal. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Bi, X.; Du, C.; Wang, X.; Wang, X.Y.; Han, W.; Wang, Y.; Qiao, Y.; Zhu, Y.; Ran, L.; Liu, Y.; et al. Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv. Sci. 2021, 8, 2002738. [Google Scholar] [CrossRef]
- Afsar, B.; Hornum, M.; Afsar, R.E.; Ertuglu, L.A.; Ortiz, A.; Covic, A.; van Raalte, D.H.; Cherney, D.Z.I.; Kanbay, M. Mitochondrion-driven nephroprotective mechanisms of novel glucose lowering medications. Mitochondrion 2021, 58, 72–82. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Jiang, H.; Pan, J.; Huang, X.R.; Wang, Y.C.; Huang, H.F.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y.; Chen, J.H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Faivre, A.; Katsyuba, E.; Verissimo, T.; Lindenmeyer, M.; Rajaram, R.D.; Naesens, M.; Heckenmeyer, C.; Mottis, A.; Feraille, E.; Cippa, P.; et al. Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhong, Y.; Li, X.; Chen, H.; Jim, B.; Zhou, M.M.; Chuang, P.Y.; He, J.C. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 2014, 63, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, H.; Hasegawa, K.; Sakamaki, Y.; Minakuchi, H.; Kawaguchi, T.; Yasuda, I.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Role of Nampt-Sirt6 Axis in Renal Proximal Tubules in Extracellular Matrix Deposition in Diabetic Nephropathy. Cell Rep. 2019, 27, 199–212.e195. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, I.; Hasegawa, K.; Sakamaki, Y.; Muraoka, H.; Kawaguchi, T.; Kusahana, E.; Ono, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; et al. Pre-emptive Short-term Nicotinamide Mononucleotide Treatment in a Mouse Model of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1355–1370. [Google Scholar] [CrossRef]
- Zhen, X.; Zhang, S.; Xie, F.; Zhou, M.; Hu, Z.; Zhu, F.; Nie, J. Nicotinamide Supplementation Attenuates Renal Interstitial Fibrosis via Boosting the Activity of Sirtuins. Kidney Dis. 2021, 7, 186–199. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work, G. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Takahashi, R.; Kanda, T.; Komatsu, M.; Itoh, T.; Minakuchi, H.; Urai, H.; Kuroita, T.; Shigaki, S.; Tsukamoto, T.; Higuchi, N.; et al. The significance of NAD + metabolites and nicotinamide N-methyltransferase in chronic kidney disease. Sci. Rep. 2022, 12, 6398. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- van Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef]
- Wang, W.J.; Cai, G.Y.; Chen, X.M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 2017, 8, 64520–64533. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, C.; Chen, X.; Han, M.; Li, J. Dexmedetomidine attenuates renal fibrosis via alpha2-adrenergic receptor-dependent inhibition of cellular senescence after renal ischemia/reperfusion. Life Sci. 2018, 207, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nunez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Interacting NAD(+) and Cell Senescence Pathways Complicate Antiaging Therapies. Rejuvenation Res. 2019, 22, 261–266. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarrago, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Ota, H.; Tokunaga, E.; Chang, K.; Hikasa, M.; Iijima, K.; Eto, M.; Kozaki, K.; Akishita, M.; Ouchi, Y.; Kaneki, M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 2006, 25, 176–185. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Birar, V.C.; Sheerin, A.N.; Jeynes, J.C.C.; Hooper, A.; Dawe, H.R.; Melzer, D.; Cox, L.S.; Faragher, R.G.A.; Ostler, E.L.; et al. Small molecule modulation of splicing factor expression is associated with rescue from cellular senescence. BMC Cell Biol. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Y.; Hu, T.; Wei, R.; Cai, C.; Wang, P.; Wang, L.; Qiao, W.; Feng, L. Endogenous Nampt upregulation is associated with diabetic nephropathy inflammatory-fibrosis through the NF-kappaB p65 and Sirt1 pathway; NMN alleviates diabetic nephropathy inflammatory-fibrosis by inhibiting endogenous Nampt. Exp. Ther. Med. 2017, 14, 4181–4193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Benito-Martin, A.; Ucero, A.C.; Izquierdo, M.C.; Santamaria, B.; Picatoste, B.; Carrasco, S.; Lorenzo, O.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Endogenous NAMPT dampens chemokine expression and apoptotic responses in stressed tubular cells. Biochim. Biophys. Acta 2014, 1842, 293–303. [Google Scholar] [CrossRef]
- Camp, S.M.; Ceco, E.; Evenoski, C.L.; Danilov, S.M.; Zhou, T.; Chiang, E.T.; Moreno-Vinasco, L.; Mapes, B.; Zhao, J.; Gursoy, G.; et al. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFkappaB Signaling and Inflammatory Lung Injury. Sci. Rep. 2015, 5, 13135. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Li, F.; Chen, Q.; Zhao, Z.; Liu, X.; Zhang, N.; Li, H. NAD(+) Anabolism Disturbance Causes Glomerular Mesangial Cell Injury in Diabetic Nephropathy. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Jin Kang, H.; Kim, D.K.; Mi Lee, S.; Han Kim, K.; Hee Han, S.; Hyun Kim, K.; Eun Kim, S.; Ki Son, Y.; An, W.S. Effects of low-dose niacin on dyslipidemia and serum phosphorus in patients with chronic kidney disease. Kidney Res. Clin. Pract. 2013, 32, 21–26. [Google Scholar] [CrossRef]
- DiPalma, J.R.; Thayer, W.S. Use of niacin as a drug. Annu. Rev. Nutr. 1991, 11, 169–187. [Google Scholar] [CrossRef]
- Reiten, O.K.; Wilvang, M.A.; Mitchell, S.J.; Hu, Z.; Fang, E.F. Preclinical and clinical evidence of NAD(+) precursors in health, disease, and ageing. Mech. Ageing Dev. 2021, 199, 111567. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease/Condition | NAD+ Boosters Compound | Outcome Observed after Genetic Modulation/NAD+ Boosting | Ref. |
|---|---|---|---|
| AKI | |||
| Model 1: IRI-induced mice: - Real NAD+, NADH, and QPRT (DOWN) - Renal and urinary Quin and Q:T (UP) Model 2: Renal Qprt-/+ mice: - Renal NAD+ and de novo pathway (DOWN) - sCr, AKI susceptibility, tubular necrosis, and renal and urinary Quin and Q:T (UP)Model 3: Ischemic human kidney: - Urinary Quin and Q:T (UP) Model 4: Cardiac surgery patients: - AKI susceptibility, sCr, and Troponin T (UP) | Genetically approaches by down-regulation QPRT or pharmacological approach by NAM | Model 1: Wild-type control mice: - Real NAD+, NADH, and QPRT (UP) - Renal and urinary Quin and Q:T (DOWN) Model 2: Wild-type control mice: - Renal NAD+ and de novo pathway (UP) - sCr, AKI susceptibility, tubular necrosis, and renal and urinary Quin and Q:T (DOWN) Model 3: Healthy human kidney: - Urinary Quin and Q:T (DOWN) Model 4: Cardiac surgery patients: - AKI susceptibility, sCr, and *Troponin T (DOWN) | [41] |
| Knockout of Pgc1-α in IRI-induced mice - sCr, DAGs, TAGs, tubular injury in cortex and medulla (UP) - Relative renal NAM (DOWN) | Genetically approaches by overexpression of Pgc1-α alone or in combination with NAM | - sCr, DAGs, TAGs, Tubular injury in cortex and medulla (DOWN) - Relative renal NAM, de novo NAD biosynthesis, renal NAD, #β-OHB, and #PGE2 (UP) | [107] |
| Model 1: IRI-induced mice: - pCr, BUN, tubular necrosis, dilation, cast formation, and MPO activity (UP) - GSH and NAD+ (DOWN) Model 2: Cisplatin-induced mice: - pCr, BUN, and renal KIM1 protein, tubular necrosis, dilation, edema, cast formation, and inflammatory cell (UP) - GFR and NAD+ (DOWN) | Pharmacological inhibition of ACMSD (TES-991 and TES-1025)/NAD+ Biosynthesis modulators | Model 1: IRI-induced mice: - pCr, BUN, tubular necrosis, dilation, cast formation, and MPO activity (UP) - GSH and NAD+ (DOWN) Model 2: Cisplatin-induced mice: - pCr, BUN, and renal KIM1 protein, tubular necrosis, dilation, edema, cast formation, and inflammatory cell (UP) - GFR and NAD+ (DOWN) | [37] |
| Model 1: Cisplatin-induced 20-month-old mice and 3-month-old mice: - BUN, sCr, damaged tubules, ac-FOXO-1, C-CAS 3 (UP) - Mitochondrial density, NAMPT, NMNAT1, and NMNAT3, NAD+, Sirt1 (DOWN) Model 2: Sirt1-/+, Cisplatin-induced mice:- BUN, sCr, damaged tubules, C-CAS 3 and 9, BAX, (UP) - Mitochondrial density, expression of Sirt1 (DOWN) Model 3: IRI-induced AKI mice: - BUN, sCr, damaged tubules (UP) | NMN | Model 1: 20-month-old and 3-month-old control mice: - BUN, sCr, damaged tubules, ac-FOXO-1, C-CAS 3 (DOWN) - Mitochondrial density, NAMPT, NMNAT1, and NMNAT3, NAD+, Sirt1 (UP) Model 2: Sirt+/+, Cisplatin-induced mice: - BUN, sCr, damaged tubules, C-CAS 3 and 9, BAX, (N.S.) - Mitochondrial density, expression of Sirt1 (N.S.) Model 3: IRI-induced AKI mice: - BUN, sCr, damaged tubules (DOWN) | [119] |
| CKD | |||
| UUO-induced renal interstitial fibrosis mice: Collagen, FN, α-SMA, tubular atrophy, C-CAS3, macrophage, T-cells, IL-1 beta, TNF-α (UP) | NAM | UUO-induced renal interstitial fibrosis mice: Collagen, FN, α-SMA, tubular atrophy, C-CAS3, macrophage, T-cells, IL-1 beta, TNF-α (DOWN) | [27] |
| NAD+ Precursors | Condition/ Disease | Dose Administration | Duration of Treatment | Age/ Sex | Study Title | Phase | Status | References/NCT |
|---|---|---|---|---|---|---|---|---|
| NR | AKI | 4X Basis™ capsule, BID, PO, (Each capsule: 125 mg of NR and 25 mg of PT) | 8 weeks (2 weeks pre-surgery and 6 weeks post-surgery. | 18+/ All | Protection from acute kidney injury (AKI) with Basis™ treatment | II | Recruiting | NCT04342975 |
| NR | AKI | 2X capsule, BID, PO, (Each capsule: 250 mg of NR) | 10 days | 18+/ All | Nicotinamide riboside in SARS-CoV-2 (COVID-19) patients for renal protection (NIRVANA) | II | Active, not recruiting | NCT04818216 |
| NR | AKI | 2X capsule, BID, PO,(Each capsule: 250 mg of NR and 50 mg of PT) | 2 days | 18+/ All | Pharmacokinetics, pharmacodynamics and safety of basis in acute kidney injury study (BAKIS) | N/A | Completed | NCT03176628 |
| NAM | AKI | 3 g/day NAM, PO | 3 days | 18+/ All | NAD+ augmentation in cardiac surgery associated myocardial injury trial (NACAM) | II | Recruiting | NCT04750616 |
| NAM | AKI | NAM (500 mg mixed in 50 mL of 0.9% saline), every 12 h, intravenously | 3 days | 18+/ All | Does high-dose vitamin B3 supplementation prevent major adverse kidney events during septic shock? (VITAKI) | III | Recruiting | NCT04589546 |
| NAM | AKI | 1 g or 3 g per day of NAM, PO | Baseline and days 1 through 4 | 18+/ All | Molecular effects of vitamin B3 (Niacinamide) in acute kidney injury | I | Completed | NCT02701127 |
| NA | AKI | 1X vitamin B complex, every 12 h, PO | 5 days | 18–100/All | Intravenous administration of vitamin B complex improves renal recovery in patients with AKI (VIBAKI) | IV | Recruiting | NCT04893733 |
| MIB-626 | AKI | 1 g MIB-626, BID, PO | 14 days | 18+/ All | Phase 2a MIB-626 vs. placebo COVID-19 | II | Recruiting | NCT05038488 |
| NR | CKD | 500 mg NR, BID, PO | 3 months | 35–80/ All | Nicotinamide riboside supplementation for treating arterial stiffness and elevated systolic blood pressure in patients with moderate to severe CKD | II | Recruiting | NCT04040959 |
| NR | CKD | 1 Tablet of 600 mg NR and 2 tablet of 250 mg Coenzyme Q10, BID, PO | 6 weeks | 30–79/ All | Trial of nicotinamide riboside and Co-enzyme Q10 in chronic kidney disease (CoNR) | II | Completed | NCT03579693 |
| NAM | CKD | 1 capsule NAM of 750 mg, BID, PO and 2* 500 mg capsules of lanthanum carbonate with each meal | 12 months | 18–85/ All | The COMBINE study: the CKD optimal management with BInders and Nicotinamide (COMBINE) | II | Completed | NCT02258074 |
| NA | CKD | 1000 mg/day, PO | 14 weeks | 21–80/ All | Niacin and endothelial function in early CKD | IV | Completed | NCT00852969 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morevati, M.; Fang, E.F.; Mace, M.L.; Kanbay, M.; Gravesen, E.; Nordholm, A.; Egstrand, S.; Hornum, M. Roles of NAD+ in Acute and Chronic Kidney Diseases. Int. J. Mol. Sci. 2023, 24, 137. https://doi.org/10.3390/ijms24010137
Morevati M, Fang EF, Mace ML, Kanbay M, Gravesen E, Nordholm A, Egstrand S, Hornum M. Roles of NAD+ in Acute and Chronic Kidney Diseases. International Journal of Molecular Sciences. 2023; 24(1):137. https://doi.org/10.3390/ijms24010137
Chicago/Turabian StyleMorevati, Marya, Evandro Fei Fang, Maria L. Mace, Mehmet Kanbay, Eva Gravesen, Anders Nordholm, Søren Egstrand, and Mads Hornum. 2023. "Roles of NAD+ in Acute and Chronic Kidney Diseases" International Journal of Molecular Sciences 24, no. 1: 137. https://doi.org/10.3390/ijms24010137
APA StyleMorevati, M., Fang, E. F., Mace, M. L., Kanbay, M., Gravesen, E., Nordholm, A., Egstrand, S., & Hornum, M. (2023). Roles of NAD+ in Acute and Chronic Kidney Diseases. International Journal of Molecular Sciences, 24(1), 137. https://doi.org/10.3390/ijms24010137