2.1. NMR Solution Structures of HGDC in the Absence and Presence of Denaturants
In the previous studies [
17,
18], we have predicted the region of HGDC that may be involved in its aggregation process under low-pH conditions by MD simulations using the X-ray crystal structure of HGDC as a starting structure. In the present study, we applied a similar approach to predict the unstable regions of HGDC and to examine the sequence of initial unfolding events that may lead to misfolding, and ultimately result in HGDC aggregation. We first determined the solution structures in the absence and presence of denaturants (5.0 M urea and 1.0 M GdnHCl) using NMR spectroscopy. The two-dimensional
1H-
15N-HSQC spectra of HGDC in the absence and presence of denaturants with the assigned residues are indicated in
Figure S1 (Supplementary Materials), while the experimentally measured
1D
N-HN RDCs of HGDC in the absence and presence of denaturants are shown in
Figure S2 (Supplementary Materials). We derived the three-dimensional solution structures of HGDC based on these NMR data.
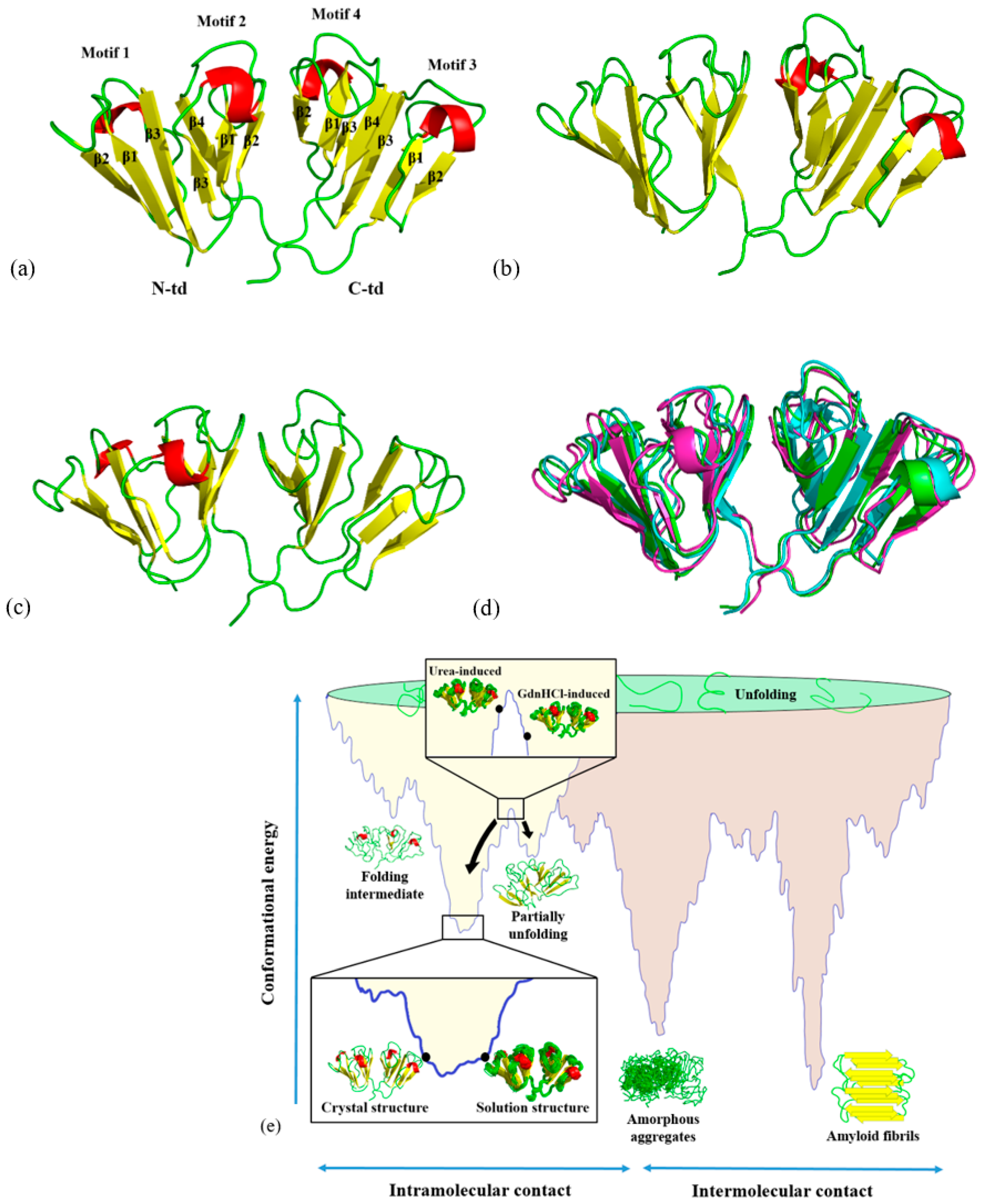
Figure 1a–c shows the three-dimensional structures of HGDC from buffer, 5.0 M urea, and 1.0 M GdnHCl solutions, respectively. An overlay of these three solution structures is shown in
Figure 1d. Although the overall three-dimensional folds of the three HGDC structures are similar, they are not completely the same.
Figure 1e shows the positions of the HGDC structures in respect to a hypothetical conformational potential energy surface [
8]. Different solution structures of HGDC can be seen positioning at different levels on its conformational potential energy surface. For example, the structure in buffer solution is positioned at the lowest point of the energy surface, while the other two denaturant-induced higher energy state structures are positioned at higher levels. For simplicity, in the subsequent sections, denaturant-induced higher energy state structures are referred to as urea-induced structure and GdnHCl-induced structure, while the solution structure in buffer is simply solution structure.
We used these NMR structures as the starting conformations for molecular dynamics (MD) simulations to understand which paths (unfolding, misfolding, or refolding) these protein structures may take when simulated in physiological solution under high temperature. By doing so, we were able to gain more insights into the relative unstable regions of the HGDC and understand how they destabilize during the initial unfolding process. Knowledge gained from this study may aid in the design of aggregation inhibitors to treat cataract.
2.2. Relative Conformational Changes between the HGDC Structures Simulated in 343K
We first performed 200-ns MD simulations in 343 K to see how the denaturant-induced higher energy structures would change when placed back in physiological solvent environment. The destabilization effects of the denaturants can be seen in the RMSD analysis of the MD simulation trajectories (
Figure S3, Supplementary Materials). The two denaturant-induced structures yielded higher RMSD values with greater fluctuations seen in the GdnHCl-induced structure. In comparison with the solution structure (with the RMSD values leveled off below 3.0 Å), both the urea- and GdnHCl-induced structures resulted in higher RMSD values with the final values leveling off just below 4.0 Å for the urea structure and fluctuating between 4.5~5.0 Å for the GdnHCl structure. Based on the PCA analysis of the essential dynamics [
19] in the simulated trajectories, we found that the ensemble of conformations for the solution structure is mainly scattered for the first 100 ns with a more distinct cluster pattern forming thereafter (
Figure 2a), which corresponds to the equilibration of the structure around this time seen in the RMSD analysis (
Figure S3, Supplementary Materials). Similarly, but more definitively, a cluster break can be seen in the urea-induced ensemble around the same time (
Figure 2b), also corresponding to the plateauing of the RMSD values. In contrast to the distinctive clustering of the solution and the urea-induced structures during the simulations, the ensemble of GdnHCl-induced conformations remained largely scattered (
Figure 2c). Although we see a bifurcation in the scatter around 50 ns, no distinctive cluster formed, and the scattering of the ensemble is consistent with the highly fluctuated RMSD values seen in
Figure S3. This indicates that there is a greater instability in the GdnHCl-induced conformation relative to the other two NMR structures.
We were interested to know what led to the isolated cluster, formed in the urea-induced ensemble, which prompted us to examine further the fluctuations within specific regions of the three NMR structures. The RMSF-per-residue analysis (
Figure 2d) shows that there are two specific regions in the urea-induced structure that fluctuated greatly and more so than either the solution or the GdnHCl-induced structures. These two regions correspond to β3-strand (G61~A64) of motif 2 and a loop (C109~F118) encompassing an α-helix in motif 3 as seen mapped onto the tertiary structure of the protein in
Figure 2e. Incidentally, the C-td loop region also encompasses part of the de novo β-strand forming region (F116~N119) discovered in our previous work on examining HGDC aggregation in low pH condition [
17]. It is interesting to note that this loop region has the potential to form β-strand in both the HGDC structure simulated under acidic condition and the urea-induced structure under physiological solution condition, as shown in
Figure S4b (Supplementary Materials). This propensity for β-strand formation in the loop region is not as evident in the GdnHCl-induced structure (
Figure S4c, Supplementary Materials) and is not observed in the solution structure (
Figure S4a, Supplementary Materials). As for the motif 2 β3-strand (G61~A64), it is evident from
Figure S4 that (in comparison to the other two NMR structures), the urea-induced structure has lost a large percentage of β-structure in this region during the simulations. The percentage of secondary structure in the motif 2 β3-strand has dropped from 85~95% range of β-structure seen in the solution and GdnHCl-induced structures down to 27~33% in the urea-induced structure.
In order to understand what happened during the simulations for the two above-mentioned regions in the urea-induced structure, we monitored residue contacts of these regions with their respective neighboring residues. It can be seen from
Figure 3 that the motif 2 β3-strand (G61~A64) contact fraction remained steady at 0.85 but declined quickly around 60 ns, showing a large decrease that was associated with the loss of β-structure and hydrogen bonds. The function of the hydrogen bonds was to keep this region attached to the neighboring anti-parallel β3-strand (C33~V38) of motif 1. Without these hydrogen bonds to keep it intact, the G61~A64 residue region detached from the intercalated anti-parallel β-sheet, as can be seen in
Figure 3 with the lower right snapshot of the protein taken at 100 ns of simulation time. As for the motif 3 loop (C109~F118), the contact fraction at the beginning of the simulation was lower (~0.75) and steadily decreased until it reached ~0.55 around 140 ns and continued to remain at about this level till the end of simulation time. The steady loss in contact fraction indicates that the long stretch of residues did not maintain its original contacts with the surrounding residues from the beginning of the simulations. In addition, this region has the propensity to form β-strand possibly due to the conformational flexibility of the region being a loop structure and its inherent dynamic nature to adopt variable conformations.
As previously mentioned, similar to the solution structure, the GdnHCl-induced structure was able to maintain a high percentage of β-structure in motif 2 β3-strand region. However, the β-strands in other parts of the protein have shortened and became wispier in comparison to the ones in the solution structure (
Figure S4a,c, Supplementary Materials). Based on these results, we can see that the denaturant has affected the integrity of some of the secondary structures, but the effects on the overall initial unfolding process are still not clear, at this point, for this particular high-energy state conformer.
2.3. The Overall Conformational Changes of the Denaturant-Induced HGDC Structures Simulated in 425 K
Previous studies have performed MD simulations in high temperature of 425 K and 8 M urea to study the stability and aggregation of HGDC [
14,
15,
20]. High-temperature MD simulations have been known to accelerate protein unfolding without affecting the course of the pathway [
21,
22]. We, therefore, increase the temperature to 425 K to speed up the process, allowing for a further detailed look at the initial steps leading to the unfolding/misfolding/refolding pathways of the two higher energy state protein structures previously induced in the NMR experiments. Hereafter, results and discussions are only aimed at these two structures, unless otherwise specified.
To get an overall picture of the protein region(s) affected the most along the initial stages of unfolding, we re-examined the RMSF per residue of the two denaturant-induced structures and found that the range of residue fluctuation has increase from nearly 6 Å, in the urea-induced structure under 343 K (
Figure 2d), to close to 7.5 Å in the GdnHCl-induced structure under 425K (
Figure S5, Supplementary Materials). Although the ranges of fluctuation in both proteins have increased in the higher temperature condition, the GdnHCl-induced structure yielded overall higher fluctuation than the urea-induced structure, suggesting that the GdnHCl-induced structure has become more destabilized. This instability is more prominent in the N-terminal domain (N-td) than the C-terminal domain (C-td) where the overall fluctuation is observed to be the highest (
Figure S5, Supplementary Materials).
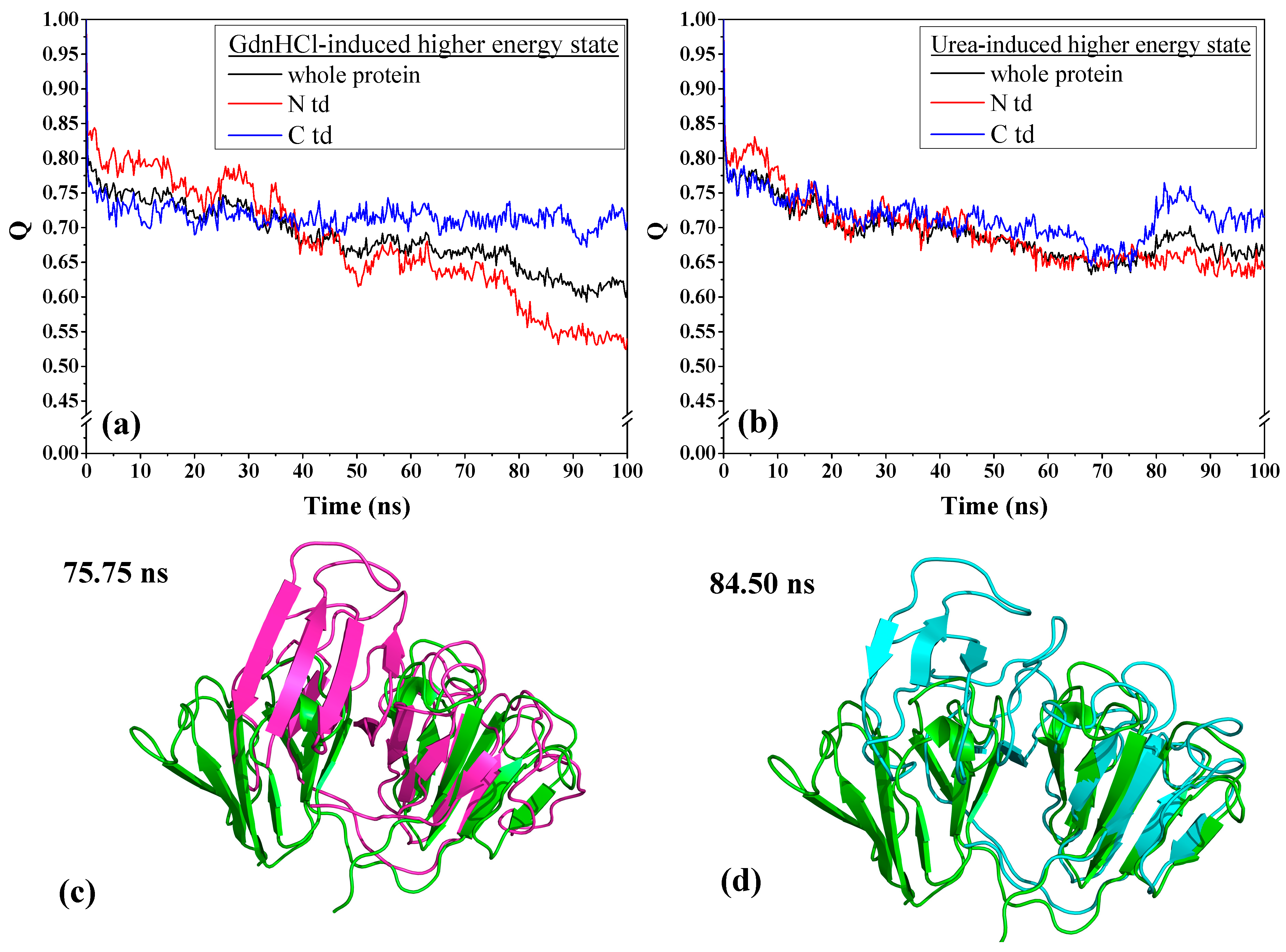
To understand the extent of the changes each domain has undergone, we examined the contact fraction for the whole protein and by protein domains, as depicted in
Figure 4. We see that the contact fraction for the GdnHCl-induced whole protein steadily decreased until reaching close to 0.60 (
Figure 4a). The contribution to this decrease by individual domains is mainly due to N-td’s loss of residue contacts. Comparing with C-td, N-td suffered the greatest loss, with contact fraction dropping down below 0.55, while it remained slightly above 0.70 in the C-td. The steepest drop in N-td residue contact occurred slightly before 80 ns, signifying that there was a drastic change in protein conformation around this time. As for the urea-induced structure, we also see a decrease in residue contacts for the whole protein and the individual domains with similar level of loss in residue contacts for the greater part of the simulations until reaching a contact fraction value of ~0.65 (
Figure 4b). However, around 75 ns of simulation time, we began to see a rise in contact fraction in the C-td, which suggests that some of the lost contacts were somehow regained and the contact fraction reached a peak of ~0.75 for the C-td after 80 ns and began to level off slightly above 0.70 till the end of simulation time.
To confirm whether the urea-induced structure really did revert back toward the original higher energy state conformation, we individually superimposed the structures at 75.75 ns and 84.50 ns, corresponding to the time of the C-td contact fraction minimum and maximum, respectively, with the initial structure at time zero of the simulation. The RMSD values for the overall protein alignment yielded 8.46 Å and 7.53 Å for the structure alignments of the conformers at 75.75 ns and 84.50 ns, respectively, showing that there is a slight decrease of RMSD at the peak time. We then superimposed the structures by domain and found that despite there was not much of a difference when superimposed by N-tds (4.83 Å and 4.85 Å for structures at 75.75 ns and 84.50 ns, respectively), superposition of C-tds at the specific time frames mentioned above (
Figure 4c,d) yielded 4.64 Å and 1.73 Å, respectively, signifying that at 84.50 ns, the C-td reverted back to a conformation closer to the original high-energy state structure. This is an additional support for the regained contact fraction observed in
Figure 4b and shows that the urea-induced structure, when transferred into a physiological solution condition, did revert back to its original higher energy state, which may possibly be a more steady-state and more preferred conformation than the GdnHCl-induced structure.
A subtractive 2D-contact map analysis (
Figure 5) was performed by subtracting the 2D residue contact map of GdnHCl-induced conformational ensembles (
Figure S6b, Supplementary Materials) from that of the urea-induced ensembles (
Figure S6a, Supplementary Materials) to reveal the relative changes between these two denaturant-induced higher state conformers during the simulations. Three distinctive regions of residue contact loss in the denaturant-induced structures can be seen in
Figure 5. The urea-induced structure lost residue contacts mainly in motif 2, indicated by region A, while contact loss was more extensive in the GdnHCl-induced structure, which included not only motif 2, but also parts of motifs 1 and 4 depicted in regions B and C. Thus, a great proportion of the N-td in the GdnHCl-induced structure suffered contact loss (region B), as well as residue contacts in the hydrophobic inner core region (region C) that encompasses the inter-domain interface between motifs 2 and 4.
2.4. Examining the Critical Interactions for Maintaining Structural Stability of HGDC
The interdomain interface has been previously shown in protein refolding experiments to be important in stabilizing the final N-td conformation after the domain has folded up [
23]. Based on several studies, these interface interactions have also been indicated as contributing to the high kinetic barriers during the early stages of HGDC unfolding [
24,
25]. Having known that the domain interface plays an important role in maintaining HGDC stability, we next examined the residue interactions in this region. Our results in
Figure S7 show that the fluctuation in the interface interactions for urea-induced structure ranges from 0.2~0.8, while, for the GdnHCl-induced structure, it extends to an even wider range of 0.1~0.9, indicating that GdnHCl’s destabilizing effect is greater in this region than that of urea’s. This large fluctuation in the inter-domain contacts of the GdnHCl-induced structure indicates that the integrity of the interface has been lost; thus, breaching the high kinetic barriers and leading to structural unfolding manifested in a greater change in conformation and loss of contacts, predominantly in the N-td (see previous section). Even though the interface contacts in the urea-induced structure also fluctuate quite a bit, its effect is not as detrimental as in the GdnHCl-induced structure and is only limited to motif 2 as seen in
Figure 5. Nevertheless, motif 2 is the first region affected in both denaturant-induced higher-energy NMR structures when the domain interface is disturbed. A previous MD simulations study of tyrosine-to-alanine substitution in X-ray HGDC structure also reached a similar conclusion, where it was stated that “the stability of motif 2 is mainly determined by the inter-domain interface” [
20].
The fact that motif 2 is the first region to be disrupted in both denaturant-induced structure prompted us to look further into how this disruption came about. According to past studies on HGDC, it was suggested that aromatic residues are the key to maintaining the lens crystallin fold and stability [
23], and some of their interactions may serve as nucleation sites for protein folding, forming “clasps” to stabilize local conformation [
26]. Aromatic residues can form pairs and clusters in β-hairpin peptides and proteins (such as HGDC) containing β-hairpins to stabilize the structural fold [
23,
27,
28]. Motif 2 in HGDC contains important aromatic residue interactions that contribute to the stability of this particular Greek key: a Tyr-pair (Y46-Y51) conserved across the βγ-crystallin superfamily and a Tyr-corner (Y63) that forms an aromatic cluster with W69 and Y56. The Greek key Tyr-pair was known to nucleate motif 2 refolding [
23], while W69 (located on a surface loop region) was found to shield an aggregation-prone stretch of residues (L54~L58), predicted by bioinformatics methods [
29]. Residue Y56 within this region is also a part of the N-td inter-domain interface contacts. The importance of aromatic clusters in stabilizing the Greek key fold has previously been explored. It was found that photo-oxidative damage to these residues by UV rays lead to the loss of aromatic interactions, which may contribute to cataract formation as the residues are known UV absorbers [
30].
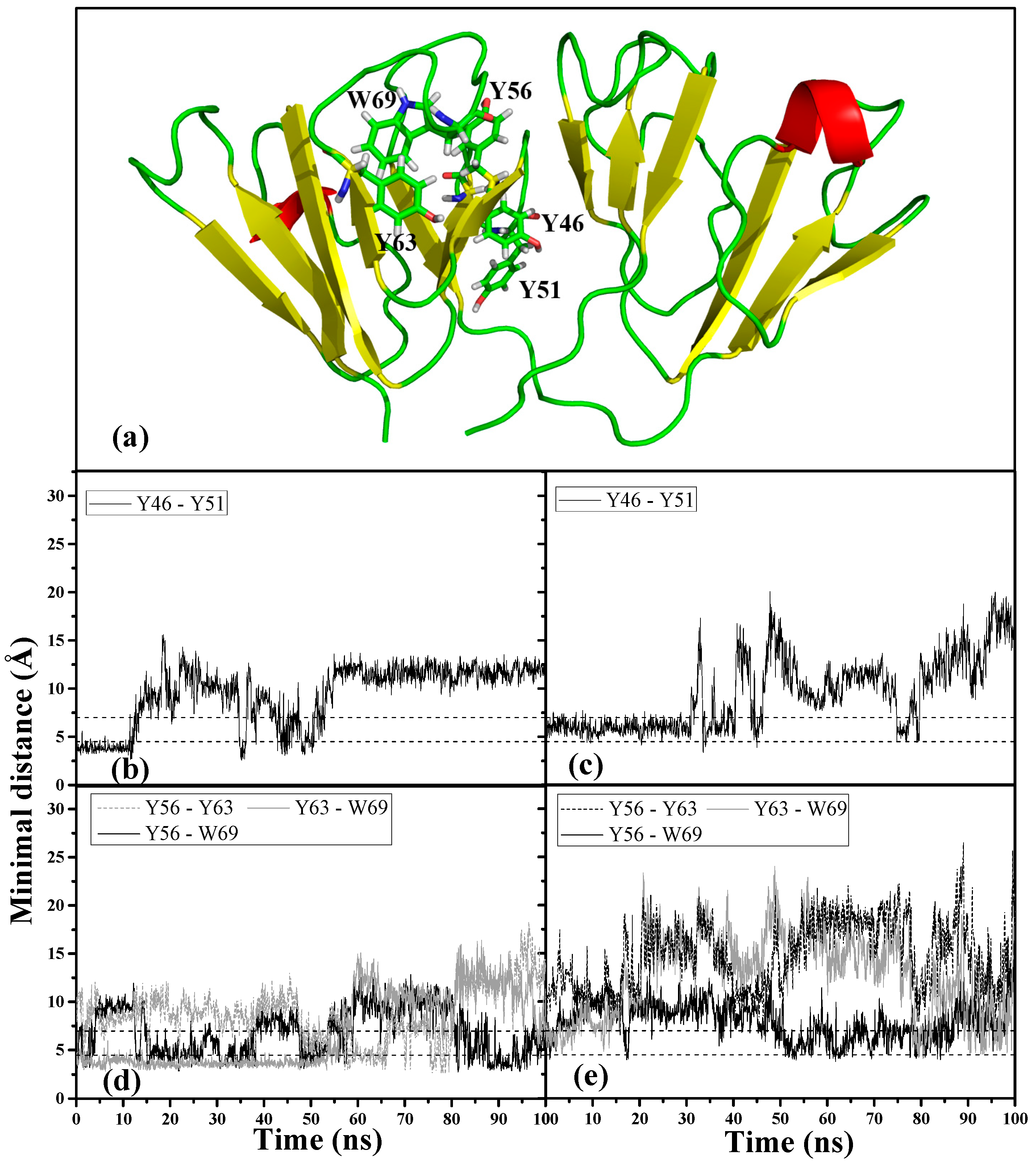
We examined the interactions of the important aromatic residues within motif 2 to understand the roles they play in the dynamic conformational changes that occurred in the two denaturant-induced structures.
Figure 6a shows the location of the motif 2 aromatic cluster (Y63-W69-Y56) and Greek key Y pair (Y46-Y51) within the 3D structure of HGDC. It has been observed that aromatic residues in proteins often form clusters and majority of the aromatic-aromatic interactions fall within the range of 4.5~7.0 Å [
26]. Based on the above criteria, we monitored these residue interactions in motif 2. Of all the aromatic interactions observed within this Greek key motif, the conserved Tyr-pair (Y46-Y51) in both denaturant-induced higher energy structures became unstable and lost its interaction much earlier on during the simulations. As can be seen in
Figure 6b,c, the Tyr-pair in the GdnHCl-induced structure started losing contact around 30 ns into the simulation, whereas the interaction in the urea structure lost contact even earlier on (~10 ns). The destabilizing effects of both denaturants were also observed to have extended to the aromatic cluster, Y63–W69–Y56, where most of the aromatic interactions were disrupted (
Figure 6d,e). It is interesting to note that, of the aromatic cluster interactions, Y56–W69 maintained the highest percentage of aromatic contacts throughout the simulation time in both denaturant-induced structures (
Table 1), despite its fluctuating quite a bit. This signifies that the interaction between Y56 and W69 is stronger than the rest of the interactions within the cluster, and more resistant to the chemical denaturation effects regardless of the denaturant type. W69 was found to shield the aggregation-prone N-td interface that encompasses Y56 [
29]. The N-td interface forms part of the interdomain interface, a crucial region that has been shown time and time again to have significant function in keeping the two domains together and preserving the overall protein stability [
31,
32,
33]. Based on the above, it is not difficult to understand (from the standpoint of evolutionary significance of HGDC’s function as a long-lived structural protein) that the interaction between these two residues would need to be the strongest in order to maintain structural stability and resist aggregation. Nevertheless, both denaturants disrupted the above-mentioned important aromatic interactions to the point that most of the interactions were lost toward the end of the simulation time. Despite the GdnHCl-induced structure having a slightly greater percentage of contacts than that of the urea structure for most of these aromatic interactions (as depicted in
Table 1), its inter-domain interface was less stable (suffering larger fluctuation and greater loss of interface contacts) as can be seen in
Figure S7.
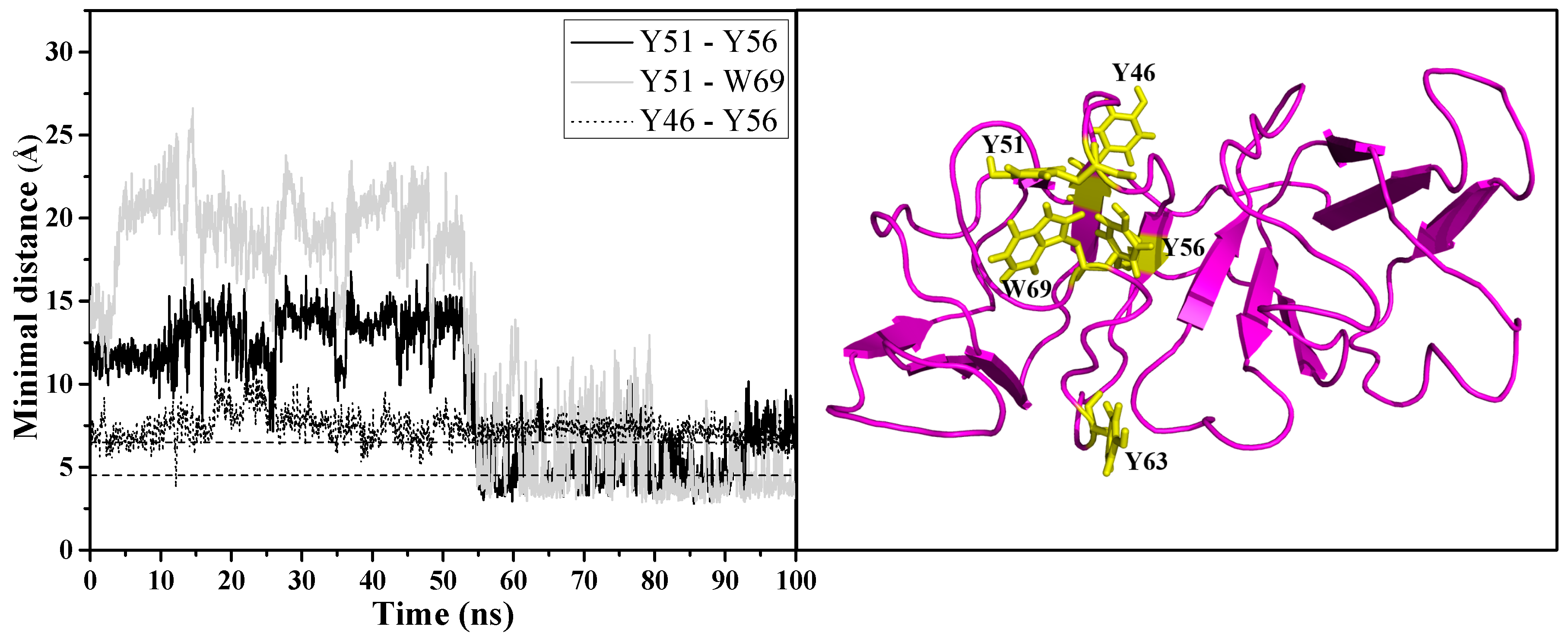
In contrast to the GdnHCl-induced structure, the urea-induced structure was found to have formed a new non-native aromatic cluster in motif 2, as seen in the right panel of
Figure 7 comprising of the following residues: Y46, Y51, Y56, and W69.
Figure 7 left panel shows that the formation of the new cluster was observed around 55 ns of simulation time and these newly formed aromatic interactions remained intact until the end of the simulation time, stabilizing motif 2 and the rest of the protein structure in a semi-intact state. Not surprisingly, the greatest percentage of contacts was maintained between the interactions involving the interface residue Y56 (Y46–Y56 and Y51–Y56) as shown in
Table 2; this is additional evidence supporting the importance of the interdomain interface in preserving the stability of N-td—in particular, motif 2. The more stable state of the urea-induced structure can be observed in
Figure 8a (left panel) where, despite large fluctuations, most of the β-strands in the urea-induced structure (with the exception of motif 2 β3-strand, spanning residues G61~A64) were retained throughout the simulation time in comparison with the GdnHCl-induced secondary structures seen in
Figure 8a (right panel). It is noteworthy to mention that the loss of β-strand in the G61~A64 residue stretch of the urea-induced structure occurred under both temperatures (343 K and 425 K) simulated in this study. This is supporting evidence indicating that the pathway that the conformer took was not altered by the higher-temperature simulation. Contrary to the urea-induced structure, we did not observe any new aromatic cluster forming in motif 2 of the GdnHCl structure. With the original aromatic interactions disrupted and without any new cluster forming to stabilize the second Greek key, motif 2 and some of the β-strands in the neighboring motifs next to it became disordered and began to lose structural integrity in the early stages of GdnHCl-induced structural unfolding.
2.5. Consequences of Losing Aromatic Interactions after Overcoming the High Kinetic Barriers Posed by the Interdomain Interactions in GdnHCl-Induced Structure
As mentioned in the previous section, the GdnHCl-induced structure has lost more β-strands than the urea-induced structure (
Figure 8a). On close observation of the right panel in
Figure 9a, this destabilization of the secondary structure was mainly observed in the N-td, starting with motif 2 β3-strand (G61~A64) around 15 ns, followed by motif 1 β3-strand (C33~V38) after 35 ns, then motif 2 β2-strand (Q55~L58) after 50 ns, finally leading to the near-complete unraveling of Greek key motif 2 after 80 ns. The sequence of the structural loss is depicted in
Figure 8b. Just like the urea-induced structure, we saw that the first region affected in motif 2 was the β3-strand. Therefore, we conclude that the second Greek key β3-strand region is the most vulnerable part of motif 2, prone to loss of β-structure after the protein has been brought to a higher energy state, irrespective of the denaturant type. Motif 1 β3-strand was the second β-strand to come undone, as this region forms an anti-parallel β-structure with motif 2 β3-strand in the native HGDC. Hence, it became unstable after losing its anti-parallel β3-strand partner in motif 2, even though the first Greek key was still able to maintain most of its secondary and tertiary structures during the entire period of the simulations (see
Figure 8a right panel and
Figure 8b). Incidentally, the motif 2 β2-strand (Q55~L58) region encompasses some of the N-td interface residues (Q55 and F57) important for keeping the two domains intact. F57 has been found to be 80% conserved across the γ-crystallins and is crucial in maintaining protein stability in both bovine GDC and HGDC based on Ala substitution site-mutagenesis experiments [
5,
35]. Thus, losing the secondary β-structure encompassing this residue may be the key to overcoming the high kinetic barrier of the interdomain interface in the GdnHCl-induced structure and causing it to spiral down toward the subsequent unfolding and possibly misfolding pathway.
Previous studies have proposed several hypotheses along the same line about the aggregation of partially unfolded HGDC, starting from domain swapped dimers as an initial process for cataract formation. One MD simulation study proposed that three β-strands from motif 4 of the C-td interact with the N-td of another monomer [
15], while another based on single-molecule force spectroscopy suggested that β1 and β2 strands from the extruded β-hairpin loop in N-td motif 1 swap with adjacent monomers [
36]. A third study, combining simulations and experiments to examine mutant HGDCs, proposed that the β1-strand of motif 1 can form anti-parallel hydrogen bonds with the β2-strand (part of the inter-domain interface) from motif 4 of C-td to form domain-swapped dimers [
37].
Despite the fact that we were unable to see past the unfolding of motif 2 in either of the denaturant-induced structures examined in our study, we were able to deduce from our results that the destabilization and unfolding of the second Greek key led to the destabilization of the first Greek key. We have already witnessed this destabilizing process with motif 1 β3-strand losing secondary structure after the loss of adjacent motif 2 β3-strand (see
Figure 8). Hence, our results suggest that the destabilization of motif 1 begins with the loss of β3-strand that may eventually translate into the extension of the motif 1 β-hairpin loop causing β1- and/or β2-strand to swap with an adjacent monomer [
36] in the high-protein-concentration setting of the human lens. During the process, anti-parallel hydrogen bonds are formed between the extended region(s) of motif 1 and motif 4 β2-strand (part of the inter-domain interface) of the C-td [
37]. This exchange of β-strands leads to the formation of domain-swapped dimers, as proposed by Garcia-Manyes et al. (2016) [
36] and Serebryany et al. (2016) [
37].
2.7. Relative Positions of the Simulated HGDCs in the Energy Model of the Initial Stages of Unfolding
There has been no direct evidence in the past literature on the sequence of how the HGDC N-td unfolds prior to its complete unfolding. Our study fills in this gap by exploring the much earlier unfolding process of the N-td. We were able to do so because our starting structures for the MD simulations originated from the more dynamic conformers that have undergone structural disturbance induced by denaturants in the solution NMR experiments. These disturbed conformational states (on the brink of unfolding) provided us starting points in the simulations that revealed in greater detail of the initial unfolding mechanism, allowing us to tease out the very beginning stages of HGDC unfolding. Our results showed that motif 2 in the N-td was disturbed first in the very early stages of unfolding, which was previously speculated through MD simulations of the X-ray HGDC structure (PDB: 1HK0) with Ala substitution of selective residues. Our study provided additional evidence that supported the theory of the domain interface being the key element in stabilizing the HGDC structure and showed that if the interface was disturbed enough to break the kinetic energy barrier that kept it intact (as in the GdnHCl-induced conformer), the N-td would begin to destabilize starting with the second Greek key. This finding is consistent with that of the previous Ala mutagenesis simulation study, which has proposed that the integrity of the interdomain interface is the main determinant of motif 2 stability [
20]. On the other hand, the interface of the urea-induced structure was not disturbed enough to cross the high-kinetic-energy barrier necessary for unfolding to occur; therefore, it remained in a semi-intact state.
We calculated the changes in potential energy between the proteins at the beginning and the end of simulation time to understand the relative changes in potential energy experienced by the two denaturant-induced higher energy state structures simulated under 425K in a physiological solution devoid of denaturants. Based on our potential energy calculation and the observation of changes in molecular details revealed by MD simulations, we were able to deduce a more thorough potential energy model for the two higher-energy-state structures, as depicted in
Figure 10. The change in potential energy from the beginning to the end of simulations was slightly positive (44.1 kcal/mol) for the urea-induced structure, while that of the GdnHCl-induced structure was a much larger negative value (−220.58 kcal/mol). This signifies that the urea-induced structure had not traversed far from its initial conformation at the beginning of the MD simulations, which was exemplified by the predominantly intact secondary structure (
Figure 8a) and the two fairly well-preserved domains in their near-native conformations were maintained up till the end of the simulation time. The fact that the contact fraction at the domain interface experienced less fluctuation (
Figure S7) and the overall residue contact loss was less extensive than in the GdnHCl-induced structure (
Figure 5) suggested that the interface stability was still somewhat retained in the urea structure; therefore, the protein did not proceed toward the unfolding pathway. Another way to see it is that, although 5M urea may have brought the protein up to a higher-energy state, it was still not enough to break the kinetic barrier leading to the melt-down of the domain interface (
Figure 10). Hence, the urea-induced higher-energy structure fell into a local minimum well and could not get out. Rolling around within the potential well, it slightly reverted back toward the original higher-energy conformation near the end of the simulation time (
Figure 4a,d), thereby yielding a small positive potential energy change seen in
Figure 10.
In contrast, the interdomain interface of the GdnHCl-induced structure was disturbed enough that it overcame the high-kinetic-energy barrier, leading to the destabilization of this important region; thus, causing the protein to quickly shift away from the unfavorable high-energy state to find another transitioning point, in the process leading to a great disruption in motif 2. A previous study found that the integrity of the domain interface is critical for maintaining the kinetic stability of the N-td core under physiological solution condition [
33]. The GdnHCl-induced structure has evidently lost the integrity of the inter-domain interface; thereby, resulting in a great disorder in N-td, starting with motif 2. The change in overall conformation of the GdnHCl-induced higher energy structure during the MD simulations was accompanied by a negative change in potential energy, with the protein transitioning into a lower-energy state as seen in
Figure 10.
At first glance, it may seem counterintuitive that the more-disturbed GdnHCl-induced structure yielded a greater negative change in energy value (−220.58 kcal/mol) than the less-disrupted urea-induced structure (44.1 kcal/mol), as it is traditionally believed that the lower (the more negative) the energy value, the more stable the conformational state [
39]. However, if we extend the general concept of the free energy landscape and correlate it with our potential energy results we can explain this contradiction from the standpoint of the structural disturbance being relative to the initial GdnHCl-induced higher-energy state and that our sampling of the simulated structure at 100 ns may have captured a transitioning point from the disordered to the ultimately more ordered state of a misfolded conformation further along the pathway that serves as a fundamental building block of HGDC aggregation. Thus, the decrease in energy value signifies that the transitioning structure is moving downhill toward another basin in the energy landscape that is separated energetically from the original, natively folded state.
Despite the fact that the denaturant-induced structures were artificially synthesized from in vitro experiments and not the naturally occurring states present in the human lens, one cannot rule out that in the natural lens, disruptive environmental factors may possibly produce unstable HGDC conformers not unlike the denaturant-induced ones seen in our study. The 1M GdnHCl concentration used in this study was not enough to denature the protein, but only to bring it up to a slightly higher energy state (
Figure 1e), possibly to the brink of unfolding. Previous refolding experiments have found that HGDC can be refolded into a native-like state in physiological conditions without the presence of chaperones when diluted from 5 M to 1M GdnHCl. However, once the concentration drops below 1 M, the partially unfolded HGDCs began to form high-molecular-weight aggregates [
40]. In our study, we extracted the 1 M GdnHCl-induced higher-energy-state NMR structure of HGDC and submitted it to MD simulations in physiological solution conditions (pH 7, 136.7 mM NaCl). In contrast to the much milder dilution refolding experiments, the drastic environmental change (from 1M GdnHCl to physiological solution conditions) created in our study pushed the higher-energy conformer even further away from the native-fold. Based on our results, we believe that if the initial unfolding conformer (similar to the GdnHCl-induced structure observed in our study) does exist at some point in the lifetime of a HGDC within the high protein concentration environment of the human lens, we would expect to see it to either continue down the unfolding or misfolding pathway. Along the path, the non-native structure has the potential to form intermolecular association with neighboring crystallin proteins, and ultimately aggregating within the lens leading to cataract formation.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}