
Slipknot or Crystallographic Error: A Computational Analysis of the Plasmodium falciparum DHFR Structural Folds

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Multiple Sequence Alignment Highlights Two Plasmodium Species Specific Inserts in DHFR
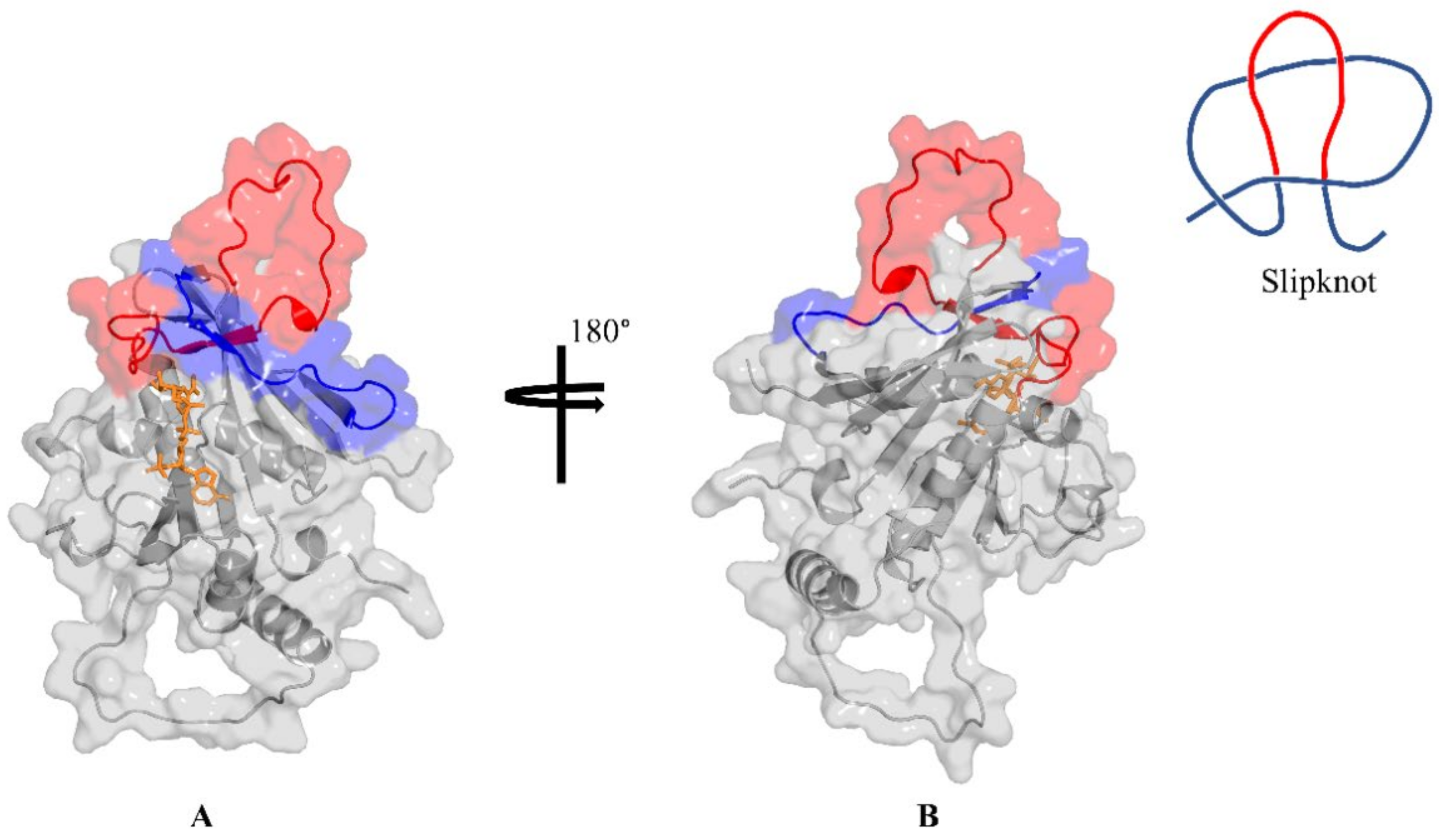
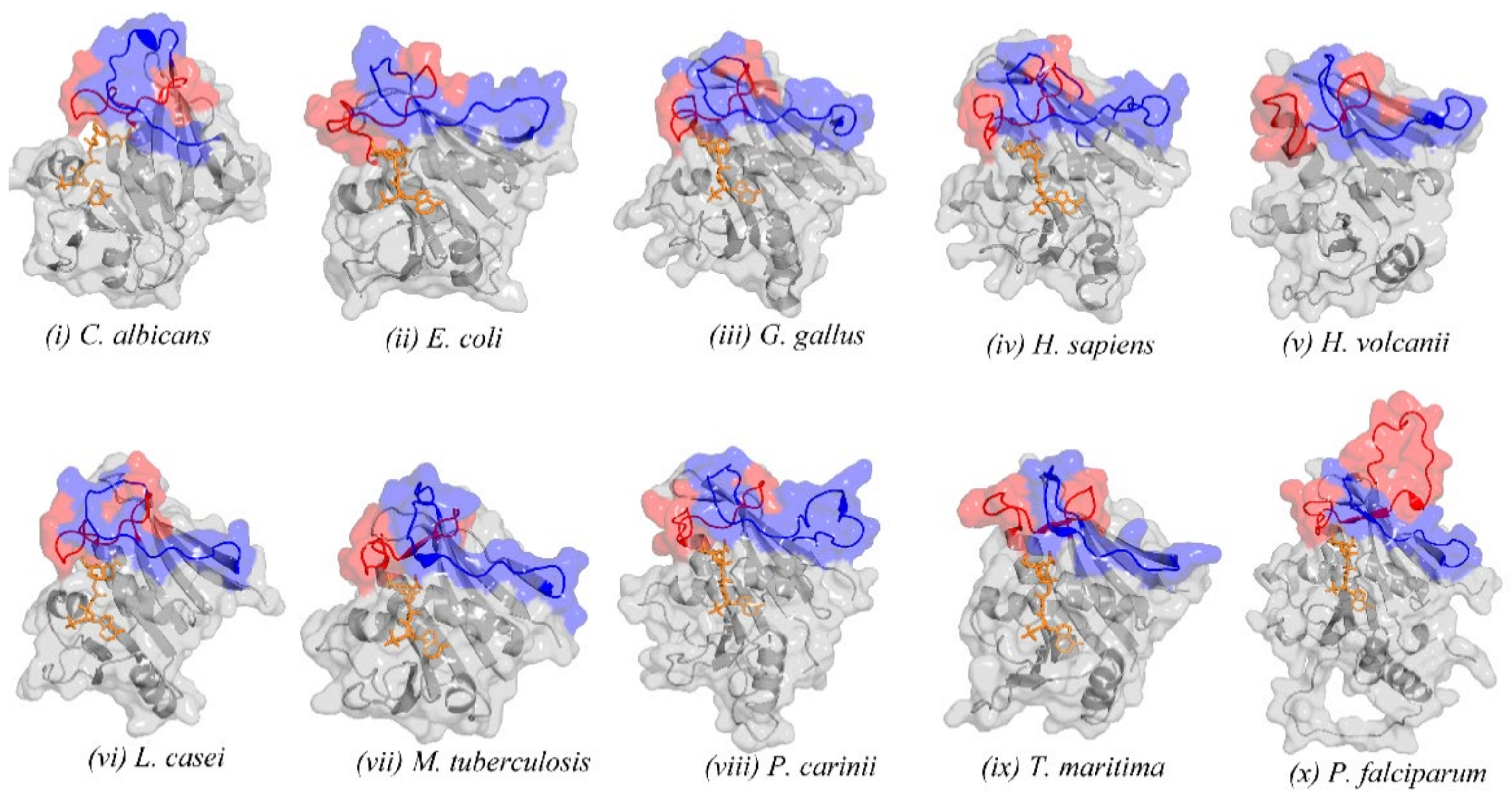
2.2. Retrieved DHFR Structures from the PDB Point to a Potentially Misplaced Loop Orientation, Which Is Exacerbated by Insert 1 of Plasmodium Species
2.3. Remodelling the DHFR Structure Using Ab Initio Modelling Programs to Verify Loop Topologies
2.4. Evaluating the Conformational Dynamics of the Slipknot-like Loops Using MD Simulations
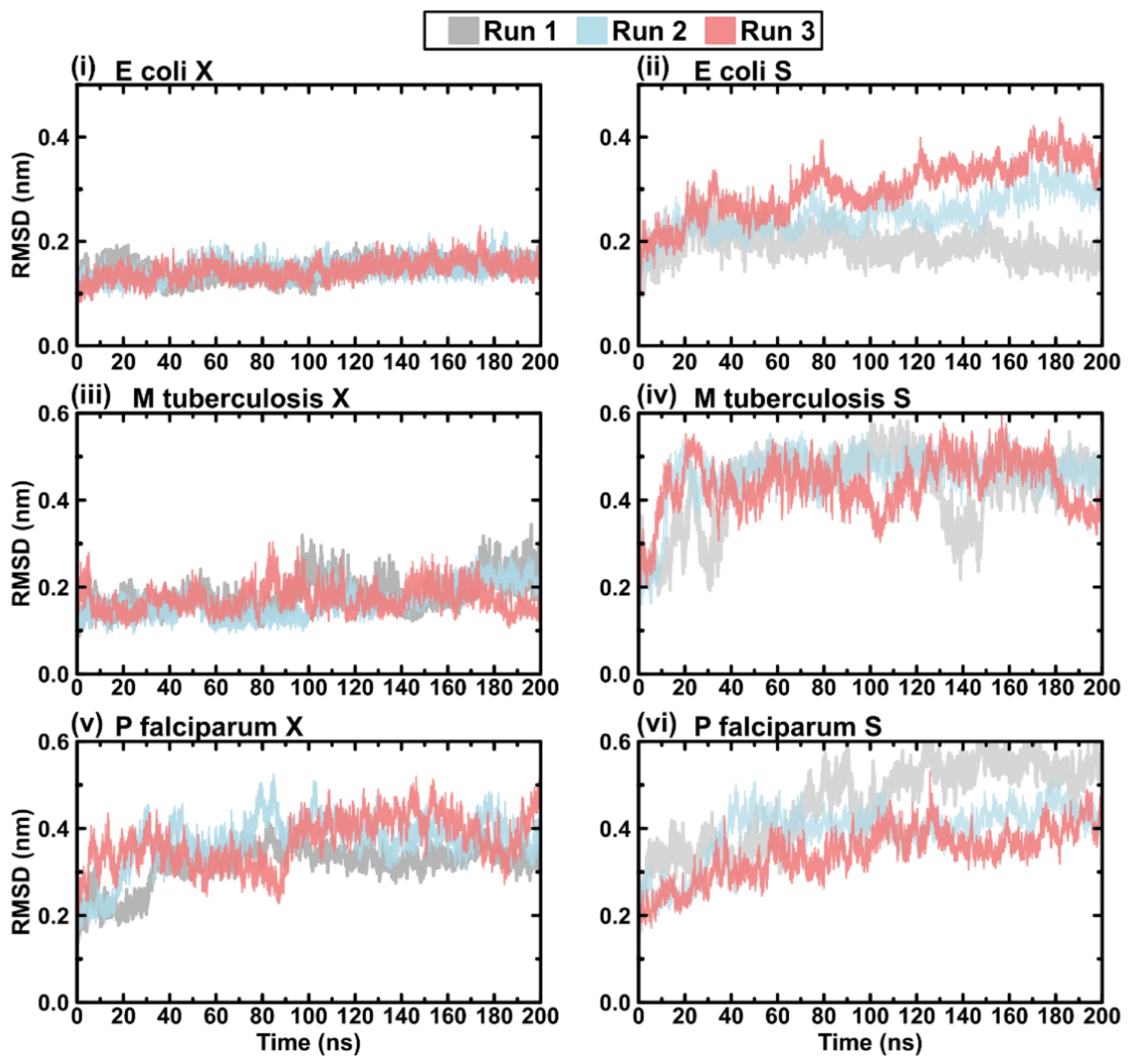
2.5. Protein RMSD
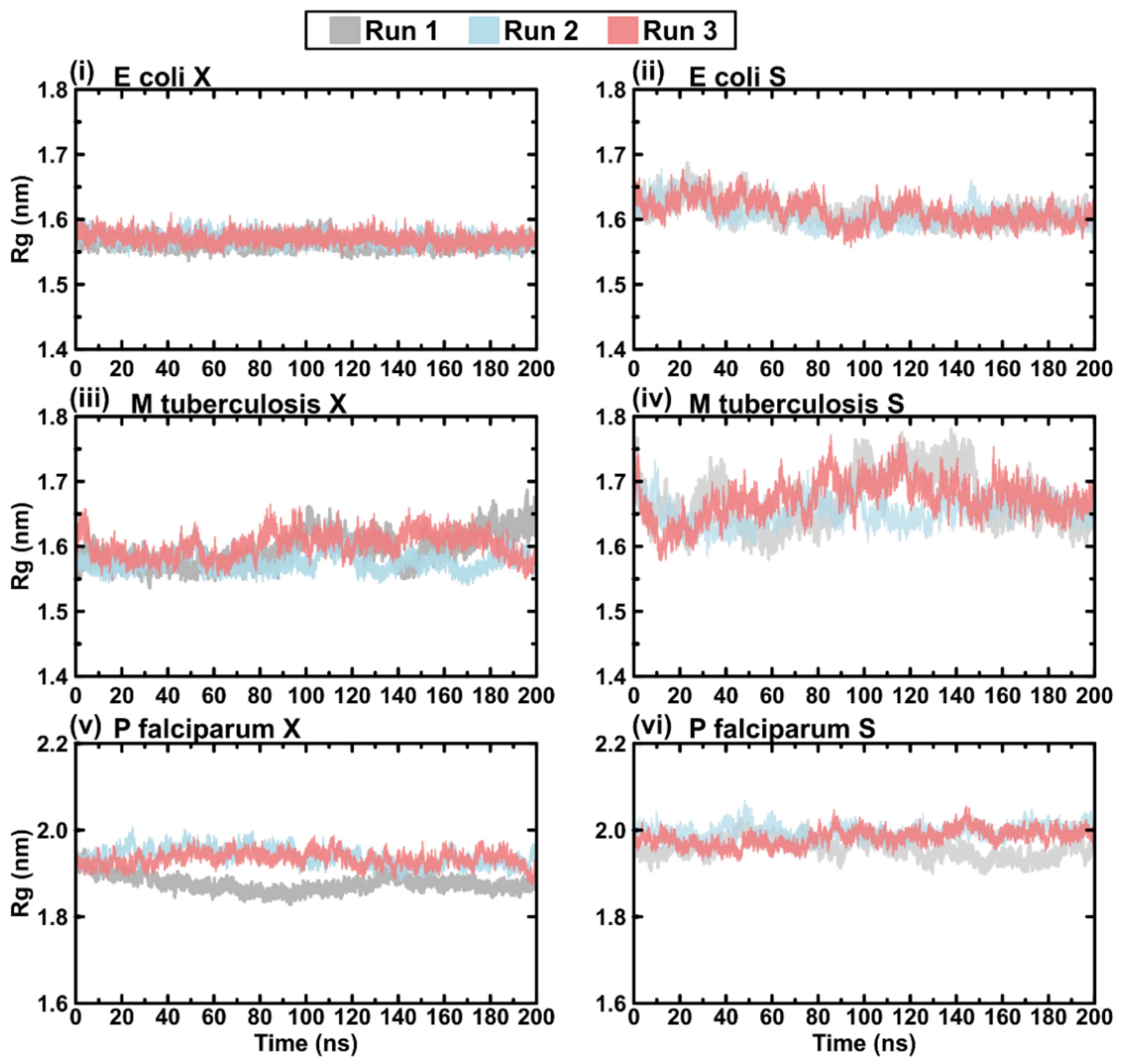
2.6. Protein Rg
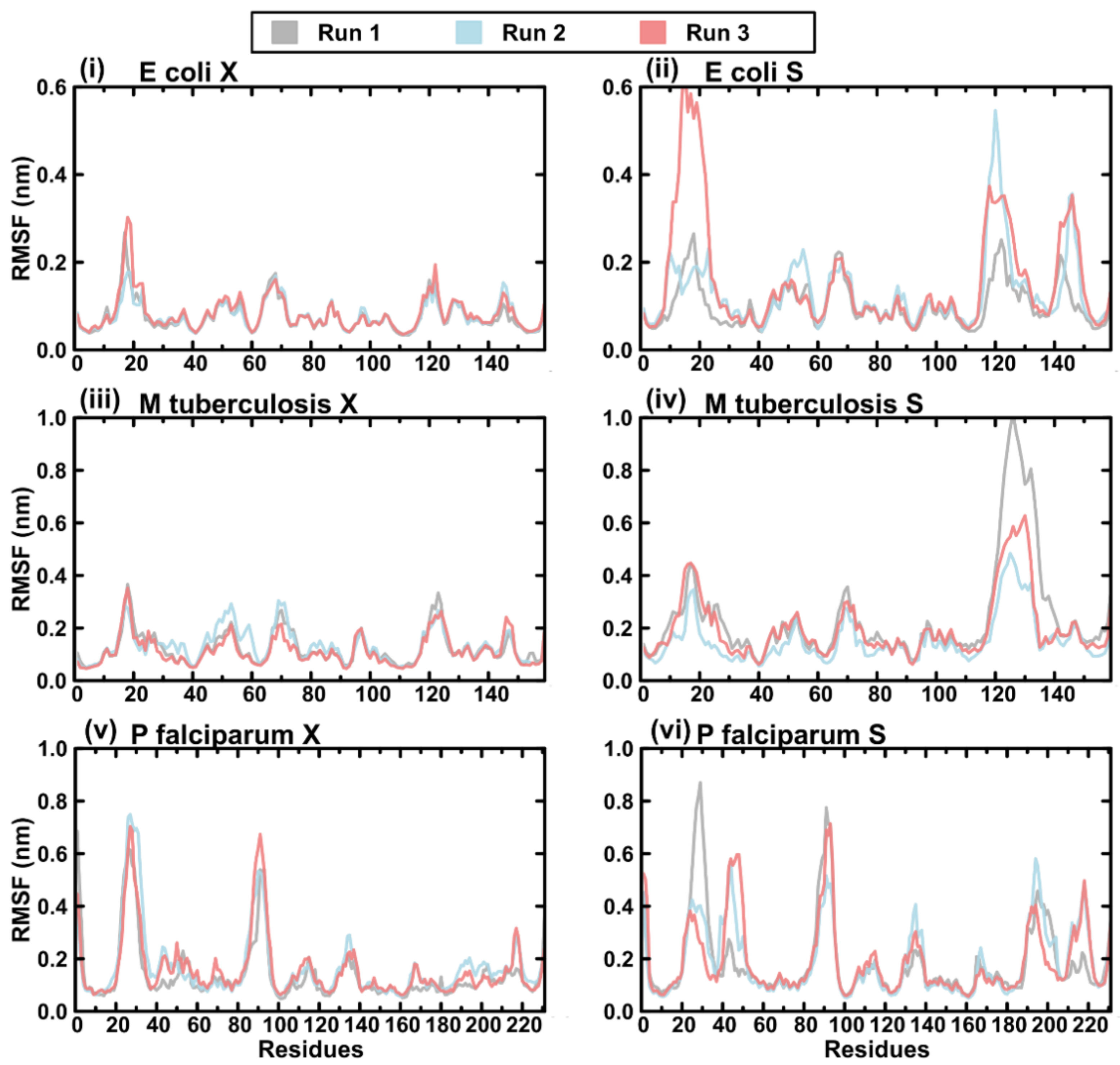
2.7. Protein RMSF
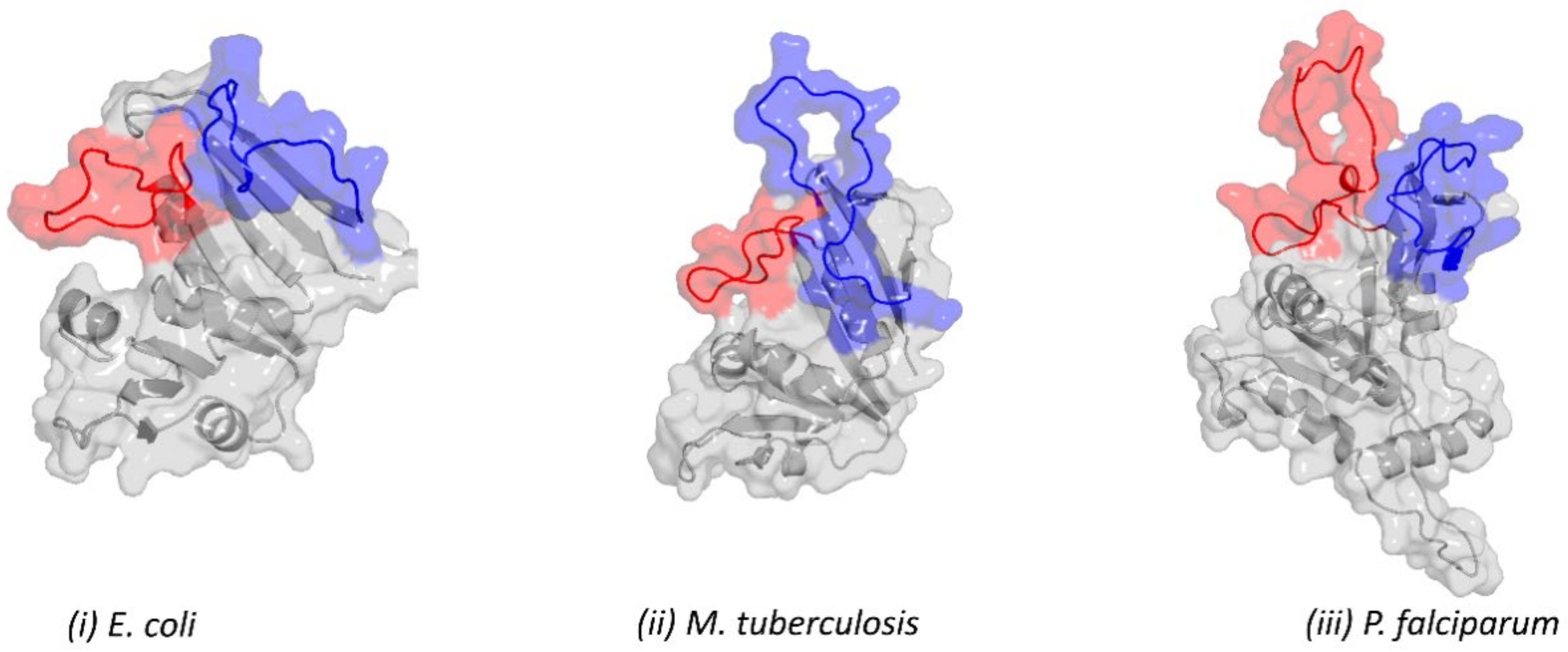
2.8. Post MD Structure Evaluation
3. Materials and Methods
3.1. Sequence Retrieval and Multiple Sequence Alignment
3.2. Search for the Presence of Knots
3.3. Loop Relaxation Using Homology Modelling
3.4. Structure Quality Evaluation
3.5. Molecular Dynamic Simulations
3.6. Trajectory Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zardecki, C.; Dutta, S.; Goodsell, D.S.; Voigt, M.; Burley, S.K. RCSB Protein Data Bank: A Resource for Chemical, Biochemical, and Structural Explorations of Large and Small Biomolecules. J. Chem. Educ. 2016, 93, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Faísca, P.F.N. Knotted Proteins: A Tangled Tale of Structural Biology. Comput. Struct. Biotechnol. J. 2015, 13, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamroz, M.; Niemyska, W.; Rawdon, E.J.; Stasiak, A.; Millett, K.C.; Sułkowski, P.; Sulkowska, J.I. KnotProt: A Database of Proteins with Knots and Slipknots. Nucleic Acids Res. 2015, 43, D306–D314. [Google Scholar] [CrossRef]
- Dabrowski-Tumanski, P.; Rubach, P.; Goundaroulis, D.; Dorier, J.; Sułkowski, P.; Millett, K.C.; Rawdon, E.J.; Stasiak, A.; Sulkowska, J.I. KnotProt 2.0: A Database of Proteins with Knots and Other Entangled Structures. Nucleic Acids Res. 2019, 47, D367–D375. [Google Scholar] [CrossRef] [Green Version]
- King, N.P.; Yeates, E.O.; Yeates, T.O. Identification of Rare Slipknots in Proteins and Their Implications for Stability and Folding. J. Mol. Biol. 2007, 373, 153–166. [Google Scholar] [CrossRef]
- Wlodawer, A.; Dauter, Z.; Porebski, P.J.; Minor, W.; Stanfield, R.; Jaskolski, M.; Pozharski, E.; Weichenberger, C.X.; Rupp, B. Detect, Correct, Retract: How to Manage Incorrect Structural Models. FEBS J. 2018, 285, 444–466. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in Protein Structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; et al. Malarial Dihydrofolate Reductase as a Paradigm for Drug Development against a Resistance-Compromised Target. Proc. Natl. Acad. Sci. USA 2012, 109, 16823–16828. [Google Scholar] [CrossRef] [Green Version]
- Müller, I.B.; Hyde, J.E. Folate Metabolism in Human Malaria Parasites—75 Years On. Mol. Biochem. Parasitol. 2013, 188, 63–77. [Google Scholar] [CrossRef]
- Yuvaniyama, J.; Chitnumsub, P.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Sirawaraporn, W.; Taylor, P.; Walkinshaw, M.D.; Yuthavong, Y. Insights into Antifolate Resistance from Malarial DHFR-TS Structures. Nat. Struct. Mol. Biol. 2003, 10, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Tata, R.B.; Tastan Bishop, Ö. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules 2020, 25, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin Resistance in Plasmodium Falciparum Malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Batzoglou, S. Multiple Sequence Alignment. Curr. Opin. Struct. Biol. 2006, 16, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C. Recent Progress in Multiple Sequence Alignment: A Survey. Pharmacogenomics 2002, 3, 131–144. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. UniProtKB/Swiss-Prot. Methods Mol. Biol. 2007, 406, 89–112. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Grishin, N.V. PROMALS3D: Multiple Protein Sequence Alignment Enhanced with Evolutionary and 3-Dimensional Structural Information. Methods Mol. Biol. 2014, 1079, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, T.; Sonnhammer, E.L.L. Automatic Assessment of Alignment Quality. Nucleic Acids Res. 2005, 33, 7120–7128. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of Loops in Protein Structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Dessì, A.; Dallocchio, R.; Tsamesidis, I.; Pau, M.C.; Turrini, F.M.; Pantaleo, A. Syk Inhibitors: New Computational Insights into Their Intraerythrocytic Action in Plasmodium Falciparum Malaria. Int. J. Mol. Sci. 2020, 21, 7009. [Google Scholar] [CrossRef] [PubMed]
- Tsamesidis, I.; Reybier, K.; Marchetti, G.; Pau, M.C.; Virdis, P.; Fozza, C.; Nepveu, F.; Low, P.S.; Turrini, F.M.; Pantaleo, A. Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium Falciparum-Parasitized Erythrocytes. Antioxidants 2020, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Sanyanga, T.A.; Nizami, B.; Tastan Bishop, Ö. Mechanism of Action of Non-Synonymous Single Nucleotide Variations Associated with α-Carbonic Anhydrase II Deficiency. Molecules 2019, 24, 3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhabha, G.; Ekiert, D.C.; Jennewein, M.; Zmasek, C.M.; Tuttle, L.M.; Kroon, G.; Dyson, H.J.; Godzik, A.; Wilson, I.A.; Wright, P.E. Divergent Evolution of Protein Conformational Dynamics in Dihydrofolate Reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Amamuddy, O.S.; Glenister, M.; Tastan Bishop, Ö. MDM-TASK-Web: A Web Platform for Protein Dynamic Residue Networks and Modal Analysis. bioRxiv 2021, 19, 5059–5071. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; et al. The Universal Protein Resource (UniProt): An Expanding Universe of Protein Information. Nucleic Acids Res. 2006, 34, D187–D191. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Sali, A. Statistical Potential for Assessment and Prediction of Protein Structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; Groot, B.L.d.; Grubmüller, H. More Bang for Your Buck: Improved Use of GPU Nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [Green Version]
- Paul Robustelli. 2021. Force-Fields. GitHub. Available online: https://github.com/paulrobustelli/Force-Fields (accessed on 14 September 2021).
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Conformation | Inner Pocket Volume (Druggability Score) | Outer Pocket Volume (Druggability Score) |
|---|---|---|---|
| P. falciparum | OC (run 1) | 397.167 (0.003) | 617.387 (0.025) |
| CC (run 1) | 627.592 (0.964) | 283.281 (0.006) | |
| E. coli | OC (run 3) | 1126.757 (0.895) | - |
| CC (run 1) | 421.624 (0.678) | - | |
| M. tuberculosis | OC (run 3) | 576.231 (0.851) | - |
| CC (run 1) | 169.615 (0.001) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tata, R.B.; Alsulami, A.F.; Sheik Amamuddy, O.; Blundell, T.L.; Tastan Bishop, Ö. Slipknot or Crystallographic Error: A Computational Analysis of the Plasmodium falciparum DHFR Structural Folds. Int. J. Mol. Sci. 2022, 23, 1514. https://doi.org/10.3390/ijms23031514
Tata RB, Alsulami AF, Sheik Amamuddy O, Blundell TL, Tastan Bishop Ö. Slipknot or Crystallographic Error: A Computational Analysis of the Plasmodium falciparum DHFR Structural Folds. International Journal of Molecular Sciences. 2022; 23(3):1514. https://doi.org/10.3390/ijms23031514
Chicago/Turabian StyleTata, Rolland B., Ali F. Alsulami, Olivier Sheik Amamuddy, Tom L. Blundell, and Özlem Tastan Bishop. 2022. "Slipknot or Crystallographic Error: A Computational Analysis of the Plasmodium falciparum DHFR Structural Folds" International Journal of Molecular Sciences 23, no. 3: 1514. https://doi.org/10.3390/ijms23031514
APA StyleTata, R. B., Alsulami, A. F., Sheik Amamuddy, O., Blundell, T. L., & Tastan Bishop, Ö. (2022). Slipknot or Crystallographic Error: A Computational Analysis of the Plasmodium falciparum DHFR Structural Folds. International Journal of Molecular Sciences, 23(3), 1514. https://doi.org/10.3390/ijms23031514