
Micronutrient Improvement of Epithelial Barrier Function in Various Disease States: A Case for Adjuvant Therapy


 and
and 
Abstract
1. Introduction
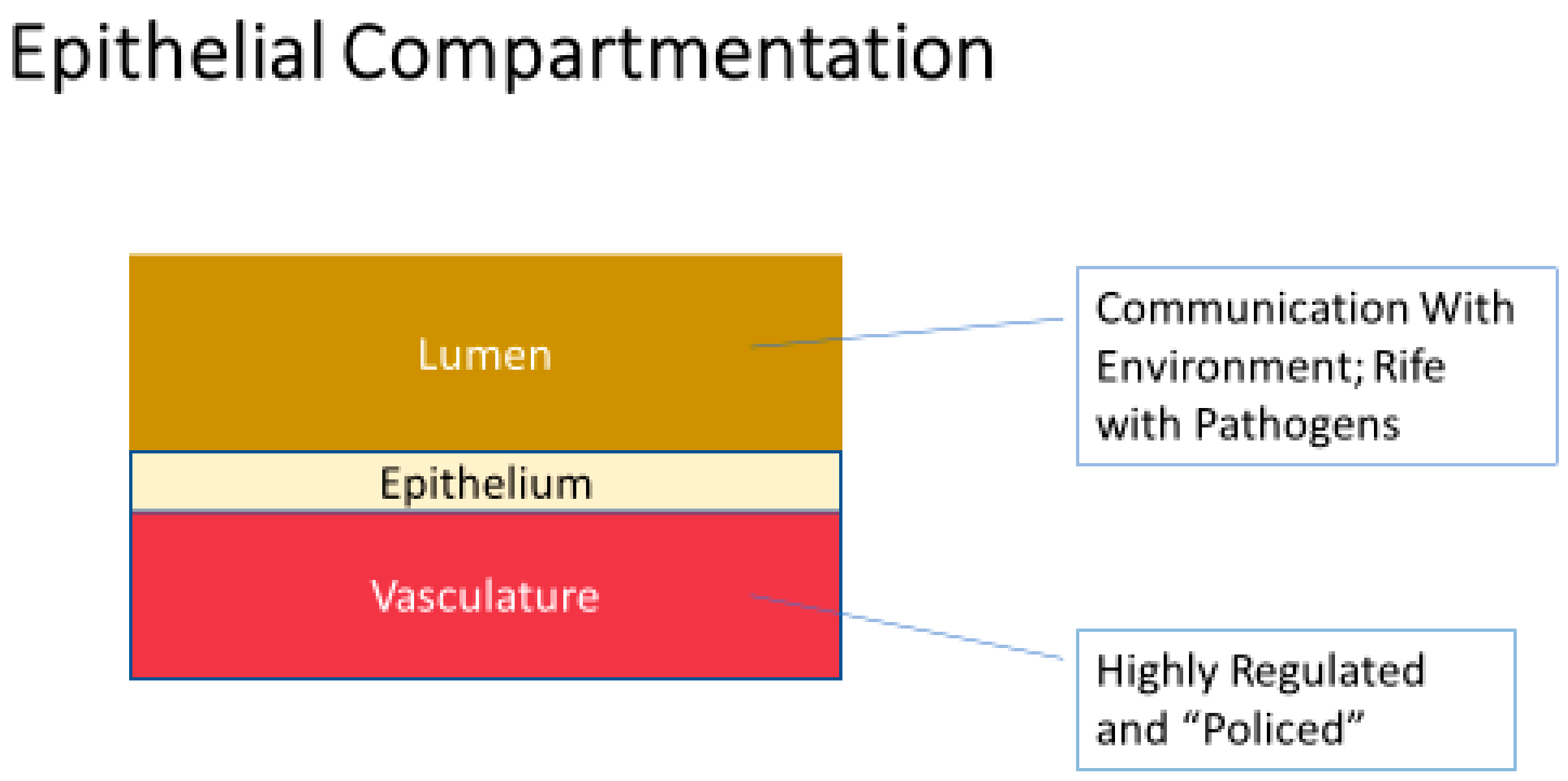
2. Is Epithelial Barrier Compromise a Common Occurrence in Disease?
2.1. Systemic Inflammation
2.2. Inflammatory Bowel Disease
2.3. Cancer
2.4. Celiac Disease
2.5. Infectious Disease
2.5.1. Gastrointestinal Bacteria
2.5.2. Non-Gastrointestinal Bacteria
2.5.3. Viral Pathogens
Rotaviruses
Flaviviruses
Influenza Viruses
2.6. Diabetes
2.7. Dust Mites
3. Does Micronutrient Deficiency Lead to Barrier Compromise and Exacerbate Disease?
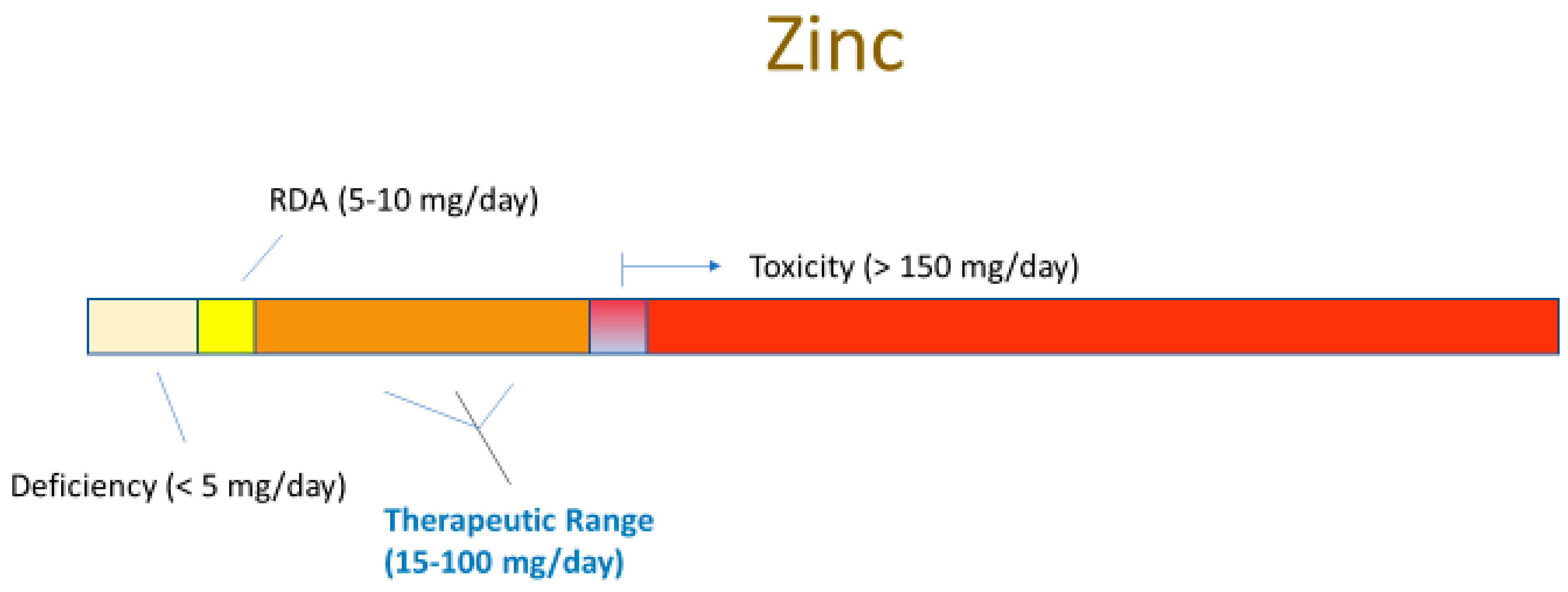
3.1. Zinc Deficiency
3.2. Vitamin A Deficiency
3.3. Vitamin D Deficiency
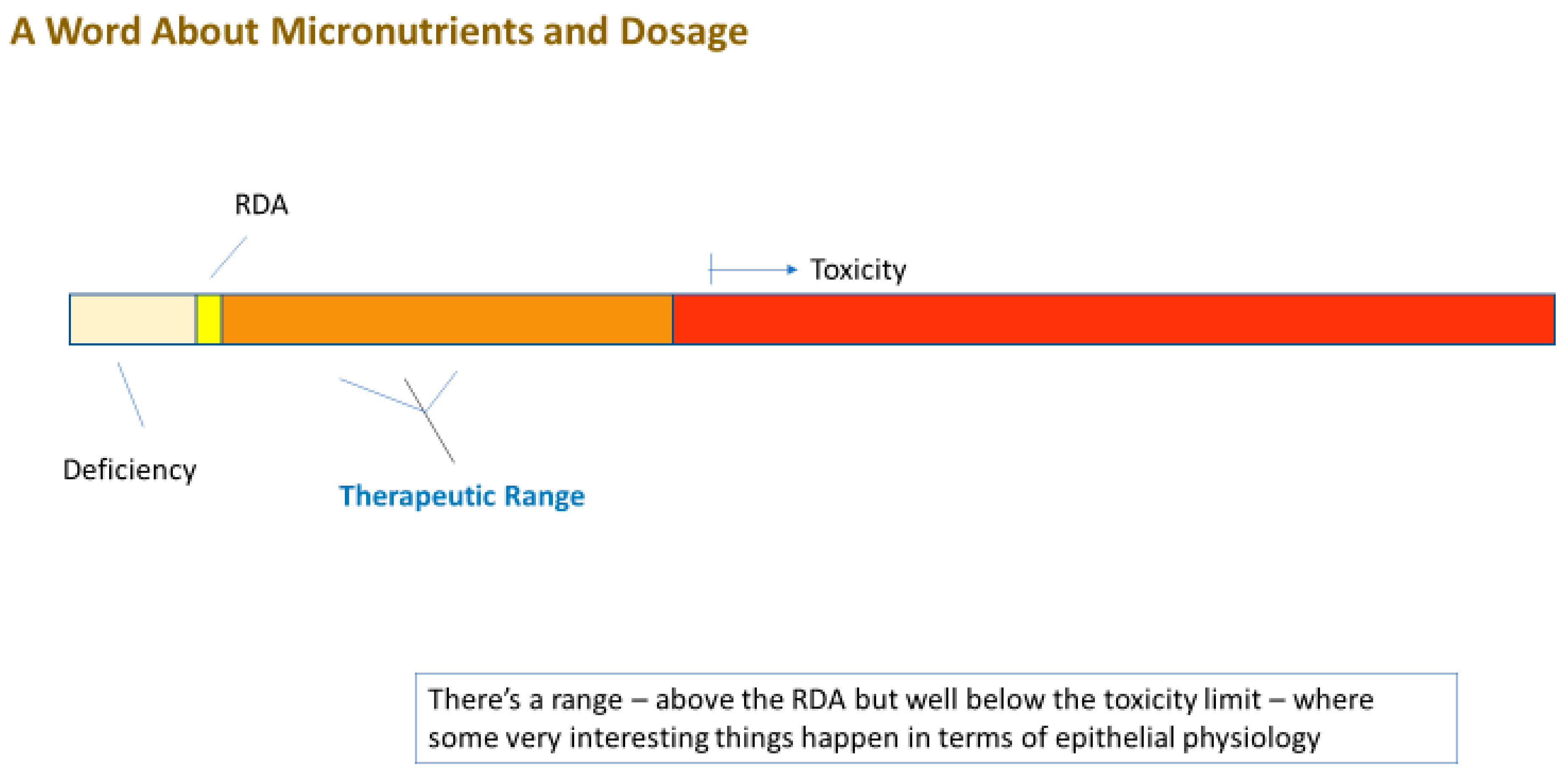
4. Can Elevated Micronutrient Levels (Supplementation) Improve Barrier Function?
4.1. Zinc Supplementation
4.2. Vitamin A Supplementation
4.3. Vitamin D Supplementation
5. Clinical Evidence for Elevated Micronutrient Levels as Therapeutic Strategies: Patient-Based Studies
6. Current Nutrition Guidelines and Consideration of Micronutrient Elevation as an Adjuvant Therapeutic

{kind=link}
{kind=link}
{kind=link}
| Recommended Oral Requirements | Recommended Parenteral Requirements | Content Provided in Multi-Vitamin/Trace Products Currently Available in the US | |
|---|---|---|---|
| Vitamin A | Male: 900 mcg or 3000 IU Female: 700 mcg or 2333 IU | 990 mcg or 3300 IU | 3300 IU per 10 mL in MVI |
| Vitamin D | Age 19–70 years: 15 mcg or 600 IU | 5 mcg or 200 IU | 200 IU per 10 mL in MVI |
| Zinc | Male: 11 mg Female: 8 mg | 3–5 mg | varies between 3–5 mg based on product |
7. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Briefel, R.R.; Bialostosky, K.; Kennedy-Stephenson, J.; McDowell, M.A.; Ervin, R.B.; Wright, J.D. Zinc Intake of the U.S. Population: Findings from the Third National Health and Nutrition Examination Survey, 1988–1994. J. Nutr. 2000, 130 (Suppl. S5), 1367S–1373S. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.S.; Hess, S.Y.; Hotz, C.; Brown, K.H. Indicators of zinc status at the population level: A review of the evidence. Br. J. Nutr. 2008, 99, S14–S23. [Google Scholar] [CrossRef] [PubMed]
- Rybakovsky, E.; Valenzano, M.C.; Deis, R.; DiGuilio, K.M.; Thomas, S.; Mullin, J.M. Improvement of Human-Oral-Epithelial-Barrier Function and of Tight Junctions by Micronutrients. J. Agric. Food Chem. 2017, 65, 10950–10958. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Valenzano, M.C.; Mercado, J.M.; Zurbach, E.P.; Mullin, J.M. Zinc Supplementation Modifies Tight Junctions and Alters Barrier Function of CACO-2 Human Intestinal Epithelial Layers. Dig. Dis. Sci. 2012, 58, 77–87. [Google Scholar] [CrossRef]
- Hu, C.; Song, J.; Li, Y.; Luan, Z.; Zhu, K. Diosmectite–zinc oxide composite improves intestinal barrier function, modulates expression of pro-inflammatory cytokines and tight junction protein in early weaned pigs. Br. J. Nutr. 2013, 110, 681–688. [Google Scholar] [CrossRef]
- Heinemann, U.; Schuetz, A. Structural Features of Tight-Junction Proteins. Int. J. Mol. Sci. 2019, 20, 6020. [Google Scholar] [CrossRef]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef]
- Prot-Bertoye, C.; Houillier, P. Claudins in Renal Physiology and Pathology. Genes 2020, 11, 290. [Google Scholar] [CrossRef]
- Slifer, Z.M.; Blikslager, A.T. The Integral Role of Tight Junction Proteins in the Repair of Injured Intestinal Epithelium. Int. J. Mol. Sci. 2020, 21, 972. [Google Scholar] [CrossRef]
- Dong, D.; Xie, W.; Liu, M. Alteration of cell junctions during viral infection. Thorac. Cancer 2020, 11, 519–525. [Google Scholar] [CrossRef]
- Cong, X.; Kong, W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell. Signal. 2020, 66, 109485. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Kaunitz, J.D. The “Leaky Gut”: Tight Junctions but Loose Associations? Dig. Dis. Sci. 2020, 65, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Rolles, B.; Slusarenko, A.J.; Rink, L. Zinc deficiency as a possible risk factor for increased susceptibility and severe progression of Corona Virus Disease 19. Br. J. Nutr. 2021, 127, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, P.J.; Ferrick, B.; Rybakovsky, E.; Thomas, S.; Mullin, J.M. Epithelial barrier function properties of the 16HBE14o- human bronchial epithelial cell culture model. Biosci. Rep. 2020, 40, BSR20201532. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.-T.; Shen, L.; Zuo, L.; Shashikanth, N.; Ong, M.D.M.; Wu, L.; Zha, J.; Edelblum, K.L.; Wang, Y.; Wang, Y.; et al. Inflammation-induced Occludin Downregulation Limits Epithelial Apoptosis by Suppressing Caspase-3 Expression. Gastroenterology 2019, 157, 1323–1337. [Google Scholar] [CrossRef]
- Rybakovsky, E.; Buleza, N.B.; Hoxha, K.; DiGuilio, K.M.; McCluskey, E.S.; Friday, C.L.; Callaghan, P.J.; Moskalenko, D.V.; Zuo, B.; Thomas, S.; et al. Spontaneous and cytokine-induced hole formation in epithelial cell layers: Implications for barrier function studies with the gingival cell culture, Gie-3B11, and other epithelial models. Trends Cell Mol. Biol. 2018, 13, 99–114. [Google Scholar]
- Al-Sadi, R.; Guo, S.; Ye, D.; Rawat, M.; Ma, T.Y. TNF-α Modulation of Intestinal Tight Junction Permeability Is Mediated by NIK/IKK-α Axis Activation of the Canonical NF-κB Pathway. Am. J. Pathol. 2016, 186, 1151–1165. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Guo, S.; Ye, D.; Ma, T.Y. TNF-α Modulation of Intestinal Epithelial Tight Junction Barrier Is Regulated by ERK1/2 Activation of Elk-1. Am. J. Pathol. 2013, 183, 1871–1884. [Google Scholar] [CrossRef]
- Petecchia, L.; Sabatini, F.; Usai, C.; Caci, E.; Varesio, L.; Rossi, G.A. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab. Investig. 2012, 92, 1140–1148. [Google Scholar] [CrossRef]
- Mullin, J.; Snock, K.V. Effect of tumor necrosis factor on epithelial tight junctions and transepithelial permeability. Cancer Res. 1990, 50, 2172–2176. [Google Scholar]
- Fink, M.P. Intestinal epithelial hyperpermeability: Update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr. Opin. Crit. Care 2003, 9, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P.; Delude, R.L. Epithelial Barrier Dysfunction: A Unifying Theme to Explain the Pathogenesis of Multiple Organ Dysfunction at the Cellular Level. Crit. Care Clin. 2005, 21, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, Q.; Shi, X.; Cong, X.; Zhang, Y.; Wu, L.; Yu, G.; Xiang, R. Disruption of tight junctions contributes to hyposalivation of salivary glands in a mouse model of type 2 diabetes. J. Anat. 2020, 237, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Mei, X.-L.; Zhao, Y.-N. Sepsis and Cerebral Dysfunction: BBB Damage, Neuroinflammation, Oxidative Stress, Apoptosis and Autophagy as Key Mediators and the Potential Therapeutic Approaches. Neurotox. Res. 2021, 39, 489–503. [Google Scholar] [CrossRef]
- Gonzales, J.; Lucas, R.; Verin, A. The Acute Respiratory Distress Syndrome: Mechanisms and Perspective Therapeutic Approaches. Austin J. Vasc. Med. 2015, 2, 1009. [Google Scholar]
- Rodrigues, S.F.; Granger, D.N. Blood cells and endothelial barrier function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef]
- Mariano, F.; Cantaluppi, V.; Stella, M.; Romanazzi, G.M.; Assenzio, B.; Cairo, M.; Biancone, L.; Triolo, G.; Ranieri, V.M.; Camussi, G. Circulating plasma factors induce tubular and glomerular alterations in septic burns patients. Crit. Care 2008, 12, R42. [Google Scholar] [CrossRef]
- Zhao, G.-J.; Li, D.; Zhao, Q.; Lian, J.; Hu, T.-T.; Hong, G.-L.; Yao, Y.-M.; Lu, Z. Prognostic Value of Plasma Tight-Junction Proteins for Sepsis in Emergency Department: An Observational Study. Shock 2016, 45, 326–332. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Akinosoglou, K.; de Lastic, A.-L.; Skintzi, A.; Mouzaki, A.; Gogos, C.A. The Prognostic Value of Endotoxemia and Intestinal Barrier Biomarker ZO-1 in Bacteremic Sepsis. Am. J. Med. Sci. 2020, 359, 100–107. [Google Scholar] [CrossRef]
- Mankertz, J.; Schulzke, J.-D. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383. [Google Scholar] [CrossRef]
- Fakhoury, M.; Negrulj, R.; Mooranian, A.; Al-Salami, H. Inflammatory bowel disease: Clinical aspects and treatments. J. Inflamm. Res. 2014, 7, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D. Crohn’s disease--a permeability disorder of the tight junction? Gut 1988, 29, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Schulzke, J.D.; Ploeger, S.; Amasheh, M.; Fromm, A.; Zeissig, S.; Troeger, H.; Richter, J.; Bojarski, C.; Schumann, M.; Fromm, M. Epithelial tight junctions in intestinal inflammation. Ann. N. Y. Acad. Sci. 2009, 1165, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Luettig, J.; Rosenthal, R.; Barmeyer, C.; Schulzke, J.D. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 2015, 3, e977176. [Google Scholar] [CrossRef] [PubMed]
- Teshima, C.; Dieleman, L.A.; Meddings, J.B. Abnormal intestinal permeability in Crohn’s disease pathogenesis. Ann. N. Y. Acad. Sci. 2012, 1258, 159–165. [Google Scholar] [CrossRef]
- Takeuchi, K.; Maiden, L.; Bjarnason, I. Genetic aspects of intestinal permeability in inflammatory bowel disease. Novartis Found Symp. 2004, 263, 151–158; discussion 159–163, 211–218. [Google Scholar]
- Luissint, A.-C.; Parkos, C.A.; Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte–Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 2016, 151, 616–632. [Google Scholar] [CrossRef]
- Bruewer, M.; Samarin, S.; Nusrat, A. Inflammatory Bowel Disease and the Apical Junctional Complex. Ann. N. Y. Acad. Sci. 2006, 1072, 242–252. [Google Scholar] [CrossRef]
- Shen, L.; Turner, J.R. Role of Epithelial Cells in Initiation and Propagation of Intestinal Inflammation. Eliminating the static: Tight junction dynamics exposed. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G577–G582. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. Intestinal Permeability Defects: Is It Time to Treat? Clin. Gastroenterol. Hepatol. 2013, 11, 1075–1083. [Google Scholar] [CrossRef]
- Zhu, L.; Han, J.; Li, L.; Wang, Y.; Li, Y.; Zhang, S. Claudin Family Participates in the Pathogenesis of Inflammatory Bowel Diseases and Colitis-Associated Colorectal Cancer. Front. Immunol. 2019, 10, 1441. [Google Scholar] [CrossRef] [PubMed]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Fries, W.; Belvedere, A.; Vetrano, S. Sealing the broken barrier in IBD: Intestinal permeability, epithelial cells and junctions. Curr. Drug Targets 2013, 14, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Marin, M.L.; Greenstein, A.J.; Geller, S.A.; Gordon, R.E.; Aufses, A.H., Jr. A freeze fracture study of Crohn’s disease of the terminal ileum: Changes in epithelial tight junction organization. Am. J. Gastroenterol. 1983, 78, 537–547. [Google Scholar]
- Schmitz, H.; Barmeyer, C.; Fromm, M.; Runkel, N.; Foss, H.-D.; Bentzel, C.J.; Riecken, E.-O.; Schulzke, J.-D. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology 1999, 116, 301–309. [Google Scholar] [CrossRef]
- Gitter, A.H.; Wullstein, F.; Fromm, M.; Schulzke, J.D. Epithelial barrier defects in ulcerative colitis: Characterization and quantification by electrophysiological imaging. Gastroenterology 2001, 121, 1320–1328. [Google Scholar] [CrossRef]
- Oshima, T.; Miwa, H.; Joh, T. Changes in the expression of claudins in active ulcerative colitis. J. Gastroenterol. Hepatol. 2008, 23, S146–S150. [Google Scholar] [CrossRef]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Das, P.; Goswami, P.; Das, T.K.; Nag, T.; Sreenivas, V.; Ahuja, V.; Panda, S.K.; Gupta, S.D.; Makharia, G.K. Comparative tight junction protein expressions in colonic Crohn’s disease, ulcerative colitis, and tuberculosis: A new perspective. Virchows Arch. 2012, 460, 261–270. [Google Scholar] [CrossRef]
- Lameris, A.L.; Huybers, S.; Kaukinen, K.; Mäkelä, T.H.; Bindels, R.J.; Hoenderop, J.G.; Nevalainen, P.I. Expression profiling of claudins in the human gastrointestinal tract in health and during inflammatory bowel disease. Scand. J. Gastroenterol. 2013, 48, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-M.; Li, X.-M.; Qin, B.-Z.; Liu, B. Effect of tight junction protein of intestinal epithelium and permeability of colonic mucosa in pathogenesis of injured colonic barrier during chronic recovery stage of rats with inflammatory bowel disease. Asian Pac. J. Trop. Med. 2016, 9, 148–152. [Google Scholar] [CrossRef]
- Zwiers, A.; Fuss, I.J.; Leijen, S.; Mulder, C.J.; Kraal, G.; Bouma, G. Increased expression of the tight junction molecule claudin-18 A1 in both experimental colitis and ulcerative colitis. Inflamm. Bowel Dis. 2008, 14, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Erakovic Haber, V.; Cuzic, S.; Dominis Kramaric, M.; Hrvacic, B.; Polancec, D.; Banic, M.; Glojnaric, I. Claudin expression in animal models of IBD and human disease. New Horiz. Transl. Med. 2015, 2, 66–67. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva Med. 2019, 110, 95–100. [Google Scholar] [CrossRef]
- Arrieta, M.C.; Madsen, K.; Doyle, J.; Meddings, J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut 2008, 58, 41–48. [Google Scholar] [CrossRef]
- Olson, T.S.; Reuter, B.K.; Scott, K.G.-E.; Morris, M.A.; Wang, X.-M.; Hancock, L.N.; Burcin, T.L.; Cohn, S.M.; Ernst, P.; Cominelli, F.; et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J. Exp. Med. 2006, 203, 541–552. [Google Scholar] [CrossRef]
- Poritz, L.S.; Garver, K.I.; Green, C.; Fitzpatrick, L.; Ruggiero, F.; Koltun, W.A. Loss of the Tight Junction Protein ZO-1 in Dextran Sulfate Sodium Induced Colitis. J. Surg. Res. 2007, 140, 12–19. [Google Scholar] [CrossRef]
- Poritz, L.S.; Harris, L.R., 3rd; Kelly, A.A.; Koltun, W.A. Increase in the Tight Junction Protein Claudin-1 in Intestinal Inflammation. Dig. Dis. Sci. 2011, 56, 2802–2809. [Google Scholar] [CrossRef]
- Kucharzik, T.; Walsh, S.V.; Chen, J.; Parkos, C.A.; Nusrat, A. Neutrophil Transmigration in Inflammatory Bowel Disease Is Associated with Differential Expression of Epithelial Intercellular Junction Proteins. Am. J. Pathol. 2001, 159, 2001–2009. [Google Scholar] [CrossRef]
- Soderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzén, L.; Lindmark, T.; Wirén, M.; Tagesson, C.; Sjödahl, R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 2002, 50, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.; Vogelsang, H.; Hübl, W.; Waldhoer, T.; Lochs, H. Intestinal permeability and the prediction of relapse in Crohri’s disease. Lancet 1993, 341, 1437–1439. [Google Scholar] [CrossRef]
- D’Incà, R.; Di Leo, V.; Corrao, G.; Martines, D.; D’Odorico, A.; Mestriner, C.; Venturi, C.; Longo, G.; Sturniolo, G.C. Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am. J. Gastroenterol. 1999, 94, 2956–2960. [Google Scholar] [CrossRef]
- Arnott, I.D.R.; Kingstone, K.; Ghosh, S. Abnormal Intestinal Permeability Predicts Relapse in Inactive Crohn Disease. Scand. J. Gastroenterol. 2000, 35, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Leong, R.W.; Wasinger, V.C.; Ip, M.; Yang, M.; Phan, T.G. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients with Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology 2017, 153, 723–731.e1. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Vadheim, C.M.; Brettholz, E.; Petersen, G.M.; Delahunty, T.; Rotter, J.I. Increased Intestinal Permeability in Patients with Crohn’s Disease and Their Relatives. A possible etiologic factor. Ann. Intern. Med. 1986, 105, 883–885. [Google Scholar] [CrossRef]
- Teshima, C.W.; Goodman, K.J.; El-Kalla, M.; Turk, S.; El-Matary, W.; Valcheva, R.; Danchak, R.; Gordon, M.; Ho, P.; Mullins, A.; et al. Increased Intestinal Permeability in Relatives of Patients with Crohn’s Disease Is Not Associated with Small Bowel Ulcerations. Clin. Gastroenterol. Hepatol. 2017, 15, 1413–1418.e1. [Google Scholar] [CrossRef]
- May, G.R.; Sutherland, L.R.; Meddings, J.B. Is small intestinal permeability really increased in relatives of patients with Crohn’s disease? Gastroenterology 1993, 104, 1627–1632. [Google Scholar] [CrossRef]
- Peeters, M.; Geypens, B.; Claus, D.; Nevens, H.; Ghoos, Y.; Verbeke, G.; Baert, F.; Vermeire, S.; Vlietinck, R.; Rutgeerts, P. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology 1997, 113, 802–807. [Google Scholar] [CrossRef]
- Turpin, W.; Lee, S.H.; Raygoza Garay, J.A.; Madsen, K.L.; Meddings, J.B.; Bedrani, L.; Power, N.; Espin-Garcia, O.; Xu, W.; Smith, M.I.; et al. Increased Intestinal Permeability Is Associated with Later Development of Crohn’s Disease. Gastroenterology 2020, 159, 2092.e5–2100.e5. [Google Scholar] [CrossRef]
- Irvine, E.; Marshall, J. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology 2000, 119, 1740–1744. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.-P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.-P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Buhner, S.; Buning, C.; Genschel, J.; Kling, K.; Herrmann, D.; Dignass, A.; Kuechler, I.; Krueger, S.; Schmidt, H.H.-J.; Lochs, H. Genetic basis for increased intestinal permeability in families with Crohn’s disease: Role of CARD15 3020insC mutation? Gut 2006, 55, 342–347. [Google Scholar] [CrossRef]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G367–G376. [Google Scholar] [CrossRef]
- Mankertz, J.; Amasheh, M.; Krug, S.M.; Fromm, A.; Hillenbrand, B.; Tavalali, S.; Fromm, M.; Schulzke, J.D. TNFα up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009, 336, 67–77. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M. Interleukin-13 Is the Key Effector Th2 Cytokine in Ulcerative Colitis That Affects Epithelial Tight Junctions, Apoptosis, and Cell Restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Blair, S.A.; Kane, S.V.; Clayburgh, D.; Turner, J.R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Investig. 2006, 86, 191–201. [Google Scholar] [CrossRef]
- Zolotarevsky, Y.; Hecht, G.; Koutsouris, A.; Gonzalez, D.E.; Quan, C.; Tom, J.; Mrsny, R.J.; Turner, J.R. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002, 123, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Gluth, M.; Pape, U.-F.; Wiedenmann, B.; Theuring, F.; Baumgart, D.C. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G970–G979. [Google Scholar] [CrossRef]
- Prasad, S.; Mingrino, R.; Kaukinen, K.; Hayes, K.L.; Powell, R.M.; Macdonald, T.T.; Collins, J. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab. Investig. 2005, 85, 1139–1162. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Kuiper, I.; Lamers, C.B.H.W.; Verspaget, H.W. Intestinal oxidative damage in inflammatory bowel disease: Semi-quantification, localization, and association with mucosal antioxidants. J. Pathol. 2003, 201, 28–36. [Google Scholar] [CrossRef]
- Strus, M.; Gosiewski, T.; Fyderek, K.; Wedrychowicz, A.; Kowalska-Duplaga, K.; Kochan, P.; Adamski, P.; Heczko, P.B. A role of hydrogen peroxide producing commensal bacteria present in colon of adolescents with inflammatory bowel disease in perpetuation of the inflammatory process. J. Physiol. Pharmacol. 2009, 60 (Suppl. S6), 49–54. [Google Scholar] [PubMed]
- Morgan, X.C.; Tickle, T.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; Leleiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Fraumeni, J.F.; Hoover, R.N.; Devasa, S.S.; Kinlen, J.L. Epidemiology of Cancer. In Cancer: Principles and Practice of Oncology; Devita, V.T., Hellmann, S., Rosenberg, S.A., Eds.; Lippincott: Philadelphia, PA, USA, 1989; pp. 196–209. [Google Scholar]
- Saito, Y.; Desai, R.R.; Muthuswamy, S.K. Reinterpreting polarity and cancer: The changing landscape from tumor suppression to tumor promotion. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Hinck, L.; Näthke, I. Changes in cell and tissue organization in cancer of the breast and colon. Curr. Opin. Cell Biol. 2014, 26, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Palomo, A. Ultrastructural modifications of intercellular junctions between tumor cells. In Vitro 1970, 6, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J. Tight junctions adjacent to tumor stromal interface in human invasive transitional cell carcinomas. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1979, 30, 289–296. [Google Scholar] [CrossRef]
- Polak-Charcon, S.; Shoham, J.; Ben-Shaul, Y. Tight Junctions in Epithelial Cells of Human Fetal Hindgut, Normal Colon, and Colon Adenocarcinoma. J. Natl. Cancer Inst. 1980, 65, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Robenek, H.; Schöpper, C.; Fasske, E.; Fetting, R.; Themann, H. Structure and function of the junctional complement of spontaneous and transplanted murine mammary carcinomas. J. Submicrosc. Cytol. 1981, 13, 347–363. [Google Scholar] [PubMed]
- Swift, J.G.; Mukherjee, T.M.; Rowland, R. Intercellular junctions in hepatocellular carcinoma. J. Submicrosc. Cytol. 1983, 15, 799–810. [Google Scholar]
- Zhong, Y.; Enomoto, K.; Tobioka, H.; Konishi, Y.; Satoh, M.; Mori, M. Sequential Decrease in Tight Junctions as Revealed by 7H6 Tight Junction-associated Protein during Rat Hepatocarcinogenesis. Jpn. J. Cancer Res. 1994, 85, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Soler, A.P.; Miller, R.; Laughlin, K.V.; Carp, N.Z.; Klurfeld, D.; Mullin, J. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 1999, 20, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Jesaitis, L.A.; Goodenough, D.A. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J. Cell Biol. 1994, 124, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Willott, E.; Balda, M.; Fanning, A.S.; Jameson, B.; Van Itallie, C.; Anderson, J. The tight junction protein ZO-1 is homologous to the Drosophila discs-large tumor suppressor protein of septate junctions. Proc. Natl. Acad. Sci. USA 1993, 90, 7834–7838. [Google Scholar] [CrossRef]
- Nakamura, T.; Blechman, J.; Tada, S.; Rozovskaia, T.; Itoyama, T.; Bullrich, F.; Mazo, A.; Croce, C.M.; Geiger, B.; Canaani, E. huASH1 protein, a putative transcription factor encoded by a human homologue of the Drosophila ash1 gene, localizes to both nuclei and cell-cell tight junctions. Proc. Natl. Acad. Sci. USA 2000, 97, 7284–7289. [Google Scholar] [CrossRef]
- Kage, H.; Flodby, P.; Zhou, B.; Borok, Z. Dichotomous roles of claudins as tumor promoters or suppressors: Lessons from knockout mice. Cell Mol Life Sci. 2019, 76, 4663–4672. [Google Scholar] [CrossRef]
- Boutwell, R.K.; Sivak, A. The Function and Mechanism of Promoters of Carcinogenesis. CRC Crit. Rev. Toxicol. 1974, 2, 419–443. [Google Scholar] [CrossRef]
- Mullin, J.; Soler, A.; Laughlin, K.; Kampherstein, J.; Russo, L.; Saladik, D.; George, K.; Shurina, R.; O’Brien, T. Chronic Exposure of LLC-PK1Epithelia to the Phorbol Ester TPA Produces Polyp-like Foci with Leaky Tight Junctions and Altered Protein Kinase C-α Expression and Localization. Exp. Cell Res. 1996, 227, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Dodane, V.; Kachar, B. Identification of Isoforms of G Proteins and PKC that Colocalize with Tight Junctions. J. Membr. Biol. 1996, 149, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.; Marano, C.W.; Soler, A.P.; Mullin, J.M. Modification of tight junction function by protein kinase C isoforms. Adv. Drug Deliv. Rev. 2000, 41, 283–301. [Google Scholar] [CrossRef]
- Buse, P.; Woo, P.L.; Alexander, D.B.; Cha, H.H.; Reza, A.; Sirota, N.D.; Firestone, G.L. Transforming Growth Factor-α Abrogates Glucocorticoid-stimulated Tight Junction Formation and Growth Suppression in Rat Mammary Epithelial Tumor Cells. J. Biol. Chem. 1995, 270, 6505–6514. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Muthuswamy, S.; Li, D.; Lelievre, S.; Bissell, M.J.; Brugge, J.S. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 2001, 3, 785–792. [Google Scholar] [CrossRef]
- Mullin, J.M. Epithelial Barriers, Compartmentation, and Cancer. Sci. STKE 2004, 216, pe2. [Google Scholar] [CrossRef]
- Pozzi, A.; Zent, R. ZO-1 and ZONAB Interact to Regulate Proximal Tubular Cell Differentiation. J. Am. Soc. Nephrol. 2010, 21, 388–390. [Google Scholar] [CrossRef]
- Balda, M.S.; Garrett, M.D.; Matter, K. The ZO-1–associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. [Google Scholar] [CrossRef]
- Kohno, T.; Konno, T.; Kojima, T. Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3555. [Google Scholar] [CrossRef]
- Bhat, A.A.; Syed, N.; Therachiyil, L.; Nisar, S.; Hashem, S.; Macha, M.; Yadav, S.K.; Krishnankutty, R.; Muralitharan, S.; Al-Naemi, H.; et al. Claudin-1, A Double-Edged Sword in Cancer. Int. J. Mol. Sci. 2020, 21, 569. [Google Scholar] [CrossRef]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef] [PubMed]
- González-Mariscal, L.; Lechuga, S.; Garay, E. Role of tight junctions in cell proliferation and cancer. Prog. Histochem. Cytochem. 2007, 42, 1–57. [Google Scholar] [CrossRef] [PubMed]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Anwer, S.; Szászi, K. Claudin-2: Roles beyond Permeability Functions. Int. J. Mol. Sci. 2019, 20, 5655. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Silva, D.; Delbue, D.; Itzlinger, A.; Moerkens, R.; Withoff, S.; Branchi, F.; Schumann, M. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients 2019, 11, 2325. [Google Scholar] [CrossRef]
- Asri, N.; Rostami-Nejad, M.; Rezaei-Tavirani, M.; Razzaghi, M.; Asadzadeh-Aghdaei, H.; Zali, M.R. Novel Therapeutic Strategies for Celiac Disease. Middle East J. Dig. Dis. 2020, 12, 229–237. [Google Scholar]
- Valitutti, F.; Fasano, A. Breaking Down Barriers: How Understanding Celiac Disease Pathogenesis Informed the Development of Novel Treatments. Dig. Dis. Sci. 2019, 64, 1748–1758. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Schumann, M.; Siegmund, B.; Schulzke, J.D.; Fromm, M. Celiac Disease: Role of the Epithelial Barrier. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 150–162. [Google Scholar] [CrossRef]
- Barmeyer, C.; Fromm, M.; Schulzke, J.-D. Active and passive involvement of claudins in the pathophysiology of intestinal inflammatory diseases. Pflügers Arch. Eur. J. Physiol. 2017, 469, 15–26. [Google Scholar] [CrossRef]
- Barmeyer, C.; Schulzke, J.D.; Fromm, M. Claudin-related intestinal diseases. Semin. Cell Dev. Biol. 2015, 42, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Schumann, M.; Kamel, S.; Pahlitzsch, M.-L.; Lebenheim, L.; May, C.; Krauss, M.; Hummel, M.; Daum, S.; Fromm, M.; Schulzke, J.-D. Defective tight junctions in refractory celiac disease. Ann. N. Y. Acad. Sci. 2012, 1258, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, H.; Schwarzenhofer, M.; Oberhuber, G. Changes in Gastrointestinal Permeability in Celiac Disease. Dig. Dis. 1998, 16, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gutierrez-Achury, J.; Kanduri, K.; Almeida, R.; Hrdlickova, B.; Zhernakova, D.V.; Westra, H.-J.; Karjalainen, J.; Ricaño-Ponce, I.; Li, Y.; et al. Systematic annotation of celiac disease loci refines pathological pathways and suggests a genetic explanation for increased interferon-gamma levels. Hum. Mol. Genet. 2015, 24, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Ricaño-Ponce, I.; Kumar, V.; Deelen, P.; Szperl, A.; Trynka, G.; Gutierrez-Achury, J.; Kanterakis, A.; Westra, H.-J.; Franke, L.; et al. Fine mapping of the celiac disease-associated LPP locus reveals a potential functional variant. Hum. Mol. Genet. 2013, 23, 2481–2489. [Google Scholar] [CrossRef]
- Jauregi-Miguel, A.; Fernandez-Jimenez, N.; Irastorza, I.; Plaza-Izurieta, L.; Vitoria, J.C.; Bilbao, J.R. Alteration of Tight Junction Gene Expression in Celiac Disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 762–767. [Google Scholar] [CrossRef]
- Kohl, D.; Ashkenazi, A.; Ben-Shaul, Y.; Bacher, A. Tight junctions of jejunal surface and crypt cells in celiac disease: A freeze-fracture study. J. Pediatr. Gastroenterol. Nutr. 1987, 6, 57–65. [Google Scholar]
- Schulzke, J.-D.; Bentzel, C.J.; Schulzke, I.; Riecken, E.-O.; Fromm, M. Epithelial Tight Junction Structure in the Jejunum of Children with Acute and Treated Celiac Sprue. Pediatr. Res. 1998, 43 Pt 1, 435–441. [Google Scholar] [CrossRef]
- Reims, A.; Strandvik, B.; Sjövall, H. Epithelial Electrical Resistance as a Measure of Permeability Changes in Pediatric Duodenal Biopsies. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 619–623. [Google Scholar] [CrossRef]
- Mishra, A.; Prakash, S.; Sreenivas, V.; Das, T.K.; Ahuja, V.; Gupta, S.D.; Makharia, G.K. Structural and Functional Changes in the Tight Junctions of Asymptomatic and Serology-negative First-degree Relatives of Patients with Celiac Disease. J. Clin. Gastroenterol. 2016, 50, 551–560. [Google Scholar] [CrossRef]
- Sowińska, A.; Morsy, Y.; Czarnowska, E.; Oralewska, B.; Konopka, E.; Woynarowski, M.; Szymańska, S.; Ejmont, M.; Scharl, M.; Bierła, J.B.; et al. Transcriptional and Ultrastructural Analyses Suggest Novel Insights into Epithelial Barrier Impairment in Celiac Disease. Cells 2020, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Khatib, K.; Guo, S.; Ye, D.; Youssef, M.; Ma, T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1054–G1064. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Finamore, A.; Ara, C.; Di Sabatino, A.; Mengheri, E.; Corazza, G.R. Altered Expression, Localization, and Phosphorylation of Epithelial Junctional Proteins in Celiac Disease. Am. J. Clin. Pathol. 2006, 125, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Pizzuti, D.; Bortolami, M.; Mazzon, E.; Buda, A.; Guariso, G.; D’Odorico, A.; Chiarelli, S.; D’Incà, R.; De Lazzari, F.; Martines, D. Transcriptional downregulation of tight junction protein ZO-1 in active coeliac disease is reversed after a gluten-free diet. Dig. Liver Dis. 2004, 36, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Das, P.; Verma, A.K.; Prakash, S.; Das, T.K.; Nag, T.C.; Ahuja, V.; Gupta, S.D.; Makharia, G.K. Are alterations of tight junctions at molecular and ultrastructural level different in duodenal biopsies of patients with celiac disease and Crohn’s disease? Virchows Arch. 2014, 465, 521–530. [Google Scholar] [CrossRef]
- Szakál, D.N.; Győrffy, H.; Arato, A.; Cseh, A.; Molnár, K.; Papp, M.; Dezsőfi, A.; Veres, G. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Arch. 2010, 456, 245–250. [Google Scholar] [CrossRef]
- Hollon, J.; Puppa, E.L.; Greenwald, B.; Goldberg, E.; Guerrerio, A.; Fasano, A. Effect of Gliadin on Permeability of Intestinal Biopsy Explants from Celiac Disease Patients and Patients with Non-Celiac Gluten Sensitivity. Nutrients 2015, 7, 1565–1576. [Google Scholar] [CrossRef]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin Induces an Increase in Intestinal Permeability and Zonulin Release by Binding to the Chemokine Receptor CXCR3. Gastroenterology 2008, 135, 194.e3–204.e3. [Google Scholar] [CrossRef]
- Sander, G.R.; Cummins, A.G.; Powell, B.C. Rapid disruption of intestinal barrier function by gliadin involves altered expression of apical junctional proteins. FEBS Lett. 2005, 579, 4851–4855. [Google Scholar] [CrossRef]
- Menard, S.; Lebreton, C.; Schumann, M.; Matysiak-Budnik, T.; Dugave, C.; Bouhnik, Y.; Malamut, G.; Cellier, C.; Allez, M.; Crenn, P.; et al. Paracellular versus Transcellular Intestinal Permeability to Gliadin Peptides in Active Celiac Disease. Am. J. Pathol. 2012, 180, 608–615. [Google Scholar] [CrossRef]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- Troisi, J.; Venutolo, G.; Terracciano, C.; Carri, M.D.; Di Micco, S.; Landolfi, A.; Fasano, A. The Therapeutic use of the Zonulin Inhibitor AT-1001 (Larazotide) for a Variety of Acute and Chronic Inflammatory Diseases. Curr. Med. Chem. 2021, 28, 5788–5807. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.L.; Nielsen, H.; Ejlertsen, T.; Engberg, J.; Günzel, D.; Zeitz, M.; Hering, N.A.; Fromm, M.; Schulzke, J.-D.; Bücker, R. Oral and Fecal Campylobacter concisus Strains Perturb Barrier Function by Apoptosis Induction in HT-29/B6 Intestinal Epithelial Cells. PLoS ONE 2011, 6, e23858. [Google Scholar] [CrossRef] [PubMed]
- Bücker, R.; Krug, S.; Fromm, A.; Nielsen, H.L.; Fromm, M.; Nielsen, H.; Schulzke, J.-D. Campylobacter fetusimpairs barrier function in HT-29/B6 cells through focal tight junction alterations and leaks. Ann. N. Y. Acad. Sci. 2017, 1405, 189–201. [Google Scholar] [CrossRef]
- Bücker, R.; Krug, S.; Moos, V.; Bojarski, C.; Schweiger, M.R.; Kerick, M.; Fromm, A.; Janßen, S.; Fromm, M.; Hering, N.A.; et al. Campylobacter jejuni impairs sodium transport and epithelial barrier function via cytokine release in human colon. Mucosal Immunol. 2017, 11, 474–485. [Google Scholar] [CrossRef]
- de Sá, F.D.L.; Backert, S.; Nattramilarasu, P.K.; Mousavi, S.; Sandle, G.I.; Bereswill, S.; Heimesaat, M.M.; Schulzke, J.-D.; Bücker, R. Vitamin D Reverses Disruption of Gut Epithelial Barrier Function Caused by Campylobacter jejuni. Int. J. Mol. Sci. 2021, 22, 8872. [Google Scholar] [CrossRef]
- Bücker, R.; Troeger, H.; Kleer, J.; Fromm, M.; Schulzke, J.-D. Arcobacter butzleriInduces Barrier Dysfunction in Intestinal HT-29/B6 Cells. J. Infect. Dis. 2009, 200, 756–764. [Google Scholar] [CrossRef]
- Karadas, G.; Bücker, R.; Sharbati, S.; Schulzke, J.-D.; Alter, T.; Gölz, G. Arcobacter butzleri isolates exhibit pathogenic potential in intestinal epithelial cell models. J. Appl. Microbiol. 2016, 120, 218–225. [Google Scholar] [CrossRef]
- Hering, N.A.; Richter, J.F.; Krug, S.M.; Günzel, D.; Fromm, A.; Bohn, E.; Rosenthal, R.; Bücker, R.; Fromm, M.; Troeger, H.; et al. Yersinia enterocolitica induces epithelial barrier dysfunction through regional tight junction changes in colonic HT-29/B6 cell monolayers. Lab. Investig. 2010, 91, 310–324. [Google Scholar] [CrossRef]
- Bücker, R.; Krug, S.M.; Rosenthal, R.; Günzel, D.; Fromm, A.; Zeitz, M.; Chakraborty, T.; Fromm, M.; Epple, H.-J.; Schulzke, J.-D. Aerolysin from Aeromonas hydrophila Perturbs Tight Junction Integrity and Cell Lesion Repair in Intestinal Epithelial HT-29/B6 Cells. J. Infect. Dis. 2011, 204, 1283–1292. [Google Scholar] [CrossRef]
- Bücker, R.; Zakrzewski, S.S.; Wiegand, S.; Pieper, R.; Fromm, A.; Fromm, M.; Günzel, D.; Schulzke, J.-D. Zinc prevents intestinal epithelial barrier dysfunction induced by alpha-hemolysin-producing Escherichia coli 536 infection in porcine colon. Vet. Microbiol. 2020, 243, 108632. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, S.; Zakrzewski, S.S.; Eichner, M.; Schulz, E.; Günzel, D.; Pieper, R.; Rosenthal, R.; Barmeyer, C.; Bleich, A.; Dobrindt, U.; et al. Zinc treatment is efficient against Escherichia coli α-haemolysin-induced intestinal leakage in mice. Sci. Rep. 2017, 7, srep45649. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Bosque, A.; Amat, C.; Polo, J.; Campbell, J.M.; Crenshaw, J.; Russell, L.; Moretó, M. Spray-Dried Animal Plasma Prevents the Effects of Staphylococcus aureus Enterotoxin B on Intestinal Barrier Function in Weaned Rats. J. Nutr. 2006, 136, 2838–2843. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, T.; Nakata, M.; Higashino, M.; Yamaguchi, M.; Kawabata, S. Group A Streptococcus exploits human plasminogen for bacterial translocation across epithelial barrier via tricellular tight junctions. Sci. Rep. 2016, 6, 20069. [Google Scholar] [CrossRef]
- Pujol, C.; Eugène, E.; Martin, L.D.S.; Nassif, X. Interaction of Neisseria meningitidis with a polarized monolayer of epithelial cells. Infect. Immun. 1997, 65, 4836–4842. [Google Scholar] [CrossRef]
- Shrestha, A.; Uzal, F.A.; McClane, B.A. The interaction of Clostridium perfringens enterotoxin with receptor claudins. Anaerobe 2016, 41, 18–26. [Google Scholar] [CrossRef]
- Gohari, I.M.; Li, J.; Navarro, M.; Uzal, F.; McClane, B.; Gohari, M.; Uzal, L. Effects of Claudin-1 on the Action of Clostridium perfringens Enterotoxin in Caco-2 Cells. Toxins 2019, 11, 582. [Google Scholar] [CrossRef]
- Eichner, M.; Augustin, C.; Fromm, A.; Piontek, A.; Walther, W.; Bücker, R.; Fromm, M.; Krause, G.; Schulzke, J.-D.; Günzel, D.; et al. In Colon Epithelia, Clostridium perfringens Enterotoxin Causes Focal Leaks by Targeting Claudins Which are Apically Accessible Due to Tight Junction Derangement. J. Infect. Dis. 2017, 217, 147–157. [Google Scholar] [CrossRef]
- Vecchio, A.J.; Rathnayake, S.S.; Stroud, R.M. Structural basis for Clostridium perfringens enterotoxin targeting of claudins at tight junctions in mammalian gut. Proc. Natl. Acad. Sci. USA 2021, 118, e2024651118. [Google Scholar] [CrossRef]
- Stephens, D.S.; Farley, M.M. Pathogenic Events during Infection of the Human Nasopharynx with Neisseria meningitidis and Haemophilus influenzae. Rev. Infect. Dis. 1991, 13, 22–33. [Google Scholar] [CrossRef]
- Clarke, T.B.; Francella, N.; Huegel, A.; Weiser, J.N. Invasive Bacterial Pathogens Exploit TLR-Mediated Downregulation of Tight Junction Components to Facilitate Translocation across the Epithelium. Cell Host Microbe 2011, 9, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.; Roscioli, E.; Murphy, J.; Ou, J.; Bassiouni, A.; Wormald, P.-J.; Vreugde, S. Staphylococcus aureusimpairs the airway epithelial barrier in vitro. Int. Forum Allergy Rhinol. 2015, 5, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Martens, K.; Seys, S.F.; Alpizar, Y.A.; Schrijvers, R.; Bullens, D.M.A.; Breynaert, C.; Lebeer, S.; Steelant, B. Staphylococcus aureus enterotoxin B disrupts nasal epithelial barrier integrity. Clin. Exp. Allergy 2021, 51, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ohnemus, U.; Kohrmeyer, K.; Houdek, P.; Rohde, H.; Wladykowski, E.; Vidal, S.; Horstkotte, M.A.; Aepfelbacher, M.; Kirschner, N.; Behne, M.J.; et al. Regulation of Epidermal Tight-Junctions (TJ) during Infection with Exfoliative Toxin-Negative Staphylococcus Strains. J. Investig. Dermatol. 2008, 128, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Tirado, C.; Maisey, K.; Rodríguez, F.E.; Reyes-Cerpa, S.; Reyes-López, F.E.; Imarai, M. Neisseria gonorrhoeae induced disruption of cell junction complexes in epithelial cells of the human genital tract. Microbes Infect. 2012, 14, 290–300. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, A.; Rochfort, K.D.; McDonnell, C.J.; Kerrigan, S.W.; Cummins, P.M. Staphylococcus aureus-mediated blood-brain barrier injury: Anin vitrohuman brain microvascular endothelial cell model. Cell. Microbiol. 2016, 19, e12664. [Google Scholar] [CrossRef]
- Liu, T.; Milia, E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J. Cell. Physiol. 2012, 227, 1438–1445. [Google Scholar] [CrossRef]
- McHale, T.M.; Garciarena, C.D.; Fagan, R.P.; Smith, S.G.J.; Martin-Loches, I.; Curley, G.; Fitzpatrick, F.; Kerrigan, S.W. Inhibition of Vascular Endothelial Cell Leak Following Escherichia coli Attachment in an Experimental Model of Sepsis. Crit. Care Med. 2018, 46, e805–e810. [Google Scholar] [CrossRef]
- Ye, P.; Harty, D.; Commandeur, Z.; Hunter, N. Binding of Streptococcus gordonii to oral epithelial monolayers increases paracellular barrier function. Microb. Pathog. 2013, 56, 53–59. [Google Scholar] [CrossRef]
- Takai, T.; Ikeda, S. Barrier Dysfunction Caused by Environmental Proteases in the Pathogenesis of Allergic Diseases. Allergol. Int. 2011, 60, 25–35. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial–Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell. Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Hadizamani, Y.; Gonzales, J.; Gorshkov, B.; Bodmer, T.; Berthiaume, Y.; Moehrlen, U.; Lode, H.; Huwer, H.; Hudel, M.; et al. Impact of Bacterial Toxins in the Lungs. Toxins 2020, 12, 223. [Google Scholar] [CrossRef] [PubMed]
- Freimuth, P.; Philipson, L.; Carson, S.D. The coxsackievirus and adenovirus receptor. Curr. Top. Microbiol. Immunol. 2008, 323, 67–87. [Google Scholar] [PubMed]
- Torres-Flores, J.M.; Arias, C.F. Tight Junctions Go Viral! Viruses 2015, 7, 5145–5154. [Google Scholar] [CrossRef]
- Obert, G.; Peiffer, I.; Servin, A.L. Rotavirus-induced structural and functional alterations in tight junctions of polarized intestinal caco-2 cell monolayers. J. Virol. 2000, 74, 4645–4651. [Google Scholar] [CrossRef]
- Nava, P.; Lopez, S.; Arias, C.F.; Islas, S.; Gonzalez-Mariscal, L. The rotavirus surface protein VP8 modulates the gate and fence function of tight junctions in epithelial cells. J. Cell Sci. 2004, 117, 5509–5519. [Google Scholar] [CrossRef]
- Torres-Flores, J.M.; Silva-Ayala, D.; Espinoza, M.A.; Lopez, S.; Arias, C.F. The tight junction protein JAM-A functions as coreceptor for rotavirus entry into MA104 cells. Virology 2015, 475, 172–178. [Google Scholar] [CrossRef]
- Tafazoli, F.; Zeng, C.Q.; Estes, M.K.; Magnusson, K.E.; Svensson, L. NSP4 enterotoxin of rotavirus induces paracellular leakage in polarized epithelial cells. J. Virol. 2001, 75, 1540–1546. [Google Scholar] [CrossRef]
- Zhao, Y.; Ran, Z.; Jiang, Q.; Hu, N.; Yu, B.; Zhu, L.; Shen, L.; Zhang, S.; Chen, L.; Chen, H.; et al. Vitamin D Alleviates Rotavirus Infection through a Microrna-155-5p Mediated Regulation of the TBK1/IRF3 Signaling Pathway In Vivo and In Vitro. Int. J. Mol. Sci. 2019, 20, 3562. [Google Scholar] [CrossRef]
- Colgate, E.R.; Haque, R.; Dickson, D.M.; Carmolli, M.P.; Mychaleckyj, J.C.; Nayak, U.; Qadri, F.; Alam, M.; Walsh, M.C.; Diehl, S.A.; et al. Delayed Dosing of Oral Rotavirus Vaccine Demonstrates Decreased Risk of Rotavirus Gastroenteritis Associated with Serum Zinc: A Randomized Controlled Trial. Clin. Infect. Dis. 2016, 63, 634–641. [Google Scholar] [CrossRef]
- Talavera, D.; Castillo, A.M.; Dominguez, M.C.; Gutierrez, A.E.; Meza, I. IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J. Gen. Virol. 2004, 85 Pt 7, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [PubMed]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7, 304ra141. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598.e8–1613.e8. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Hirsch, A.J.; Brien, J.D.; Uhrlaub, J.L.; Mason, P.W.; Wiley, C.; Nikolich-Zugich, J.; Nelson, J.A. West nile virus capsid degradation of claudin proteins disrupts epithelial barrier function. J. Virol. 2009, 83, 6125–6134. [Google Scholar] [CrossRef]
- Xu, Z.; Waeckerlin, R.; Urbanowski, M.D.; van Marle, G.; Hobman, T.C. West Nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS ONE 2012, 7, e37886. [Google Scholar] [CrossRef]
- Miranda, J.; Martin-Tapia, D.; Valdespino-Vazquez, Y.; Alarcon, L.; Espejel-Nunez, A.; Guzman-Huerta, M.; Muñoz-Medina, J.E.; Shibayama, M.; Chávez-Munguía, B.; Estrada-Gutiérrez, G.; et al. Syncytiotrophoblast of Placentae from Women with Zika Virus Infection Has Altered Tight Junction Protein Expression and Increased Paracellular Permeability. Cells 2019, 8, 1174. [Google Scholar] [CrossRef]
- Leda, A.; Bertrand, L.; Andras, I.E.; El-Hage, N.; Nair, M.; Toborek, M. Selective Disruption of the Blood–Brain Barrier by Zika Virus. Front. Microbiol. 2019, 10, 2158. [Google Scholar] [CrossRef]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Molina-Jiménez, F.; Barreiro, O.; Maldonado-Rodríguez, A.; Prieto, J.; Moreno-Otero, R.; Aldabe, R.; López-Cabrera, M.; Majano, P.L. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 2008, 48, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Molina-Jiménez, F.; Bartosch, B.; Cosset, F.L.; Lavillette, D.; Prieto, J.; Moreno-Otero, R.; Valenzuela-Fernández, A.; Aldabe, R.; López-Cabrera, M.; et al. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009, 83, 8012–8020. [Google Scholar] [CrossRef] [PubMed]
- Baktash, Y.; Madhav, A.; Coller, K.E.; Randall, G. Single Particle Imaging of Polarized Hepatoma Organoids upon Hepatitis C Virus Infection Reveals an Ordered and Sequential Entry Process. Cell Host Microbe 2018, 23, e5–e94. [Google Scholar] [CrossRef]
- Gupta, S.; Read, S.A.; Shackel, N.A.; Hebbard, L.; George, J.; Ahlenstiel, G. The Role of Micronutrients in the Infection and Subsequent Response to Hepatitis C Virus. Cells 2019, 8, 603. [Google Scholar] [CrossRef]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The avian influenza virus NS1 ESEV PDZ binding motif associates with Dlg1 and Scribble to disrupt cellular tight junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar] [CrossRef]
- Short, K.R.; Kasper, J.; Van Der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef]
- Abioye, A.I.; Bromage, S.; Fawzi, W. Effect of micronutrient supplements on influenza and other respiratory tract infections among adults: A systematic review and meta-analysis. BMJ Glob. Health 2021, 6, e003176. [Google Scholar] [CrossRef]
- Cowley, T.J.; Weiss, S.R. Murine coronavirus neuropathogenesis: Determinants of virulence. J. NeuroVirol. 2010, 16, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bleau, C.; Filliol, A.; Samson, M.; Lamontagne, L. Brain Invasion by Mouse Hepatitis Virus Depends on Impairment of Tight Junctions and Beta Interferon Production in Brain Microvascular Endothelial Cells. J. Virol. 2015, 89, 9896–9908. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Gao, J.; Zhu, L.; Yang, Q. Transmissible gastroenteritis virus and porcine epidemic diarrhoea virus infection induces dramatic changes in the tight junctions and microfilaments of polarized IPEC-J2 cells. Virus Res. 2014, 192, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Guo, L.; Zhang, J.; Xu, Y.; Gu, W.; Feng, L.; Wang, Y. Tight Junction Protein Occludin Is a Porcine Epidemic Diarrhea Virus Entry Factor. J. Virol. 2017, 91, e00202-17. [Google Scholar] [CrossRef]
- Langel, S.; Paim, F.C.; Alhamo, M.A.; Lager, K.M.; Vlasova, A.N.; Saif, L.J. Oral vitamin A supplementation of porcine epidemic diarrhea virus infected gilts enhances IgA and lactogenic immune protection of nursing piglets. Vet. Res. 2019, 50, 1–16. [Google Scholar] [CrossRef]
- Yang, J.; Tian, G.; Chen, D.; Mao, X.; He, J.; Zheng, P.; Yu, J.; Luo, Y.; Luo, J.; Huang, Z.; et al. 1,25-Dihydroxyvitamin D3 inhibits porcine epidemic diarrhea virus replication by regulating cell cycle resumption in IPEC-J2 porcine epithelial cells. Microb. Pathog. 2021, 158, 105017. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, T.; Li, S.; Meng, Y.; Tan, Z.; Wu, M.; Yi, D.; Wang, L.; Zhao, D.; Hou, Y. Protective Effect of Zinc Oxide and Its Association with Neutrophil Degranulation in Piglets Infected with Porcine Epidemic Diarrhea Virus. Oxidative Med. Cell. Longev. 2021, 2021, 3055810. [Google Scholar] [CrossRef]
- Chai, W.; Zakrzewski, S.S.; Günzel, D.; Pieper, R.; Wang, Z.; Twardziok, S.; Janczyk, P.; Osterrieder, N.; Burwinkel, M. High-dose dietary zinc oxide mitigates infection with transmissible gastroenteritis virus in piglets. BMC Vet. Res. 2014, 10, 75. [Google Scholar] [CrossRef]
- Wei, Z.; Burwinkel, M.; Palissa, C.; Ephraim, E.; Schmidt, M.F. Antiviral activity of zinc salts against transmissible gastroenteritis virus in vitro. Vet. Microbiol. 2012, 160, 468–472. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.T.; Siu, Y.L.; Chan, W.L.; Schlüter, M.A.; Liu, C.J.; Peiris, J.M.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell. 2010, 21, 3838–3852. [Google Scholar] [CrossRef] [PubMed]
- Toto, A.; Ma, S.; Malagrinò, F.; Visconti, L.; Pagano, L.; Stromgaard, K.; Gianni, S. Comparing the binding properties of peptides mimicking the Envelope protein of SARS-CoV and SARS-CoV-2 to the PDZ domain of the tight junction-associated PALS1 protein. Protein Sci. 2020, 29, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- De Maio, F.; Cascio, E.L.; Babini, G.; Sali, M.; Della Longa, S.; Tilocca, B.; Roncada, P.; Arcovito, A.; Sanguinetti, M.; Scambia, G.; et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect. 2020, 22, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Cascio, E.L.; Toto, A.; Babini, G.; De Maio, F.; Sanguinetti, M.; Mordente, A.; Della Longa, S.; Arcovito, A. Structural determinants driving the binding process between PDZ domain of wild type human PALS1 protein and SLiM sequences of SARS-CoV E proteins. Comput. Struct. Biotechnol. J. 2021, 19, 1838–1847. [Google Scholar] [CrossRef]
- Shepley-McTaggart, A.; Sagum, C.A.; Oliva, I.; Rybakovsky, E.; DiGuilio, K.; Liang, J.; Bedford, M.T.; Cassel, J.; Sudol, M.; Mullin, J.M.; et al. SARS-CoV-2 Envelope (E) Protein Interacts with PDZ-Domain-2 of Host Tight Junction Protein ZO1. PLoS ONE 2021, 16, e0251955. [Google Scholar] [CrossRef]
- Bae, J.H.; Choe, H.J.; Holick, M.F.; Lim, S. Association of vitamin D status with COVID-19 and its severity. Rev. Endocr. Metab. Disord. 2022, 23, 1–21. [Google Scholar] [CrossRef]
- Qayyum, S.; Mohammad, T.; Slominski, R.M.; Hassan, I.; Tuckey, R.C.; Raman, C.; Slominski, A.T. Vitamin D and lumisterol novel metabolites can inhibit SARS-CoV-2 replication machinery enzymes. Am. J. Physiol. Metab. 2021, 321, E246–E251. [Google Scholar] [CrossRef]
- Shalayel, M.H.; Al-Mazaideh, G.M.; Aladaileh, S.H.; Al-Swailmi, F.K.; Al-Thiabat, M.G. Vitamin D is a potential inhibitor of COVID-19: In silico molecular docking to the binding site of SARS-CoV-2 endoribonuclease Nsp15. Pak. J. Pharm. Sci. 2020, 33, 2179–2186. [Google Scholar]
- Panchariya, L.; Khan, W.A.; Kuila, S.; Sonkar, K.; Sahoo, S.; Ghoshal, A.; Kumar, A.; Verma, D.K.; Hasan, A.; Khan, M.A.; et al. Zinc2+ ion inhibits SARS-CoV-2 main protease and viral replication in vitro. Chem. Commun. 2021, 57, 10083–10086. [Google Scholar] [CrossRef] [PubMed]
- Sosula, L. Retinal capillary junctions: Ultrastructural tight junction artefacts induced by sodium ions and membrane reduction in streptozotocin diabetes. Cell Tissue Res. 1975, 161, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: Vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 1998, 47, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.; Antonetti, D. Alterations to the Blood–Retinal Barrier in Diabetes: Cytokines and Reactive Oxygen Species. Antioxid. Redox Signal. 2011, 15, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Hughes, J.M.; Vogels, I.M.; Schalkwijk, C.G.; Van Noorden, C.J.; Schlingemann, R.O. Altered expression of genes related to blood–retina barrier disruption in streptozotocin-induced diabetes. Exp. Eye Res. 2009, 89, 4–15. [Google Scholar] [CrossRef]
- Jiang, Q.-W.; Kaili, D.; Freeman, J.; Lei, C.-Y.; Geng, B.-C.; Tan, T.; He, J.-F.; Shi, Z.; Ma, J.-J.; Luo, Y.-H.; et al. Diabetes inhibits corneal epithelial cell migration and tight junction formation in mice and human via increasing ROS and impairing Akt signaling. Acta Pharmacol. Sin. 2019, 40, 1205–1211. [Google Scholar] [CrossRef]
- Banks, W.A. The Blood-Brain Barrier Interface in Diabetes Mellitus: Dysfunctions, Mechanisms and Approaches to Treatment. Curr. Pharm. Des. 2020, 26, 1438–1447. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar]
- Taïlé, J.; Patché, J.; Veeren, B.; Gonthier, M.-P. Hyperglycemic Condition Causes Pro-Inflammatory and Permeability Alterations Associated with Monocyte Recruitment and Deregulated NFκB/PPARγ Pathways on Cerebral Endothelial Cells: Evidence for Polyphenols Uptake and Protective Effect. Int. J. Mol. Sci. 2021, 22, 1385. [Google Scholar] [CrossRef]
- Yoo, D.Y.; Yim, H.S.; Jung, H.Y.; Nam, S.M.; Kim, J.W.; Choi, J.H.; Seong, J.K.; Yoon, Y.S.; Kim, D.W.; Hwang, I.K. Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J. Vet. Med. Sci. 2016, 78, 957–962. [Google Scholar] [CrossRef]
- Nascimento, J.C.; Matheus, V.A.; Oliveira, R.B.; Tada, S.F.S.; Collares-Buzato, C.B. High-Fat Diet Induces Disruption of the Tight Junction-Mediated Paracellular Barrier in the Proximal Small Intestine Before the Onset of Type 2 Diabetes and Endotoxemia. Dig. Dis. Sci. 2021, 66, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Lundeen, T.F.; Norwood, K.M.; Brooks, H.L.; Egleton, R.D. Increased blood–brain barrier permeability and altered tight junctions in experimental diabetes in the rat: Contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 2006, 50, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.-E.; Vaillant, J.-C.; Oppert, J.-M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Horton, F.; Wright, J.; Smith, L.; Hinton, P.J.; Robertson, M.D. Increased intestinal permeability to oral chromium (51Cr)-EDTA in human Type 2 diabetes. Diabet. Med. 2013, 31, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Kanazawa, A.; Watada, H. Type 2 Diabetes and Bacteremia. Ann. Nutr. Metab. 2017, 71 (Suppl. S1), 17–22. [Google Scholar] [CrossRef]
- Neu, J.; Reverte, C.M.; Mackey, A.D.; Liboni, K.; Tuhacek-Tenace, L.M.; Hatch, M.; Li, N.; Caicedo, R.A.; Schatz, D.A.; Atkinson, M. Changes in Intestinal Morphology and Permeability in the BioBreeding Rat Before the Onset of Type 1 Diabetes. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 589–595. [Google Scholar] [CrossRef]
- Shi, Y.; Qian, J.; Zhang, F.; Jia, B.; Liu, X.; Hu, Y.; Zhang, Q.; Yang, Y.; Sun, D.; Jiang, L. Low molecular weight heparin (nadroparin) improves placental permeability in rats with gestational diabetes mellitus via reduction of tight junction factors. Mol. Med. Rep. 2019, 21, 623–630. [Google Scholar] [CrossRef]
- Wan, H.; Winton, H.L.; Soeller, C.; Taylor, G.W.; Gruenert, D.C.; Thompson, P.J.; Cannell, M.B.; Stewart, G.A.; Garrod, D.R.; Robinson, C. The transmembrane protein occludin of epithelial tight junctions is a functional target for serine peptidases from faecal pellets of Dermatophagoides pteronyssinus. Clin. Exp. Allergy 2001, 31, 279–294. [Google Scholar] [CrossRef]
- Wan, H.; Winton, H.L.; Soeller, C.; Tovey, E.R.; Gruenert, D.C.; Thompson, P.J.; Stewart, G.A.; Taylor, G.W.; Garrod, D.R.; Cannell, M.B.; et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Investig. 1999, 104, 123–133. [Google Scholar] [CrossRef]
- Baker, S.F.; Yin, Y.; Runswick, S.K.; Stewart, G.A.; Thompson, P.J.; Garrod, D.R.; Robinson, C. Peptidase allergen Der p 1 initiates apoptosis of epithelial cells independently of tight junction proteolysis. Mol. Membr. Biol. 2003, 20, 71–81. [Google Scholar] [CrossRef]
- Henriquez, O.A.; Bs, K.D.B.; Hoddeson, E.K.; Parkos, C.A.; Nusrat, A.; Wise, S.K. House dust mite allergen Der p 1 effects on sinonasal epithelial tight junctions. Int. Forum Allergy Rhinol. 2013, 3, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Steelant, B.; Farre, R.; Wawrzyniak, P.; Belmans, J.; Dekimpe, E.; Vanheel, H.; Van Gerven, L.; Krohn, I.K.; Bullens, D.M.A.; Ceuppens, J.; et al. Impaired barrier function in patients with house dust mite–induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J. Allergy Clin. Immunol. 2016, 137, 1043–1053.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-C.; Tsai, J.-J.; Kuo, C.-Y.; Chen, H.-M.; Kao, S.-H. Non-proteolytic house dust mite allergen, Der p 2, upregulated expression of tight junction molecule claudin-2 associated with Akt/GSK-3β/β-catenin signaling pathway. J. Cell. Biochem. 2011, 112, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Tulic, M.K.; Vivinus-Nébot, M.; Rekima, A.; Medeiros, S.R.; Bonnart, C.; Shi, H.; Walker, A.; Dainese, R.; Boyer, J.; Vergnolle, N.; et al. Presence of commensal house dust mite allergen in human gastrointestinal tract: A potential contributor to intestinal barrier dysfunction. Gut 2016, 65, 757–766. [Google Scholar] [CrossRef]
- Ma, S.W.; Ende, J.A.; Alvarado, R.; Christensen, J.M.; Kalish, L.; Sacks, R.; Campbell, R.; Rimmer, J.; Harvey, R. Topical Vitamin D May Modulate Human Sinonasal Mucosal Responses to House Dust Mite Antigen. Am. J. Rhinol. Allergy 2020, 34, 471–481. [Google Scholar] [CrossRef]
- Bates, P.J.; Farr, S.J.; Nicholls, P.J. Effect of Cotton, Hemp, and Flax Dust Extracts on Lung Permeability in the Guinea Pig. Exp. Lung Res. 1995, 21, 643–665. [Google Scholar] [CrossRef]
- Robinson, C.; Baker, S.F.; Garrod, D.R. Peptidase allergens, occludin and claudins. Do their interactions facilitate the development of hypersensitivity reactions at mucosal surfaces? Clin. Exp. Allergy 2001, 31, 186–192. [Google Scholar] [CrossRef]
- Prasad, A.S. Discovery of Human Zinc Deficiency: Its Impact on Human Health and Disease. Adv. Nutr. 2013, 4, 176–190. [Google Scholar] [CrossRef]
- Davidson, G.; Kritas, S.; Butler, R. Stressed mucosa. Nutr. Support Infants Child. Risk 2007, 59, 133–142; discussion 143–146. [Google Scholar] [CrossRef]
- Finamore, A.; Massimi, M.; Devirgiliis, L.C.; Mengheri, E. Zinc Deficiency Induces Membrane Barrier Damage and Increases Neutrophil Transmigration in Caco-2 Cells. J. Nutr. 2008, 138, 1664–1670. [Google Scholar] [CrossRef]
- Ranaldi, G.; Ferruzza, S.; Canali, R.; Leoni, G.; Zalewski, P.D.; Sambuy, Y.; Perozzi, G.; Murgia, C. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J. Nutr. Biochem. 2013, 24, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, H.; Kashiwaya, M.; Shinoki, A.; Lee, J.-S.; Hayashi, K.; Hara, H.; Ishizuka, S. Marginal Zinc Deficiency Exacerbates Experimental Colitis Induced by Dextran Sulfate Sodium in Rats. J. Nutr. 2011, 141, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, G.J.; Aydemir, T.B.; Troche, C.; Martin, A.B.; Chang, S.-M.; Cousins, R.J. Influence of ZIP14 (slc39A14) on intestinal zinc processing and barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G171–G178. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; McClain, C.J.; Cave, M.; Kang, Y.J.; Zhou, Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G625–G633. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Mehta, A.; Jabber, W.S.; Fan, X.; Guidot, D.M. Zinc Deficiency Mediates Alcohol-Induced Alveolar Epithelial and Macrophage Dysfunction in Rats. Am. J. Respir. Cell Mol. Biol. 2009, 41, 207–216. [Google Scholar] [CrossRef]
- Ozutemiz, A.O.; Aydin, H.H.; Isler, M.; Celik, H.A.; Batur, Y. Effect of omeprazole on plasma zinc levels after oral zinc administration. Indian J. Gastroenterol. 2002, 21, 216–218. [Google Scholar]
- Farrell, C.P.; Morgan, M.; Rudolph, D.S.; Hwang, A.; Albert, N.E.; Valenzano, M.C.; Wang, X.; Mercogliano, G.; Mullin, J.M. Proton Pump Inhibitors Interfere with Zinc Absorption and Zinc Body Stores. Gastroenterol. Res. 2011, 4, 243–251. [Google Scholar] [CrossRef]
- Hara, H.; Konishi, A.; Kasai, T. Contribution of the cecum and colon to zinc absorption in rats. J. Nutr. 2000, 130, 83–89. [Google Scholar] [CrossRef]
- Serfaty-Lacrosniere, C.; Wood, R.J.; Voytko, D.; Saltzman, J.R.; Pedrosa, M.; Sepe, T.E.; Russell, R.R. Hypochlorhydria from short-term omeprazole treatment does not inhibit intestinal absorption of calcium, phosphorus, magnesium or zinc from food in humans. J. Am. Coll. Nutr. 1995, 14, 364–368. [Google Scholar] [CrossRef]
- Joshaghani, H.; Amiriani, T.; Vaghari, G.; Besharat, S.; Molana, A.; Badeleh, M.; Roshandel, G. Effects of omeprazole consumption on serum levels of trace elements. J. Trace Elem. Med. Biol. 2012, 26, 234–237. [Google Scholar] [CrossRef]
- Saka, Y.; Naruse, T.; Matsumoto, J.; Takeda, Y.; Onogi, C.; Yokoi, J.; Kato, A.; Tawada, N.; Noda, Y.; Niwa, S.; et al. Low Serum Zinc Concentration Is Associated with Infection Particularly in Patients with Stage 5 Chronic Kidney Disease Medicated with Proton Pump Inhibitors. J. Ren. Nutr. 2021, 31, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Snyder, L.; Arora, J. Vitamin A and vitamin D regulate the microbial complexity, barrier function, and the mucosal immune responses to ensure intestinal homeostasis. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Yang, Q.; Tan, B.; Dong, X.; Chi, S.; Liu, H.; Zhang, S. Dietary vitamin A deficiency reduces growth performance, immune function of intestine, and alters tight junction proteins of intestine for juvenile hybrid grouper (Epinephelus fuscoguttatus ♀ × Epinephelus lanceolatus ♂). Fish Shellfish Immunol. 2020, 107 Pt A, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.-D.; Zhou, X.-Q.; Zhang, L.; Liu, Y.; Wu, P.; Jiang, J.; Kuang, S.-Y.; Tang, L.; Tang, W.-N.; Zhang, Y.-A.; et al. Vitamin A deficiency impairs intestinal physical barrier function of fish. Fish Shellfish Immunol. 2019, 87, 546–558. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Cui, T.; Yang, T.; Liu, L.; Li, T.; Chen, J. Retinoic Acid Facilitates Toll-Like Receptor 4 Expression to Improve Intestinal Barrier Function through Retinoic Acid Receptor Beta. Cell. Physiol. Biochem. 2017, 42, 1390–1406. [Google Scholar] [CrossRef]
- Movahedan, A.; Afsharkhamseh, N.; Sagha, H.M.; Shah, J.R.; Milani, B.Y.; Milani, F.Y.; Logothetis, H.D.; Chan, C.-C.; Djalilian, A.R. Loss of Notch1 Disrupts the Barrier Repair in the Corneal Epithelium. PLoS ONE 2013, 8, e69113. [Google Scholar] [CrossRef]
- Chung, S.S.; Choi, C.; Wang, X.; Hallock, L.; Wolgemuth, D.J. Aberrant distribution of junctional complex components in retinoic acid receptor alpha-deficient mice. Microsc. Res. Tech. 2009, 73, 583–596. [Google Scholar] [CrossRef]
- Huang, H.F.; Yang, C.S.; Meyenhofer, M.; Gould, S.; Boccabella, A.V. Disruption of Sustentacular (Sertoli) Cell Tight Junctions and Regression of Spermatogenesis in Vitamin-A-Defícient Rats. Acta Anat. 1988, 133, 10–15. [Google Scholar] [CrossRef]
- Rearick, J.I.; Jetten, A.M. Effect of substratum and retinoids upon the mucosecretory differentiation of airway epithelial cells in vitro. Environ. Health Perspect. 1989, 80, 229–237. [Google Scholar] [CrossRef]
- Xiao, L.; Cui, T.; Liu, S.; Chen, B.; Wang, Y.; Yang, T.; Li, T.; Chen, J. Vitamin A supplementation improves the intestinal mucosal barrier and facilitates the expression of tight junction proteins in rats with diarrhea. Nutrition 2019, 57, 97–108. [Google Scholar] [CrossRef]
- Feng, D.; Chen, B.; Zeng, B.; Xiao, L.; Yan, J.; Yang, T.; Zhu, J.; Li, T.; Wang, L.; Wei, H.; et al. Fecal microbiota from children with vitamin A deficiency impair colonic barrier function in germ-free mice: The possible role of alterative bile acid metabolites. Nutrition 2021, 90, 111274. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Morales, C.R. Effects of vitamin A deficiency on the inter?Sertoli cell tight junctions and on the germ cell population. Microsc. Res. Tech. 1992, 20, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Gorodeski, G.I.; Eckert, R.L.; Pal, D.; Utian, W.H.; Rorke, E.A. Retinoids regulate tight junctional resistance of cultured human cervical cells. Am. J. Physiol. 1997, 273, C1707–C1713. [Google Scholar] [CrossRef] [PubMed]
- Hetta, H.F.; Muhammad, K.; El-Masry, E.A.; Taha, A.E.; Ahmed, E.A.; Phares, C.; Kader, H.A.; Waheed, Y.; Zahran, A.M.; Yahia, R.; et al. The interplay between vitamin D and COVID-19: Protective or bystander? Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2131–2145. [Google Scholar]
- Liu, N.; Sun, J.; Wang, X.; Zhang, T.; Zhao, M.; Li, H. Low vitamin D status is associated with coronavirus disease 2019 outcomes: A systematic review and meta-analysis. Int. J. Infect. Dis. 2021, 104, 58–64. [Google Scholar] [CrossRef]
- Sizar, O.; Khare, S.; Goyal, A.; Bansal, P.; Givler, A. Vitamin D Deficiency. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ames, B.N.; Grant, W.B.; Willett, W.C. Does the High Prevalence of Vitamin D Deficiency in African Americans Contribute to Health Disparities? Nutrients 2021, 13, 499. [Google Scholar] [CrossRef]
- Liu, F.H.; Li, S.S.; Li, X.X.; Wang, S.; Li, M.G.; Guan, L.; Luan, T.G.; Liu, Z.G.; Liu, Z.J.; Yang, P.C. Vitamin D3 induces vitamin D receptor and HDAC11 binding to relieve the promoter of the tight junction proteins. Oncotarget 2017, 8, 58781–58789. [Google Scholar] [CrossRef]
- Assa, A.; Vong, L.; Pinnell, L.J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D Deficiency Promotes Epithelial Barrier Dysfunction and Intestinal Inflammation. J. Infect. Dis. 2014, 210, 1296–1305. [Google Scholar] [CrossRef]
- Yang, K.; Zhu, J.; Wu, J.; Zhong, Y.; Shen, X.; Ms, B.P.; Cai, W. Maternal Vitamin D Deficiency Increases Intestinal Permeability and Programs Wnt/β-Catenin Pathway in BALB/C Mice. J. Parenter. Enter. Nutr. 2021, 45, 102–114. [Google Scholar] [CrossRef]
- Kellermann, L.; Jensen, K.B.; Bergenheim, F.; Gubatan, J.; Chou, N.D.; Moss, A.; Nielsen, O.H. Mucosal vitamin D signaling in inflammatory bowel disease. Autoimmun. Rev. 2020, 19, 102672. [Google Scholar] [CrossRef]
- Yeung, C.-Y.; Chiau, J.-S.C.; Cheng, M.-L.; Chan, W.-T.; Jiang, C.-B.; Chang, S.-W.; Liu, C.-Y.; Chang, C.-W.; Lee, H.-C. Effects of Vitamin D-Deficient Diet on Intestinal Epithelial Integrity and Zonulin Expression in a C57BL/6 Mouse Model. Front. Med. 2021, 8, 649818. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Li, X.; Du, J.; Ge, X.; Sun, Y.; Li, X.; Xun, Z.; Liu, W.; Wang, Z.-Y.; Li, Y.C. Vitamin D Deficiency Exacerbates Colonic Inflammation Due to Activation of the Local Renin–Angiotensin System in the Colon. Dig. Dis. Sci. 2021, 66, 3813–3821. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, I.; Turan, N.; Stein, D.G.; Wali, B. Vitamin D deficiency increases blood-brain barrier dysfunction after ischemic stroke in male rats. Exp. Neurol. 2019, 312, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Solidoro, P.; Bellocchia, M.; Facchini, F. The immunobiological and clinical role of vitamin D in obstructive lung diseases. Minerva Med. 2016, 107 (Suppl. S1), 12–19. [Google Scholar] [PubMed]
- Chen, H.; Lu, R.; Zhang, Y.-G.; Sun, J. Vitamin D Receptor Deletion Leads to the Destruction of Tight and Adherens Junctions in Lungs. Tissue Barriers 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Gorman, S.; Buckley, A.G.; Ling, K.-M.; Berry, L.J.; Fear, V.; Stick, S.; Larcombe, A.; Kicic, A.; Hart, P.H. Vitamin D supplementation of initially vitamin D-deficient mice diminishes lung inflammation with limited effects on pulmonary epithelial integrity. Physiol. Rep. 2017, 5, e13371. [Google Scholar] [CrossRef]
- Hambidge, K.M. Zinc and diarrhea. Acta Paediatr. Suppl. 1992, 81, 82–86. [Google Scholar] [CrossRef]
- Folwaczny, C. Zinc and Diarrhea in Infants. J. Trace Elem. Med. Biol. 1997, 11, 116–122. [Google Scholar] [CrossRef]
- Lamberti, L.M.; Walker, C.L.F.; Chan, K.Y.; Jian, W.-Y.; Black, R.E. Oral Zinc Supplementation for the Treatment of Acute Diarrhea in Children: A Systematic Review and Meta-Analysis. Nutrients 2013, 5, 4715–4740. [Google Scholar] [CrossRef]
- Hering, N.A.; Schulzke, J.-D. Therapeutic Options to Modulate Barrier Defects in Inflammatory Bowel Disease. Dig. Dis. 2009, 27, 450–454. [Google Scholar] [CrossRef]
- Amasheh, M.; Andres, S.; Amasheh, S.; Fromm, M.; Schulzke, J.-D. Barrier Effects of Nutritional Factors. Ann. N. Y. Acad. Sci. 2009, 1165, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Skrovanek, S.; DiGuilio, K.; Bailey, R.; Huntington, W.; Urbas, R.; Mayilvaganan, B.; Mercogliano, G.; Mullin, J.M. Zinc and gastrointestinal disease. World J. Gastrointest. Pathophysiol. 2014, 5, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, M.C.; DiGuilio, K.; Mercado, J.; Teter, M.; To, J.; Ferraro, B.; Mixson, B.; Manley, I.; Baker, V.; Moore, B.A.; et al. Remodeling of Tight Junctions and Enhancement of Barrier Integrity of the CACO-2 Intestinal Epithelial Cell Layer by Micronutrients. PLoS ONE 2015, 10, e0133926. [Google Scholar] [CrossRef]
- Shao, Y.-X.; Lei, Z.; Wolf, P.G.; Gao, Y.; Guo, Y.-M.; Zhang, B.-K. Zinc Supplementation, via GPR39, Upregulates PKCζ to Protect Intestinal Barrier Integrity in Caco-2 Cells Challenged by Salmonella enterica Serovar Typhimurium. J. Nutr. 2017, 147, 1282–1289. [Google Scholar] [CrossRef]
- Buddington, R.; Wong, T.; Howard, S. Paracellular Filtration Secretion Driven by Mechanical Force Contributes to Small Intestinal Fluid Dynamics. Med. Sci. 2021, 9, 9. [Google Scholar] [CrossRef]
- Cohen, L.Y.; Sekler, I.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, controls proliferation and differentiation of colonocytes and thereby tight junction formation in the colon. Cell Death Dis. 2014, 5, e1307. [Google Scholar] [CrossRef]
- Zhang, B.; Guo, Y. Supplemental zinc reduced intestinal permeability by enhancing occludin and zonula occludens protein-1 (ZO-1) expression in weaning piglets. Br. J. Nutr. 2009, 102, 687–693. [Google Scholar] [CrossRef]
- Grilli, E.; Tugnoli, B.; Vitari, F.; Domeneghini, C.; Morlacchini, M.; Piva, A.; Prandini, A. Low doses of microencapsulated zinc oxide improve performance and modulate the ileum architecture, inflammatory cytokines and tight junctions expression of weaned pigs. Animals 2015, 9, 1760–1768. [Google Scholar] [CrossRef]
- Peng, P.; Deng, D.; Chen, S.; Li, C.; Luo, J.; Romeo, A.; Li, T.; Tang, X.; Fang, R. The Effects of Dietary Porous Zinc Oxide Supplementation on Growth Performance, Inflammatory Cytokines and Tight Junction’s Gene Expression in Early-Weaned Piglets. J. Nutr. Sci. Vitaminol. 2020, 66, 311–318. [Google Scholar] [CrossRef]
- Zhu, C.; Lv, H.; Chen, Z.; Wang, L.; Wu, X.; Chen, Z.; Zhang, W.; Liang, R.; Jiang, Z. Dietary Zinc Oxide Modulates Antioxidant Capacity, Small Intestine Development, and Jejunal Gene Expression in Weaned Piglets. Biol. Trace Elem. Res. 2017, 175, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Van Noten, N.; DeGroote, J.; Romeo, A.; Vermeir, P.; Michiels, J. Effect of zinc oxide sources and dosages on gut microbiota and integrity of weaned piglets. J. Anim. Physiol. Anim. Nutr. 2018, 103, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Zhao, H.; Liu, G.; Chen, X.; Wu, B.; Tian, G.; Cai, J.; Jia, G. Effect of Zinc Supplementation on Growth Performance, Intestinal Development, and Intestinal Barrier-Related Gene Expression in Pekin Ducks. Biol. Trace Elem. Res. 2017, 183, 351–360. [Google Scholar] [CrossRef]
- Fan, P.; Tan, Y.; Jin, K.; Lin, C.; Xia, S.; Han, B.; Zhang, F.; Wu, L.; Ma, X. Supplemental lipoic acid relieves post-weaning diarrhoea by decreasing intestinal permeability in rats. J. Anim. Physiol. Anim. Nutr. 2017, 101, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Bzik, V.A.; Medani, M.; Baird, A.; Winter, D.C.; Brayden, D.J. Mechanisms of action of zinc on rat intestinal epithelial electrogenic ion secretion: Insights into its antidiarrhoeal actions. J. Pharm. Pharmacol. 2012, 64, 644–653. [Google Scholar] [CrossRef]
- Ranaldi, G.; Caprini, V.; Sambuy, Y.; Perozzi, G.; Murgia, C. Intracellular zinc stores protect the intestinal epithelium from Ochratoxin A toxicity. Toxicol. Vitr. 2009, 23, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Roselli, M.; Finamore, A.; Garaguso, I.; Britti, M.S.; Mengheri, E. Zinc Oxide Protects Cultured Enterocytes from the Damage Induced by Escherichia coli. J. Nutr. 2003, 133, 4077–4082. [Google Scholar] [CrossRef]
- Choudhry, N.; Scott, F.; Edgar, M.; Sanger, G.; Kelly, P. Reversal of Pathogen-Induced Barrier Defects in Intestinal Epithelial Cells by Contra-pathogenicity Agents. Dig. Dis. Sci. 2021, 66, 88–104. [Google Scholar] [CrossRef]
- Sarkar, P.; Saha, T.; Sheikh, I.A.; Chakraborty, S.; Aoun, J.; Chakrabarti, M.K.; Rajendran, V.M.; Ameen, N.A.; Dutta, S.; Hoque, K.M. Zinc ameliorates intestinal barrier dysfunctions in shigellosis by reinstating claudin-2 and -4 on the membranes. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G229–G246. [Google Scholar] [CrossRef]
- Song, Z.-H.; Ke, Y.-L.; Xiao, K.; Jiao, L.-F.; Hong, Q.-H.; Hu, C.-H. Diosmectite–zinc oxide composite improves intestinal barrier restoration and modulates TGF-β1, ERK1/2, and Akt in piglets after acetic acid challenge. J. Anim. Sci. 2015, 93, 1599–1607. [Google Scholar] [CrossRef]
- Sturniolo, G.C.; Fries, W.; Mazzon, E.; Di Leo, V.; Barollo, M.; D’Inca, R. Effect of zinc supplementation on intestinal permeability in experimental colitis. J. Lab. Clin. Med. 2002, 139, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Li, Q.; Sun, Q.; Zhang, W.; Zhang, J.; Sun, X.; Yin, X.; Zhang, X.; Zhou, Z. Preventing Gut Leakiness and Endotoxemia Contributes to the Protective Effect of Zinc on Alcohol-Induced Steatohepatitis in Rats. J. Nutr. 2015, 145, 2690–2698. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.; Darmon, N.; Chappuis, P.; Candalh, C.; Blaton, M.A.; Bouchaud, C.; Heyman, M. Intestinal paracellular permeability during malnutrition in guinea pigs: Effect of high dietary zinc. Gut 1996, 39, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wen, M.; Zhao, H.; Liu, G.; Chen, X.; Tian, G.; Cai, J.; Jia, G. Effect of zinc supplementation on growth performance, intestinal development, and intestinal barrier function in Pekin ducks with lipopolysaccharide challenge. Poult. Sci. 2021, 100, 101462. [Google Scholar] [CrossRef]
- Bortoluzzi, C.; Lumpkins, B.; Mathis, G.; França, M.; King, W.; Graugnard, D.; Dawson, K.; Applegate, T. Zinc source modulates intestinal inflammation and intestinal integrity of broiler chickens challenged with coccidia and Clostridium perfringens. Poult. Sci. 2019, 98, 2211–2219. [Google Scholar] [CrossRef]
- Davison, G.; Marchbank, T.; March, D.S.; Thatcher, R.; Playford, R.J. Zinc carnosine works with bovine colostrum in truncating heavy exercise–induced increase in gut permeability in healthy volunteers. Am. J. Clin. Nutr. 2016, 104, 526–536. [Google Scholar] [CrossRef]
- Ben Lagha, A.; Yang, Y.; Trivedi, H.M.; Masters, J.G.; Grenier, D. A Dual Zinc plus Arginine formulation protects against tumor necrosis factor-alpha-induced barrier dysfunction and enhances cell proliferation and migration in an in vitro gingival keratinocyte model. Arch. Oral Biol. 2021, 126, 105126. [Google Scholar] [CrossRef]
- Thomas, P.; Converse, A.; Berg, H. ZIP9, a novel membrane androgen receptor and zinc transporter protein. Gen. Comp. Endocrinol. 2018, 257, 130–136. [Google Scholar] [CrossRef]
- Song, Y.; Xue, Y.; Liu, X.; Wang, P.; Liu, L. Effects of acute exposure to aluminum on blood–brain barrier and the protection of zinc. Neurosci. Lett. 2008, 445, 42–46. [Google Scholar] [CrossRef]
- Qi, Z.; Liang, J.; Pan, R.; Dong, W.; Shen, J.; Yang, Y.; Zhao, Y.; Shi, W.; Luo, Y.; Ji, X.; et al. Zinc contributes to acute cerebral ischemia-induced blood–brain barrier disruption. Neurobiol. Dis. 2016, 95, 12–21. [Google Scholar] [CrossRef]
- Wang, X.; Valenzano, M.C.; Mercado, J.M.; Zurbach, E.P.; Flounders, C.J.; Mullin, J.M. Zinc enhancement of LLC-PK1 renal epithelial barrier function. Clin. Nutr. 2014, 33, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Carr, G.; Wright, J.A.; Simmons, N.L. Epithelial Barrier Resistance is Increased by the Divalent Cation Zinc in Cultured MDCKII Epithelial Monolayers. J. Membr. Biol. 2010, 237, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Jacquillet, G.; Barbier, O.; Cougnon, M.; Tauc, M.; Namorado, M.C.; Martin, D.; Reyes, J.L.; Poujeol, P. Zinc protects renal function during cadmium intoxication in the rat. Am. J. Physiol. Physiol. 2006, 290, F127–F137. [Google Scholar] [CrossRef] [PubMed]
- Mercado, J.; Valenzano, M.C.; Jeffers, C.; Sedlak, J.; Cugliari, M.K.; Papanikolaou, E.; Clouse, J.; Miao, J.; Wertan, N.E.; Mullin, J.M. Enhancement of Tight Junctional Barrier Function by Micronutrients: Compound-Specific Effects on Permeability and Claudin Composition. PLoS ONE 2013, 8, e78775. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yuan, X.; Zuo, H.; Li, X.; Sun, Y.; Feng, A. Berberine induces ZIP14 expression and modulates zinc redistribution to protect intestinal mucosal barrier during polymicrobial sepsis. Life Sci. 2019, 233, 116697. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros, P.H.Q.S.; Pinto, D.V.; De Almeida, J.Z.; Rêgo, J.M.C.; Rodrigues, F.A.P.; Lima, A.; Ângelo, M.; Bolick, D.T.; Guerrant, R.L.; Oriá, R.B. Modulation of Intestinal Immune and Barrier Functions by Vitamin A: Implications for Current Understanding of Malnutrition and Enteric Infections in Children. Nutrients 2018, 10, 1128. [Google Scholar] [CrossRef]
- Abdelhamid, L.; Luo, X.M. Retinoic Acid, Leaky Gut, and Autoimmune Diseases. Nutrients 2018, 10, 1016. [Google Scholar] [CrossRef]
- Yamada, S.; Kanda, Y. Retinoic acid promotes barrier functions in human iPSC-derived intestinal epithelial monolayers. J. Pharmacol. Sci. 2019, 140, 337–344. [Google Scholar] [CrossRef]
- Wang, P.-F.; Yao, D.-H.; Hu, Y.-Y.; Li, Y. Vitamin D Improves Intestinal Barrier Function in Cirrhosis Rats by Upregulating Heme Oxygenase-1 Expression. Biomol. Ther. 2019, 27, 222–230. [Google Scholar] [CrossRef]
- Zuo, L.; Yang, X.; Lu, M.; Hu, R.; Zhu, H.; Zhang, S.; Zhou, Q.; Chen, F.; Gui, S.; Wang, Y. All-Trans Retinoic Acid Inhibits Human Colorectal Cancer Cells RKO Migration via Downregulating Myosin Light Chain Kinase Expression through MAPK Signaling Pathway. Nutr. Cancer 2016, 68, 1225–1233. [Google Scholar] [CrossRef]
- Filteau, S.M.; Rollins, N.C.; Coutsoudis, A.; Sullivan, K.R.; Willumsen, J.F.; Tomkins, A.M. The Effect of Antenatal Vitamin A and β-Carotene Supplementation on Gut Integrity of Infants of HIV-Infected South African Women. J. Pediatr. Gastroenterol. Nutr. 2001, 32, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.A.; Soares, A.M.; Lima, N.L.; Mota, R.M.; Maciel, B.L.; Kvalsund, M.P.; Barrett, L.J.; Fitzgerald, R.P.; Blaner, W.S.; Guerrant, R.L. Effects of Vitamin A Supplementation on Intestinal Barrier Function, Growth, Total Parasitic, and Specific Giardia spp Infections in Brazilian Children: A Prospective Randomized, Double-blind, Placebo-controlled Trial. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 309–315. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hu, X.; Xiao, D.; Wu, J.; Zhou, S.; Deng, J.; Xu, S.; Huang, Y.; Peng, M.; Yang, X. Vitamin A prevents lipopolysaccharide-induced injury on tight junctions in mice. Food Sci. Nutr. 2020, 8, 1942–1948. [Google Scholar] [CrossRef]
- Xiao, S.; Li, Q.; Hu, K.; He, Y.; Ai, Q.; Hu, L.; Yu, J. Vitamin A and Retinoic Acid Exhibit Protective Effects on Necrotizing Enterocolitis by Regulating Intestinal Flora and Enhancing the Intestinal Epithelial Barrier. Arch. Med Res. 2018, 49, 1–9. [Google Scholar] [CrossRef]
- Cheng, J.; Balbuena, E.; Miller, B.; Eroglu, A. The Role of β-Carotene in Colonic Inflammation and Intestinal Barrier Integrity. Front. Nutr. 2021, 8, 723480. [Google Scholar] [CrossRef]
- Maciel, A.A.; Oriá, R.B.; Braga-Neto, M.B.; Braga, A.B.; Carvalho, E.B.; Lucena, H.B.; Brito, G.A.; Guerrant, R.L.; Lima, A.A. Role of retinol in protecting epithelial cell damage induced by Clostridium difficile toxin A. Toxicon 2007, 50, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Osanai, M.; Nishikiori, N.; Murata, M.; Chiba, H.; Kojima, T.; Sawada, N. Cellular Retinoic Acid Bioavailability Determines Epithelial Integrity: Role of Retinoic Acid Receptor α Agonists in Colitis. Mol. Pharmacol. 2006, 71, 250–258. [Google Scholar] [CrossRef]
- Molina-Jijón, E.; Rodríguez-Muñoz, R.; Namorado, M.D.C.; Bautista-García, P.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Reyes, J.L. All- trans retinoic acid prevents oxidative stress-induced loss of renal tight junction proteins in type-1 diabetic model. J. Nutr. Biochem. 2015, 26, 441–454. [Google Scholar] [CrossRef]
- Lochbaum, R.; Schilpp, C.; Nonnenmacher, L.; Frick, M.; Dietl, P.; Wittekindt, O.H. Retinoic acid signalling adjusts tight junction permeability in response to air-liquid interface conditions. Cell. Signal. 2020, 65, 109421. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Ishida, K.; Takeda, Y. Changes in cell characteristics due to retinoic acid; specifically, a decrease in the expression of claudin-1 and increase in claudin-4 within tight junctions in stratified oral keratinocytes. J. Periodontal Res. 2010, 45, 207–215. [Google Scholar] [CrossRef]
- Groeger, S.; Jarzina, F.; Windhorst, A.; Meyle, J. Influence of retinoic acid on human gingival epithelial barriers. J. Periodontal Res. 2016, 51, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, D.; Hu, D.; Liu, B.; Peng, J.; Shen, L.; Long, C.; Yu, Y.; Zhang, Y.; Liu, X.; et al. Retinoic acid: A potential therapeutic agent for cryptorchidism infertility based on investigation of flutamide-induced cryptorchid rats in vivo and in vitro. Reprod. Toxicol. 2019, 87, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zhouguang, W.; Zhang, H.; Zheng, B.; Ye, L.; Zhu, S.; Johnson, N.R.; Wang, Z.; Wei, X.; Chen, D.; Cao, G.; et al. Retinoic Acid Induced-Autophagic Flux Inhibits ER-Stress Dependent Apoptosis and Prevents Disruption of Blood-Spinal Cord Barrier after Spinal Cord Injury. Int. J. Biol. Sci. 2016, 12, 87–99. [Google Scholar] [CrossRef]
- Satoh, H.; Zhong, Y.; Isomura, H.; Saitoh, M.; Enomoto, K.; Sawada, N.; Mori, M. Localization of 7H6 Tight Junction-Associated Antigen along the Cell Border of Vascular Endothelial Cells Correlates with Paracellular Barrier Function against Ions, Large Molecules, and Cancer Cells. Exp. Cell Res. 1996, 222, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Liu, S. Effect of all-trans retinoic acid on the barrier function in human retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2011, 407, 605–609. [Google Scholar] [CrossRef]
- Kubota, H.; Chiba, H.; Takakuwa, Y.; Osanai, M.; Tobioka, H.; Kohama, G.-I.; Mori, M.; Sawada, N. Retinoid X Receptor α and Retinoic Acid Receptor γ Mediate Expression of Genes Encoding Tight-Junction Proteins and Barrier Function in F9 Cells during Visceral Endodermal Differentiation. Exp. Cell Res. 2001, 263, 163–172. [Google Scholar] [CrossRef]
- Tobioka, H.; Sawada, N.; Zhong, Y.; Mori, M. Enhanced paracellular barrier function of rat mesothelial cells partially protects against cancer cell penetration. Br. J. Cancer 1996, 74, 439–445. [Google Scholar] [CrossRef][Green Version]
- Retana, C.; Sanchez, E.; Perez-Lopez, A.; Cruz, A.; Lagunas, J.; Cruz, C.; Vital, S.; Reyes, J.L. Alterations of Intercellular Junctions in Peritoneal Mesothelial Cells from Patients Undergoing Dialysis: Effect of Retinoic Acid. Perit. Dial. Int. 2015, 35, 275–287. [Google Scholar] [CrossRef]
- Telgenhoff, D.; Ramsay, S.; Hilz, S.; Slusarewicz, P.; Shroot, B. Claudin 2 mRNA and Protein Are Present in Human Keratinocytes and May Be Regulated by All-trans-Retinoic Acid. Ski. Pharmacol. Physiol. 2008, 21, 211–217. [Google Scholar] [CrossRef]
- Elias, P.M.; Friend, D.S. Vitamin-A-induced mucous metaplasia. An in vitro system for modulating tight and gap junction differentiation. J. Cell Biol. 1976, 68, 173–188. [Google Scholar] [CrossRef]
- Gorodeski, G.I.; Pal, D.; Rorke, E.A.; Eckert, R.L.; Burfeind, P. Retinoids modulate P2Upurinergic receptor-mediated changes in transcervical paracellular permeability. Am. J. Physiol. Cell Physiol. 1998, 274, C1108–C1116. [Google Scholar] [CrossRef] [PubMed]
- Baltes, S.; Nau, H.; Lampen, A. All-trans retinoic acid enhances differentiation and influences permeability of intestinal Caco-2 cells under serum-free conditions. Dev. Growth Differ. 2004, 46, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Moss, A.C. Vitamin D in inflammatory bowel disease: More than just a supplement. Curr. Opin. Gastroenterol. 2018, 34, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Noriega, D.B.; Savelkoul, H. Vitamin D and Allergy Susceptibility during Gestation and Early Life. Nutrients 2021, 13, 1015. [Google Scholar] [CrossRef]
- Battistini, C.; Ballan, R.; Herkenhoff, M.; Saad, S.; Sun, J. Vitamin D Modulates Intestinal Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2020, 22, 362. [Google Scholar] [CrossRef]
- Du, J.; Chen, Y.; Shi, Y.; Liu, T.; Cao, Y.; Tang, Y.; Ge, X.; Nie, H.; Zheng, C.; Li, Y.C. 1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway. Inflamm. Bowel Dis. 2015, 21, 2495–2506. [Google Scholar] [CrossRef]
- Kong, J.; Zhang, Z.; Musch, M.W.; Ning, G.; Sun, J.; Hart, J.; Bissonnette, M.; Li, Y.C. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G208–G216. [Google Scholar] [CrossRef]
- Lee, C.; Lau, E.; Chusilp, S.; Filler, R.; Li, B.; Zhu, H.; Yamoto, M.; Pierro, A. Protective effects of vitamin D against injury in intestinal epithelium. Pediatr. Surg. Int. 2019, 35, 1395–1401. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, H.; Wu, H.; Li, H.; Liu, L.; Guo, J.; Li, C.; Shih, D.Q.; Zhang, X. Protective role of 1,25(OH)2vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol. 2012, 12, 57. [Google Scholar] [CrossRef]
- Liu, T.; Shi, Y.; Du, J.; Ge, X.; Teng, X.; Liu, L.; Wang, E.; Zhao, Q. Vitamin D treatment attenuates 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced colitis but not oxazolone-induced colitis. Sci. Rep. 2016, 6, 32889. [Google Scholar] [CrossRef]
- Chatterjee, I.; Zhang, Y.; Zhang, J.; Lu, R.; Xia, Y.; Sun, J. Overexpression of Vitamin D Receptor in Intestinal Epithelia Protects Against Colitis via Upregulating Tight Junction Protein Claudin 15. J. Crohn’s Colitis 2021, 15, 1720–1736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-G.; Wu, S.; Lu, R.; Zhou, D.; Zhou, J.; Carmeliet, G.; Petrof, E.; Claud, E.C.; Sun, J. Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep. 2015, 5, 10642. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meng, F.; Wang, S.; Xia, S.; Wang, R. Vitamin D3 mitigates lipopolysaccharide-induced oxidative stress, tight junction damage and intestinal inflammatory response in yellow catfish, Pelteobagrus fulvidraco. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 243, 108982. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Zhang, Z.; Yang, L.; Li, R.; Ma, Y. Combined effect of vitamin C and vitamin D3 on intestinal epithelial barrier by regulating Notch signaling pathway. Nutr. Metab. 2021, 18, 1–11. [Google Scholar] [CrossRef]
- Assa, A.; Vong, L.; Pinnell, L.J.; Rautava, J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D Deficiency Predisposes to Adherent-invasive Escherichia coli-induced Barrier Dysfunction and Experimental Colonic Injury. Inflamm. Bowel Dis. 2015, 21, 297–306. [Google Scholar] [CrossRef]
- Chen, S.-W.; Ma, Y.-Y.; Zhu, J.; Zuo, S.; Zhang, J.-L.; Chen, Z.-Y.; Chen, G.-W.; Wang, X.; Pan, Y.-S.; Liu, Y.-C.; et al. Protective effect of 1,25-dihydroxyvitamin D3 on ethanol-induced intestinal barrier injury both in vitro and in vivo. Toxicol. Lett. 2015, 237, 79–88. [Google Scholar] [CrossRef]
- Dong, S.; Singh, T.P.; Wei, X.; Yao, H.; Wang, H. Protective Effect of 1,25-Dihydroxy Vitamin D3 on Pepsin–Trypsin-Resistant Gliadin-Induced Tight Junction Injuries. Dig. Dis. Sci. 2018, 63, 92–104. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Wang, Y.; Wang, L.; Yin, Y.; Yin, L.; Yang, H.; Yin, Y. Dietary vitamin A affects growth performance, intestinal development, and functions in weaned piglets by affecting intestinal stem cells. J. Anim. Sci. 2020, 98. [Google Scholar] [CrossRef]
- Liu, X.Z.; You, B.; Zhang, Y.L.; Yang, Z.C.; Chen, P.; Shi, Y.L.; Chen, Y.; Chen, J.; Peng, Y.Z. Effects of vitamin D3 on intestinal mucosal barrier of mice with severe burns. Zhonghua Shao Shang Za Zhi 2019, 35, 284–291. [Google Scholar]
- Lee, P.; Hsieh, Y.; Huo, T.; Yang, U.; Lin, C.; Li, C.; Huang, Y.; Hou, M.; Lin, H.; Lee, K. Active Vitamin D 3 Treatment Attenuated Bacterial Translocation via Improving Intestinal Barriers in Cirrhotic Rats. Mol. Nutr. Food Res. 2021, 65, e2000937. [Google Scholar] [CrossRef]
- Fernandez-Robredo, P.; González-Zamora, J.; Recalde, S.; Bilbao-Malavé, V.; Bezunartea, J.; Hernandez, M.; Garcia-Layana, A. Vitamin D Protects against Oxidative Stress and Inflammation in Human Retinal Cells. Antioxidants 2020, 9, 838. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Kamolvit, W.; Hertting, O.; Brauner, A. Vitamin D strengthens the bladder epithelial barrier by inducing tight junction proteins during E. coli urinary tract infection. Cell Tissue Res. 2020, 380, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-Y.; Liu, T.-J.; Fu, J.-H.; Xu, W.; Wu, L.-L.; Hou, A.-N.; Xue, X.-D. Vitamin D/VDR signaling attenuates lipopolysaccharide-induced acute lung injury by maintaining the integrity of the pulmonary epithelial barrier. Mol. Med. Rep. 2016, 13, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, H.; Zhao, H.; Song, J.; Tang, H.; Yao, L.; Liu, L.; Tong, W.; Zou, M.; Zou, F.; et al. 1,25-Dihydroxyvitamin D3 prevents toluene diisocyanate-induced airway epithelial barrier disruption. Int. J. Mol. Med. 2015, 36, 263–270. [Google Scholar] [CrossRef]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D Prevents Hypoxia/Reoxygenation-Induced Blood-Brain Barrier Disruption via Vitamin D Receptor-Mediated NF-kB Signaling Pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [PubMed]
- Boguniewicz, M.; Leung, D.Y.M. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [CrossRef]
- Tudpor, K.; Teerapornpuntakit, J.; Jantarajit, W.; Krishnamra, N.; Charoenphandhu, N. 1,25-Dihydroxyvitamin D3 Rapidly Stimulates the Solvent Drag–Induced Paracellular Calcium Transport in the Duodenum of Female Rats. J. Physiol. Sci. 2008, 58, 297–307. [Google Scholar] [CrossRef]
- Fujita, H.; Sugimoto, K.; Inatomi, S.; Maeda, T.; Osanai, M.; Uchiyama, Y.; Yamamoto, Y.; Wada, T.; Kojima, T.; Yokozaki, H.; et al. Tight Junction Proteins Claudin-2 and -12 Are Critical for Vitamin D-dependent Ca2+ Absorption between Enterocytes. Mol. Biol. Cell 2008, 19, 1912–1921. [Google Scholar] [CrossRef]
- Chirayath, M.V.; Gajdzik, L.; Hulla, W.; Graf, J.; Cross, H.S.; Peterlik, M. Vitamin D increases tight-junction conductance and paracellular Ca2+ transport in Caco-2 cell cultures. Am. J. Physiol. Gastrointest. Liver Physiol. 1998, 274, G389–G396. [Google Scholar] [CrossRef]
- Kladnitsky, O.; Rozenfeld, J.; Efrati, E.; Zelikovic, I.; Azulay-Debby, H. The claudin-16 channel gene is transcriptionally inhibited by 1,25-dihydroxyvitamin D. Exp. Physiol. 2014, 100, 79–94. [Google Scholar] [CrossRef]
- Camilleri, M. What is the leaky gut? Clinical considerations in humans. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 473–482. [Google Scholar] [CrossRef] [PubMed]
- DiGuilio, K.M.; Rybakovsky, E.; Valenzano, M.C.; Lander, J.; Manganiello, A.; Leroy, K.; Bensinger, A.; Chen, W.; Chen, V.; Majumdar, S.; et al. S1354 Modification of the Tight Junctional Barrier in Human Duodenal Mucosal by Oral Zinc Administration in a Patient-Based Study: RNA-Seq and Western Immunoblot Analyses. Am. J. Gastroenterol. 2021, 116, S623. [Google Scholar] [CrossRef]
- Sturniolo, G.C.; Di Leo, V.; Ferronato, A.; D’Odorico, A.; D’Incà, R. Zinc Supplementation Tightens “Leaky Gut” in Crohn’s Disease. Inflamm. Bowel Dis. 2001, 7, 94–98. [Google Scholar] [CrossRef]
- Roy, S.K.; Behrens, R.H.; Haider, R.; Akramuzzaman, S.M.; Mahalanabis, D.; Wahed, M.A.; Tomkins, A.M. Impact of Zinc Supplementation on Intestinal Permeability in Bangladeshi Children with Acute Diarrhoea and Persistent Diarrhoea Syndrome. J. Pediatr. Gastroenterol. Nutr. 1992, 15, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.N.; Sarker, S.A.; Wahed, M.A.; Khatun, M.; Rahaman, M.M. Enteric protein loss and intestinal permeability changes in children during acute shigellosis and after recovery: Effect of zinc supplementation. Gut 1994, 35, 1707–1711. [Google Scholar] [CrossRef]
- Ryan, K.N.; Stephenson, K.B.; Trehan, I.; Shulman, R.J.; Thakwalakwa, C.; Murray, E.; Maleta, K.; Manary, M.J. Zinc or Albendazole Attenuates the Progression of Environmental Enteropathy: A Randomized Controlled Trial. Clin. Gastroenterol. Hepatol. 2014, 12, 1507–1513.e1. [Google Scholar] [CrossRef]
- Tran, C.D.; Hawkes, J.; Graham, R.D.; Kitchen, J.L.; Symonds, E.; Davidson, G.P.; Butler, R.N. Zinc-Fortified Oral Rehydration Solution Improved Intestinal Permeability and Small Intestinal Mucosal Recovery. Clin. Pediatr. 2015, 54, 676–682. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Ko, W.-S.; Hsiao, J.-L.; Pan, H.-H.; Chiou, Y.-L. Zinc sulfate improved the unbalanced T cell profiles in Der p-allergic asthma: An ex vivo study. Clin. Respir. J. 2016, 12, 563–571. [Google Scholar] [CrossRef]
- Stio, M.; Retico, L.; Annese, V.; Bonanomi, A.G. Vitamin D regulates the tight-junction protein expression in active ulcerative colitis. Scand. J. Gastroenterol. 2016, 51, 1193–1199. [Google Scholar] [CrossRef]
- Domazetovic, V.; Iantomasi, T.; Bonanomi, A.G.; Stio, M. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int. J. Color. Dis. 2020, 35, 1231–1242. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, X.; Li, J.; Wang, H.; Li, Y.; Chen, Y.; Zhang, H. Clinical evaluation of vitamin D status and its relationship with disease activity and changes of intestinal immune function in patients with Crohn’s disease in the Chinese population. Scand. J. Gastroenterol. 2021, 56, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Meckel, K.; Li, Y.C.; Lim, J.; Kocherginsky, M.; Weber, C.; Almoghrabi, A.; Chen, X.; Kaboff, A.; Sadiq, F.; Hanauer, S.B.; et al. Serum 25-hydroxyvitamin D concentration is inversely associated with mucosal inflammation in patients with ulcerative colitis. Am. J. Clin. Nutr. 2016, 104, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Dancer, R.C.A.; Parekh, D.; Lax, S.; D’Souza, V.; Zheng, S.; Bassford, C.R.; Park, D.; Bartis, D.G.; Mahida, R.; Turner, A.M.; et al. Vitamin D deficiency contributes directly to the acute respiratory distress syndrome (ARDS). Thorax 2015, 70, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Thurnham, D.I.; Northrop-Clewes, C.A.; McCullough, F.S.W.; Das, B.S.; Lunn, P.G. Innate Immunity, Gut Integrity, and Vitamin A in Gambian and Indian Infants. J. Infect. Dis. 2000, 182 (Suppl. S1), S23–S28. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.M. (Ed.) The ASPEN Adult Nutrition Support Core Curriculum, 3rd ed.; ASPEN: Silver Spring, MD, USA, 2017. [Google Scholar]
- Vanek, V.W.; Borum, P.; Buchman, A.; Fessler, T.A.; Howard, L.; Jeejeebhoy, K.; Kochevar, M.; Shenkin, A.; Valentine, C.J.; Novel Nutrient Task Force; et al. A.S.P.E.N. position paper: Recommendations for changes in commercially available parenteral multivitamin and multi-trace element products. Nutr. Clin. Pract. 2012, 27, 440–491. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academy Press: Washington, DC, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK222310/ (accessed on 3 March 2022). [CrossRef]
- Murphy, S.P.; Yates, A.A.; Atkinson, S.A.; Barr, S.I.; Dwyer, J. History of Nutrition: The Long Road Leading to the Dietary Reference Intakes for the United States and Canada. Adv. Nutr. 2016, 7, 157–168. [Google Scholar] [CrossRef]
- Vanek, V.W.; Borum, P.; Buchman, A.; Fessler, T.; Howard, L.; Shenkin, A.; Valentine, C.J.; Novel Nutrient Task Force; Parenteral Vitamin and Trace Element Working Group; American Society for Parenteral and Enteral Nutrition (ASPEN). A Call to Action to Bring Safer Parenteral Micronutrient Products to the U.S. Market. Nutr. Clin. Pract. 2015, 30, 559–569. [Google Scholar] [CrossRef]
- American Society for Parenteral and Enteral Nutrition. Appropriate Dosing for Parenteral Nutrition: ASPEN Recommendations. 2019. Updated November 2020. Available online: https://www.nutritioncare.org/uploadedFiles/Documents/Guidelines_and_Clinical_Resources/ (accessed on 17 May 2021).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DiGuilio, K.M.; Rybakovsky, E.; Abdavies, R.; Chamoun, R.; Flounders, C.A.; Shepley-McTaggart, A.; Harty, R.N.; Mullin, J.M. Micronutrient Improvement of Epithelial Barrier Function in Various Disease States: A Case for Adjuvant Therapy. Int. J. Mol. Sci. 2022, 23, 2995. https://doi.org/10.3390/ijms23062995
DiGuilio KM, Rybakovsky E, Abdavies R, Chamoun R, Flounders CA, Shepley-McTaggart A, Harty RN, Mullin JM. Micronutrient Improvement of Epithelial Barrier Function in Various Disease States: A Case for Adjuvant Therapy. International Journal of Molecular Sciences. 2022; 23(6):2995. https://doi.org/10.3390/ijms23062995
Chicago/Turabian StyleDiGuilio, Katherine M., Elizabeth Rybakovsky, Reza Abdavies, Romy Chamoun, Colleen A. Flounders, Ariel Shepley-McTaggart, Ronald N. Harty, and James M. Mullin. 2022. "Micronutrient Improvement of Epithelial Barrier Function in Various Disease States: A Case for Adjuvant Therapy" International Journal of Molecular Sciences 23, no. 6: 2995. https://doi.org/10.3390/ijms23062995
APA StyleDiGuilio, K. M., Rybakovsky, E., Abdavies, R., Chamoun, R., Flounders, C. A., Shepley-McTaggart, A., Harty, R. N., & Mullin, J. M. (2022). Micronutrient Improvement of Epithelial Barrier Function in Various Disease States: A Case for Adjuvant Therapy. International Journal of Molecular Sciences, 23(6), 2995. https://doi.org/10.3390/ijms23062995