
Molecular Advances in MAFLD—A Link between Sphingolipids and Extracellular Matrix in Development and Progression to Fibrosis
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Pathogenesis of MAFLD
2.1. Diet
2.2. Insulin Resistance
2.3. Microbiota
2.4. Genes

{kind=link}
{kind=link}
| Factor | Mechanism | References |
|---|---|---|
| METABOLIC | ||
| Obesity | increased fat accumulation in the hepatocytes | [6,7] |
| Western diet | increased supply of fructose responsible for the intensification of gluconeogenesis and de novo lipogenesis in hepatocytes increased plasma TGs levels | [8,9] |
| Modification in microbiota | increased free fatty acid absorption by increased permeability of the intestinal wall increased synthesis of SCFAs that modulate lipogenesis, production of cholesterol, and glucose homeostasis decreased level of FIAF, which leads to increased activity of the LPL andincreased release of FAs from VLDL particles, and increased lipid accumulation in the liver | [18,19,20,21] |
| Hypertriglyceridemia | increased TGs accumulation in the hepatocytes | [6,13] |
| Hyperinsulinemia | promotion of hepatic de novo lipogenesis via activation of SREBP1c and ChREBP | [14] |
| Insulin resistance | generation of free FAs and glycerol resulting from TGs degradation and their storage in the liver | [12,13] |
| INFLAMMATORY | ||
| HIV | increased metabolic comorbidities hepatotoxic effect of lifelong antiretroviral therapy | [24] |
| Hepatitis C infection | the imbalance between pro-inflammatory and anti-inflammatory bioactive lipids increased ROS production increased lipid peroxidation | [25] |
| Oxidative stress | induction of hepatocytes injury by the inhibition of the mitochondrial respiratory chain enzymes increased ROS production increased lipid peroxidation increased cytokine production | [26,27] |
| GENETIC | ||
| Polymorphism in various genes: | ||
| PNPLA3 | increased fat accumulation in the hepatocytes increased liver enzymes | [22,28] |
| TM6SF2 | increased fat accumulation in the hepatocytes | [22,29] |
| GCKR | increased fat accumulation in the hepatocytes decreased β-oxidation | [29,30] |
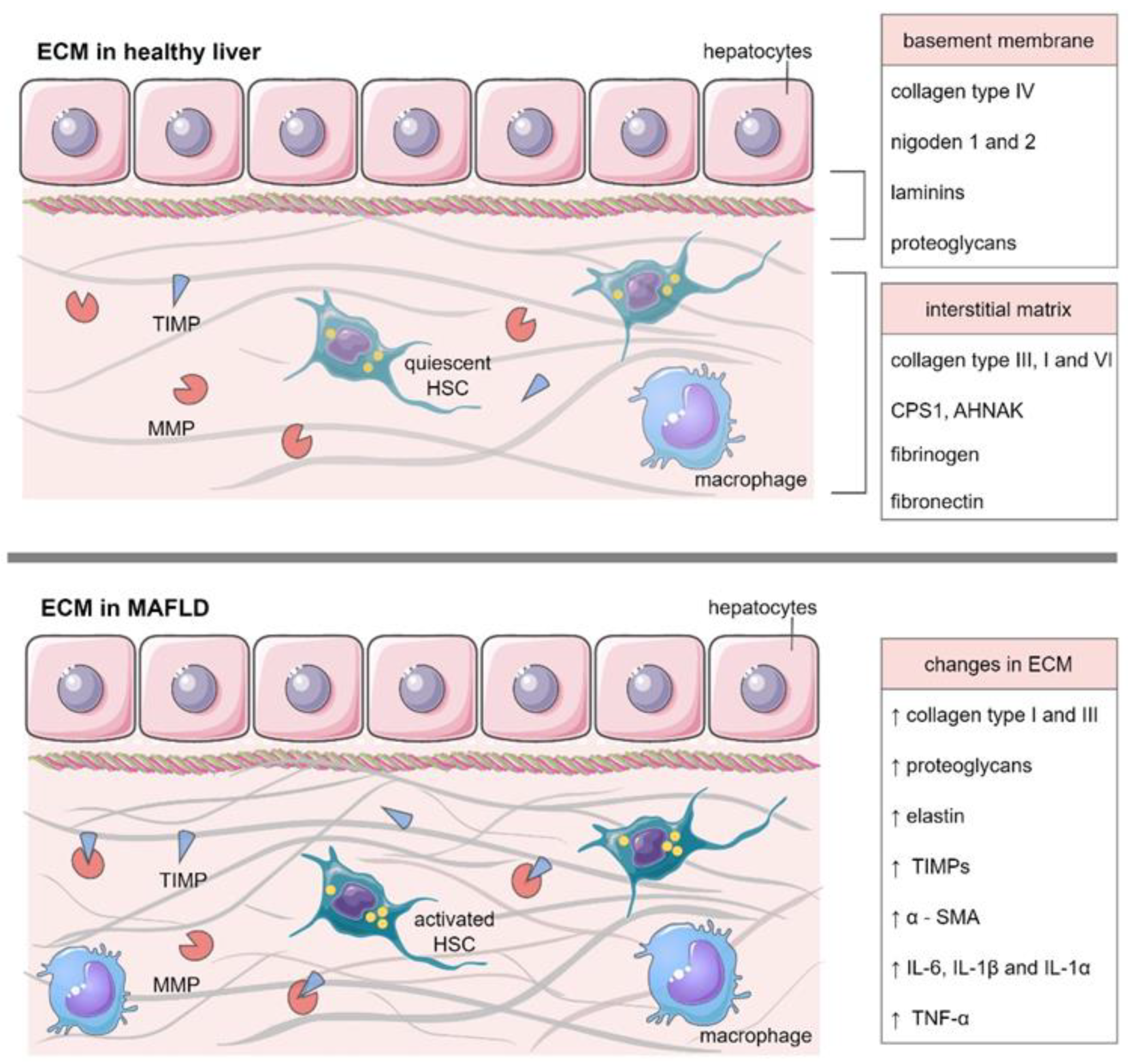
3. ECM in Healthy Liver
3.1. Collagens
3.1.1. Fibrillar Collagens
3.1.2. Basement Membrane Collagens
3.1.3. Short-Chain Collagens and FACITs
3.1.4. Changes during Aging
3.2. Glycoproteins
3.2.1. Fibrinogen and Fibronectin
3.2.2. Periostin, Tenascin C Nad X
3.2.3. Laminins
3.2.4. Changes during Aging
3.3. Proteoglycans
3.3.1. Decorin
3.3.2. Biglycan
3.3.3. Asporin
3.4. Matrix Metalloproteinases
3.4.1. Tissue Inhibitors of Metalloproteinases
3.4.2. Changes during Aging
3.5. Cytokines and Growth Factors
Changes during Aging
4. ECM in MAFLD
4.1. The Role of MMPs and TIMPs in MAFLD Deterioration
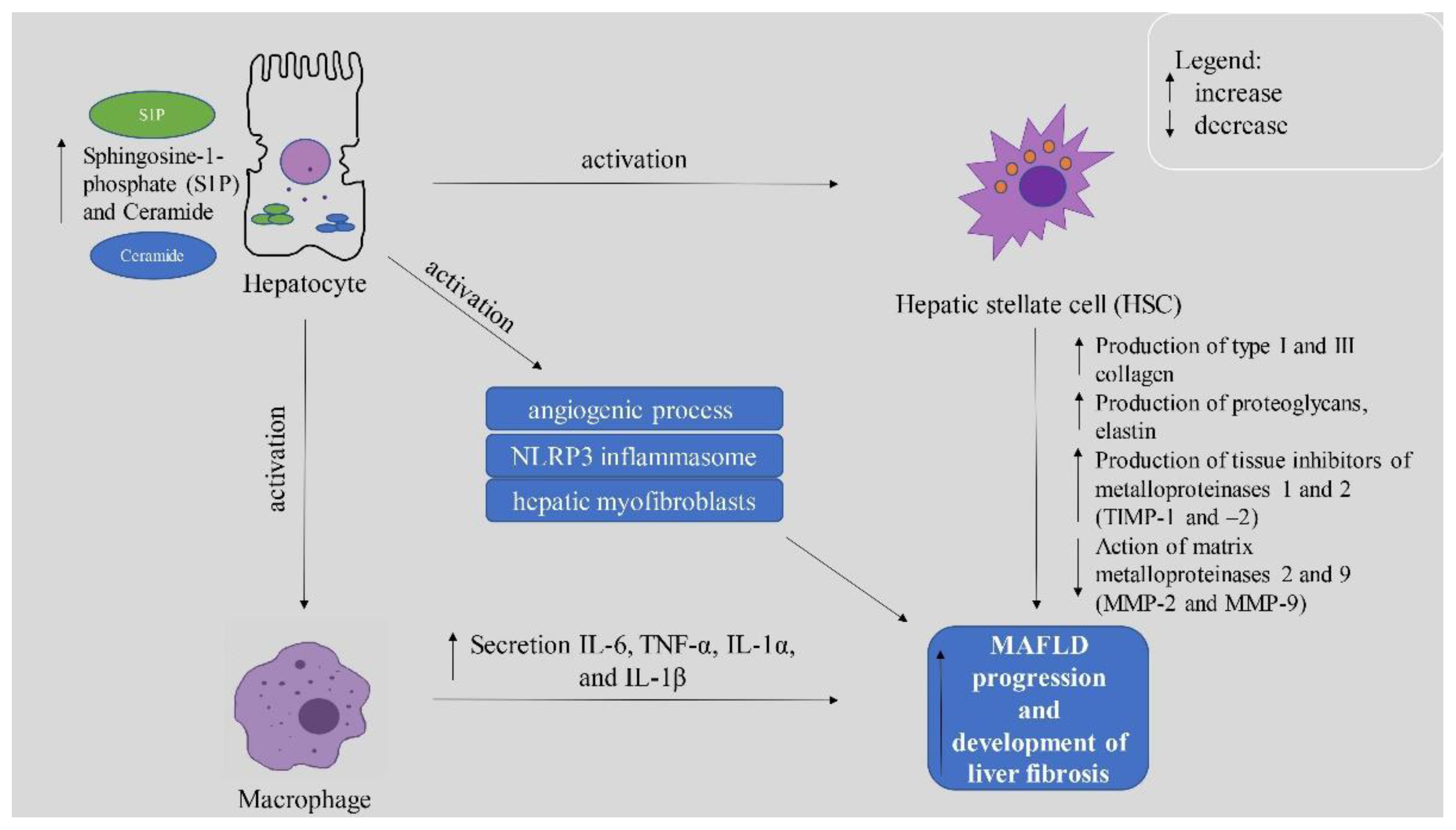
4.2. Importance of Sphingolipids in MAFLD Development and Deterioration
4.2.1. Ceramide and Its Derivatives
4.2.2. Sphingosine-1-Phosphate (S1P)
4.2.3. The Role of S1P Receptors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Lazarus, J.V.; Newsome, P.N.; Serfaty, L.; Aghemo, A.; Augustin, S.; Tsochatzis, E.; de Ledinghen, V.; Bugianesi, E.; Romero-Gomez, M.; et al. Disease burden and economic impact of diagnosed non-alcoholic steatohepatitis in five European countries in 2018, A cost-of-illness analysis. Liver Int. 2021, 41, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Eslam, M.; Kawaguchi, T.; Tsutsumi, T.; Nakano, D.; Yoshinaga, S.; Takahashi, H.; Anzai, K.; George, J.; Torimura, T. MAFLD identifies patients with significant hepatic fibrosis better than NAFLD. Liver Int. 2020, 40, 3018–3030. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xue, W.; Wang, M.; Wu, Y.; Singh, M.; Zhu, Y.; Kumar, R.; Lin, S. MAFLD Criteria May Overlook a Subtype of Patient with Steatohepatitis and Significant Fibrosis. Diabetes Metab. Syndr. Obes. 2021, 14, 3417–3425. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef]
- Chong, M.F.-F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [CrossRef]
- Berná, G.; Romero-Gomez, M. The role of nutrition in non-alcoholic fatty liver disease: Pathophysiology and management. Liver Int. 2020, 40 (Suppl. 1), 102–108. [Google Scholar] [CrossRef] [PubMed]
- Spooner, M.H.; Jump, D.B. Omega-3 fatty acids and nonalcoholic fatty liver disease in adults and children: Where do we stand? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Romauch, M.; Schreiber, R.; Grabner, G.F.; Hütter, S.; Kotzbeck, P.; Benedikt, P.; Eichmann, T.O.; Yamada, S.; Knittelfelder, O.; et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat. Commun. 2017, 8, 14859. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Anderson, N.; Borlak, J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev. 2008, 60, 311–357. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Claudel, T.; Kumari, P.; Haemmerle, G.; Pollheimer, M.J.; Stojakovic, T.; Scharnagl, H.; Halilbasic, E.; Gumhold, J.; Silbert, D.; et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology 2012, 56, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Suo, C.; Shi, O.; Lin, C.; Zhao, R.; Yuan, H.; Jin, L.; Zhang, T.; Chen, X. The Health Impact of MAFLD, a Novel Disease Cluster of NAFLD, Is Amplified by the Integrated Effect of Fatty Liver Disease-Related Genetic Variants. Clin. Gastroenterol. Hepatol. 2022, 20, e855–e875. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-T.; Liu, S.-S.; Xie, X.-J.; Liu, Q.; Xin, Y.-N.; Xuan, S.-Y. Independent and joint correlation of PNPLA3 I148M and TM6SF2 E167K variants with the risk of coronary heart disease in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 29. [Google Scholar] [CrossRef]
- Cervo, A.; Shengir, M.; Patel, K.; Sebastiani, G. NASH in HIV. Curr. HIV/AIDS Rep. 2020, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Beneficial role of bioactive lipids in the pathobiology, prevention, and management of HBV, HCV and alcoholic hepatitis, NAFLD, and liver cirrhosis: A review. J. Adv. Res. 2019, 17, 17–29. [Google Scholar] [CrossRef]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef]
- Hong, T.; Chen, Y.; Li, X.; Lu, Y. The Role and Mechanism of Oxidative Stress and Nuclear Receptors in the Development of NAFLD. Oxid. Med. Cell Longev. 2021, 2021, 6889533. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Jonas, W.; Schürmann, A. Genetic and epigenetic factors determining NAFLD risk. Mol. Metab. 2021, 50, 101111. [Google Scholar] [CrossRef]
- Rajas, F.; Gautier-Stein, A.; Mithieux, G. Glucose-6 Phosphate, A Central Hub for Liver Carbohydrate Metabolism. Metabolites 2019, 9, 282. [Google Scholar] [CrossRef]
- Ortiz, C.; Schierwagen, R.; Schaefer, L.; Klein, S.; Trepat, X.; Trebicka, J. Extracellular Matrix Remodeling in Chronic Liver Disease. Curr. Tissue Microenviron Rep. 2021, 2, 41–52. [Google Scholar] [CrossRef]
- Roderfeld, M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. 2018, 68–69, 452–462. [Google Scholar] [CrossRef]
- Saheli, M.; Sepantafar, M.; Pournasr, B.; Farzaneh, Z.; Vosough, M.; Piryaei, A.; Baharvand, H. Three-dimensional liver-derived extracellular matrix hydrogel promotes liver organoids function. J. Cell. Biochem. 2018, 119, 4320–4333. [Google Scholar] [CrossRef]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 2020, 11, 2903. [Google Scholar] [CrossRef]
- Bedossa, P.; Paradis, V. Liver extracellular matrix in health and disease. J. Pathol. 2003, 200, 504–515. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef]
- Arteel, G.E.; Naba, A. The liver matrisome–looking beyond collagens. JHEP Rep. 2020, 2, 100115. [Google Scholar] [CrossRef]
- Verstegen, M.M.A.; Willemse, J.; van den Hoek, S.; Kremers, G.J.; Luider, T.M.; van Huizen, N.A.; Willemssen, F.E.J.A.; Metselaar, H.J.; Ijzermans, J.N.M.; van der Laan, L.J.W.; et al. Decellularization of Whole Human Liver Grafts Using Controlled Perfusion for Transplantable Organ Bioscaffolds. Stem Cells Dev. 2017, 26, 1304–1315. [Google Scholar] [CrossRef]
- Arriazu, E.; Ruiz de Galarreta, M.; Cubero, F.J.; Varela-Rey, M.; Pérez de Obanos, M.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid. Redox. Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Daniels, S.J.; Holm Nielsen, S.; Bager, C.; Rasmussen, D.G.K.; Loomba, R.; Surabattula, R.; Villesen, I.F.; Luo, Y.; Shevell, D.; et al. Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int. 2020, 40, 736–750. [Google Scholar] [CrossRef]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Bella, J.; Hulmes, D.J.S. Fibrillar collagens. Subcell. Biochem. 2017, 82, 457–490. [Google Scholar] [CrossRef]
- Takai, K.K.; Hattori, S.; Irie, S. Type V collagen distribution in liver is reconstructed in coculture system of hepatocytes and stellate cells; the possible functions of type V collagen in liver under normal and pathological conditions. Cell Struct. Funct. 2001, 26, 289–302. [Google Scholar] [CrossRef]
- Brown, B.; Lindberg, K.; Reing, J.; Stolz, D.B.; Badylak, S.F. The basement membrane component of biologic scaffolds derived from extracellular matrix. Tissue Eng. 2006, 12, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef]
- Calvo, A.; Moreno, L.; Moreno, L.; Toivonen, J.; Manzano, R.; Molina, N.; de La Torre, M.; López, T.; Miana-Mena, F.; Muñoz, M.; et al. Type XIX collagen: A promising biomarker from the basement membranes. Neural Regen. Res. 2020, 15, 988–995. [Google Scholar]
- Oudart, J.B.; Brassart-Pasco, S.; Vautrin, A.; Sellier, C.; Machado, C.; Dupont-Deshorgue, A.; Brassart, B.; Baud, S.; Dauchez, M.; Monboisse, J.C.; et al. Plasmin releases the anti-tumor peptide from the NC1 domain of collagen XIX. Oncotarget 2015, 6, 3656–3668. [Google Scholar] [CrossRef]
- Gagliano, N.; Arosio, B.; Grizzi, F.; Masson, S.; Tagliabue, J.; Dioguardi, N.; Vergani, C.; Annoni, G. Reduced collagenolytic activity of matrix metalloproteinases and development of liver fibrosis in the aging rat. Mech. Ageing Dev. 2002, 123, 413–425. [Google Scholar] [CrossRef]
- Acun, A.; Oganesyan, R.; Uygun, K.; Yeh, H.; Yarmush, M.L.; Uygun, B.E. Liver donor age affects hepatocyte function through age-dependent changes in decellularized liver matrix. Biomaterials 2021, 270, 120689. [Google Scholar] [CrossRef]
- Horbett, T.A. Fibrinogen adsorption to biomaterials. J. Biomed. Mater. Res.-Part A 2018, 106, 2777–2788. [Google Scholar] [CrossRef]
- Halper, J.; Kjaer, M. Basic Components of Connective Tissues and Extracellular Matrix: Elastin, Fibrillin, Fibulins, Fibrinogen, Fibronectin, Laminin, Tenascins and Thrombospondins. In Progress in Heritable Soft Connective Tissue Diseases; Springer: Berlin/Heidelberg, Germany, 2014; pp. 31–47. [Google Scholar]
- Aziz-Seible, R.S.; Casey, C.A. Fibronectin: Functional character and role in alcoholic liver disease. World J. Gastroenterol. 2011, 17, 2482–2499. [Google Scholar] [CrossRef]
- Lenselink, E.A. Role of fibronectin in normal wound healing. Int. Wound J. 2015, 12, 313–316. [Google Scholar] [CrossRef]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. 2017, 60–61, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhong, F.; Jiang, S.; Guo, Q.; Jin, H.; Wang, F.; Li, M.; Wang, L.; Chen, A.; Zhang, F.; et al. Periostin in chronic liver diseases: Current research and future perspectives. Life Sci. 2019, 226, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Rohn, F.; Kordes, C.; Castoldi, M.; Götze, S.; Poschmann, G.; Stühler, K.; Herebian, D.; Benk, A.S.; Geiger, F.; Zhang, T.; et al. Laminin-521 promotes quiescence in isolated stellate cells from rat liver. Biomaterials 2018, 180, 36–51. [Google Scholar] [CrossRef]
- Lorenzini, S.; Bird, T.G.; Boulter, L.; Bellamy, C.; Samuel, K.; Aucott, R.; Clayton, E.; Andreone, P.; Bernardi, M.; Golding, M.; et al. Characterisation of a stereotypical cellular and extracellular adult liver progenitor cell niche in rodents and diseased human liver. Gut 2010, 59, 645–654. [Google Scholar] [CrossRef]
- Dubuquoy, L.; Louvet, A.; Lassailly, G.; Truant, S.; Boleslawski, E.; Artru, F.; Maggiotto, F.; Gantier, E.; Buob, D.; Leteurtre, E.; et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 2015, 64, 1949–1960. [Google Scholar] [CrossRef]
- Appunni, S.; Anand, V.; Khandelwal, M.; Gupta, N.; Rubens, M.; Sharma, A. Small Leucine Rich Proteoglycans (decorin, biglycan and lumican) in cancer. Clin. Chim. Acta 2019, 491, 1–7. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.B.; Hansen, N.-U.B.; Bay-Jensen, A.-C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G807–G830. [Google Scholar] [CrossRef]
- Neill, T.; Painter, H.; Buraschi, S.; Owens, R.T.; Lisanti, M.P.; Schaefer, L.; Iozzo, R.V. Decorin antagonizes the angiogenic network: Concurrent inhibition of met, hypoxia inducible factor 1α, vascular endothelial growth factor A, and induction of thrombospondin-1 and tiMP3. J. Biol. Chem. 2012, 287, 5492–5506. [Google Scholar] [CrossRef]
- Reszegi, A.; Horváth, Z.; Fehér, H.; Wichmann, B.; Tátrai, P.; Kovalszky, I.; Baghy, K. Protective Role of Decorin in Primary Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 645. [Google Scholar] [CrossRef]
- Baghy, K.; Tátrai, P.; Regős, E.; Kovalszky, I. Proteoglycans in liver cancer. World J. Gastroenterol. 2016, 22, 379–393. [Google Scholar] [CrossRef]
- Sobhy, A.; Fakhry, M.M.; AAzeem, H.; Ashmawy, A.M.; Khalifa, H.O. Significance of biglycan and osteopontin as non-invasive markers of liver fibrosis in patients with chronic hepatitis B virus and chronic hepatitis C virus. J. Investig. Med. 2019, 67, 681–685. [Google Scholar] [CrossRef]
- Zeng-Brouwers, J.; Beckmann, J.; Nastase, M.V.; Iozzo, R.V.; Schaefer, L. De novo expression of circulating biglycan evokes an innate inflammatory tissue response via MyD88/TRIF pathways. Matrix Biol. 2014, 35, 132–142. [Google Scholar] [CrossRef]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Lichtinghagen, R.; Helmbrecht, T.; Arndt, B.; Böker, K.H. Expression pattern of matrix metalloproteinases in human liver. Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 65–71. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.-H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 2002, 100, 1160–1167. [Google Scholar] [CrossRef]
- Chirco, R.; Liu, X.-W.; Jung, K.-K.; Kim, H.-R.C. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006, 25, 99–113. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Feng, B.; Edwards, D.R.; Cocuzzi, E.T.; Malyankar, U.M. Tissue inhibitor of metalloproteinases (TIMP, aka EPA): Structure, control of expression and biological functions. Pharmacol. Ther. 1993, 59, 329–341. [Google Scholar] [CrossRef]
- Murphy, F.R.; Issa, R.; Zhou, X.; Ratnarajah, S.; Nagase, H.; Arthur, M.J.P.; Benyon, C.; Iredale, J.P. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: Implications for reversibility of liver fibrosis. J. Biol. Chem. 2002, 277, 11069–11076. [Google Scholar] [CrossRef]
- Murthy, A.; Shao, Y.W.; Defamie, V.; Wedeles, C.; Smookler, D.; Khokha, R. Stromal TIMP3 regulates liver lymphocyte populations and provides protection against Th1 T cell-driven autoimmune hepatitis. J. Immunol. 2012, 188, 2876–2883. [Google Scholar] [CrossRef]
- Kim, L.B.; Shkurupy, V.A.; Putyatina, A.N. Age-Related Changes in the System Metalloproteinases/Tissue Metalloproteinase Inhibitors and Proteoglycan Components in Mouse Organs. Bull. Exp. Biol. Med. 2016, 161, 32–36. [Google Scholar] [CrossRef]
- Merline, R.; Schaefer, R.M.; Schaefer, L. The matricellular functions of small leucine-rich proteoglycans (SLRPs). J. Cell Commun. Signal. 2009, 3, 323–335. [Google Scholar] [CrossRef]
- Wijelath, E.S.; Rahman, S.; Namekata, M.; Murray, J.; Nishimura, T.; Mostafavi-Pour, Z.; Patel, Y.; Suda, Y.; Humphries, M.J.; Sobel, M. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: Enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ. Res. 2006, 99, 853–860. [Google Scholar] [CrossRef]
- Ariel, A.; Hershkoviz, R.; Cahalon, L.; Williams, D.E.; Akiyama, S.K.; Yamada, K.M.; Chen, C.; Alon, R.; Lapidot, T.; Lider, O. Induction of T cell adhesion to extracellular matrix or endothelial cell ligands by soluble or matrix-bound interleukin-7. Eur. J. Immunol. 1997, 27, 2562–2570. [Google Scholar] [CrossRef]
- Hershkoviz, R.; Goldkorn, I.; Lider, O. Tumour necrosis factor-alpha interacts with laminin and functions as a pro-adhesive cytokine. Immunology 1995, 85, 125–130. [Google Scholar]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef]
- Okazaki, I.; Noro, T.; Tsutsui, N.; Yamanouchi, E.; Kuroda, H.; Nakano, M.; Yokomori, H.; Inagaki, Y. Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs). Cancers 2014, 6, 1220–1255. [Google Scholar] [CrossRef]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef]
- Ezhilarasan, D.; Sokal, E.; Najimi, M. Hepatic fibrosis: It is time to go with hepatic stellate cell-specific therapeutic targets. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 192–197. [Google Scholar] [CrossRef]
- Vallet, S.D.; Ricard-Blum, S. Lysyl oxidases: From enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019, 63, 349–364. [Google Scholar] [CrossRef]
- Hanson, A.; Wilhelmsen, D.; DiStefano, J.K. The role of long non-coding RNAs (lncRNAs) in the development and progression of fibrosis associated with nonalcoholic fatty liver disease (NAFLD). Noncoding RNA 2018, 4, 18. [Google Scholar] [CrossRef]
- Munsterman, I.D.; Kendall, T.J.; Khelil, N.; Popa, M.; Lomme, R.; Drenth, J.P.H.; Tjwa, E.T.T.L. Extracellular matrix components indicate remodelling activity in different fibrosis stages of human non-alcoholic fatty liver disease. Histopathology 2018, 73, 612–621. [Google Scholar] [CrossRef]
- Barchuk, M.; Schreier, L.; Berg, G.; Miksztowicz, V. Metalloproteinases in non-alcoholic fatty liver disease and their behavior in liver fibrosis. Horm. Mol. Biol. Clin. Investig. 2018, 41, 37. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends Endocrinol. Metab. 2017, 28, 506–518. [Google Scholar] [CrossRef]
- Oates, J.R.; McKell, M.C.; Moreno-Fernandez, M.E.; Damen, M.S.M.A.; Deepe, G.S.J.; Qualls, J.E.; Divanovic, S. Macrophage Function in the Pathogenesis of Non-alcoholic Fatty Liver Disease: The Mac Attack. Front. Immunol. 2019, 10, 2893. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a mediator of non-alcoholic Fatty liver disease and associated atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef]
- Yue, F.; Xia, K.; Wei, L.; Xing, L.; Wu, S.; Shi, Y.; Lam, S.M.; Shui, G.; Xiang, X.; Russell, R.; et al. Effects of constant light exposure on sphingolipidomics and progression of NASH in high-fat-fed rats. J. Gastroenterol. Hepatol. 2020, 35, 1978–1989. [Google Scholar] [CrossRef]
- Hirsova, P.; Weng, P.; Salim, W.; Bronk, S.F.; Griffith, T.S.; Ibrahim, S.H.; Gores, G.J. TRAIL Deletion Prevents Liver, but Not Adipose Tissue, Inflammation during Murine Diet-Induced Obesity. Hepatol. Commun. 2017, 1, 648–662. [Google Scholar] [CrossRef]
- Török, N.J. Extracellular vesicles and ceramide: New mediators for macrophage chemotaxis? J. Lipid. Res. 2016, 57, 157–158. [Google Scholar] [CrossRef]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1α-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; de Araujo Horcel, L.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 646–663.e4. [Google Scholar] [CrossRef]
- Hernández, A.; Arab, J.P.; Reyes, D.; Lapitz, A.; Moshage, H.; Bañales, J.M.; Arrese, M. Extracellular Vesicles in NAFLD/ALD: From Pathobiology to Therapy. Cells 2020, 9, 817. [Google Scholar] [CrossRef]
- Kurek, K.; Piotrowska, D.M.; Wiesiołek-Kurek, P.; Łukaszuk, B.; Chabowski, A.; Górski, J.; Zendzian-Piotrowska, M. Inhibition of ceramide de novo synthesis reduces liver lipid accumulation in rats with nonalcoholic fatty liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2014, 34, 1074–1083. [Google Scholar] [CrossRef]
- Kim, Y.-R.; Lee, E.-J.; Shin, K.-O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.-M.; Park, J.-W.; Futerman, A.H.; Park, W.-J. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zanieri, F.; Levi, A.; Montefusco, D.; Longato, L.; de Chiara, F.; Frenguelli, L.; Omenetti, S.; Andreola, F.; Luong, T.V.; Massey, V.; et al. Exogenous Liposomal Ceramide-C6 Ameliorates Lipidomic Profile, Energy Homeostasis, and Anti-Oxidant Systems in NASH. Cells 2020, 9, 1237. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, H.; Peterson, C.; Liu, H.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim. Biophys. Acta 2002, 1582, 8–17. [Google Scholar] [CrossRef]
- Goto, H.; Miyamoto, M.; Kihara, A. Direct uptake of sphingosine-1-phosphate independent of phospholipid phosphatases. J. Biol. Chem. 2021, 296, 100605. [Google Scholar] [CrossRef]
- Rohrbach, T.; Maceyka, M.; Spiegel, S. Sphingosine kinase and sphingosine-1-phosphate in liver pathobiology. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 543–553. [Google Scholar] [CrossRef]
- Kołakowski, A.; Kurzyna, P.F.; Żywno, H.; Bzdęga, W.; Harasim-Symbor, E.; Chabowski, A.; Konstantynowicz-Nowicka, K. Vitamin K2 as a New Modulator of the Ceramide De Novo Synthesis Pathway. Molecules 2021, 26, 3377. [Google Scholar] [CrossRef]
- Jin, J.; Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Huang, Y. LPS and palmitate synergistically stimulate sphingosine kinase 1 and increase sphingosine 1 phosphate in RAW264.7 macrophages. J. Leukoc. Biol. 2018, 104, 843–853. [Google Scholar] [CrossRef]
- Al Fadel, F.; Fayyaz, S.; Japtok, L.; Kleuser, B. Involvement of Sphingosine 1-Phosphate in Palmitate-Induced Non-Alcoholic Fatty Liver Disease. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 1637–1645. [Google Scholar] [CrossRef]
- Liao, C.-Y.; Song, M.J.; Gao, Y.; Mauer, A.S.; Revzin, A.; Malhi, H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front. Immunol. 2018, 9, 2980. [Google Scholar] [CrossRef] [PubMed]
- Geng, T.; Sutter, A.; Harland, M.D.; Law, B.A.; Ross, J.S.; Lewin, D.; Palanisamy, A.; Russo, S.B.; Chavin, K.D.; Cowart, L.A. SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. J. Lipid Res. 2015, 56, 2359–2371. [Google Scholar] [CrossRef]
- Anderson, A.K.; Lambert, J.M.; Montefusco, D.J.; Tran, B.N.; Roddy, P.; Holland, W.L.; Cowart, L.A. Depletion of adipocyte sphingosine kinase 1 leads to cell hypertrophy, impaired lipolysis, and nonalcoholic fatty liver disease. J. Lipid Res. 2020, 61, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yue, S.; Yang, L.; Liu, X.; Han, Z.; Zhang, Y.; Li, L. Sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis is involved in liver fibrosis-associated angiogenesis. J. Hepatol. 2013, 59, 114–123. [Google Scholar] [CrossRef]
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Macrophage Sphingosine 1-Phosphate Receptor 2 Blockade Attenuates Liver Inflammation and Fibrogenesis Triggered by NLRP3 Inflammasome. Front. Immunol. 2020, 11, 1149. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, S.; You, H.; Liu, X.; Lin, M.; Yang, L.; Li, L. Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J. Hepatol. 2011, 54, 1205–1213. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Syn, W.-K.; Wang, Z.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Amitriptyline inhibits nonalcoholic steatohepatitis and atherosclerosis induced by high-fat diet and LPS through modulation of sphingolipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E131–E144. [Google Scholar] [CrossRef]
- Mauer, A.S.; Hirsova, P.; Maiers, J.L.; Shah, V.H.; Malhi, H. Inhibition of sphingosine 1-phosphate signaling ameliorates murine nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G300–G313. [Google Scholar] [CrossRef]
- Rohrbach, T.D.; Asgharpour, A.; Maczis, M.A.; Montefusco, D.; Cowart, L.A.; Bedossa, P.; Sanyal, A.J.; Spiegel, S. FTY720/fingolimod decreases hepatic steatosis and expression of fatty acid synthase in diet-induced nonalcoholic fatty liver disease in mice. J. Lipid Res. 2019, 60, 1311–1322. [Google Scholar] [CrossRef]
- Becker, S.; Kinny-Köster, B.; Bartels, M.; Scholz, M.; Seehofer, D.; Berg, T.; Engelmann, C.; Thiery, J.; Ceglarek, U.; Kaiser, T. Low sphingosine-1-phosphate plasma levels are predictive for increased mortality in patients with liver cirrhosis. PLoS ONE 2017, 12, e0174424. [Google Scholar] [CrossRef]
- Ding, B.-S.; Liu, C.H.; Sun, Y.; Chen, Y.; Swendeman, S.L.; Jung, B.; Chavez, D.; Cao, Z.; Christoffersen, C.; Nielsen, L.B.; et al. HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P(1)) promotes regeneration and suppresses fibrosis in the liver. JCI Insight 2016, 1, e87058. [Google Scholar] [CrossRef]
- Hajny, S.; Christoffersen, C. A Novel Perspective on the ApoM-S1P Axis, Highlighting the Metabolism of ApoM and Its Role in Liver Fibrosis and Neuroinflammation. Int. J. Mol. Sci. 2017, 18, 1636. [Google Scholar] [CrossRef]


| ECM Components | Function | |
|---|---|---|
| Collagens | 1. fibrillar collagens (I, II, III, V, XI, XXVI, XXVII) 2. basement membrane collagens (IV, VII, XXVIII) 3. short-chain collagens (VI, VIII, X) 4. collagens with multiple interruptions (IX, XII, XIV, XV, XVI, XVIII, XIX–XXII) | 1. providing structural integrity and tensile strength 2. providing support for polarized cells 3. forming extensive associations within the protein network 4. linking other collagens together and with other ECM molecules |
| Glycoproteins | 1. fibrinogen 2. fibronectin 3. periostin 4. tenascin C and X | 1. crucial role in hemostasis and binding to growth factors, fibronectin, albumin, von Willebrand factor, or fibulin 2. crucial component of a membrane-like matrix and key factor in ECM formation and maturation 3. protective effect on several tissues 4. tissue regeneration and recovery after mechanical injuries |
| Proteoglycans | 1. small leucine-rich repeat proteoglycans (SLRPs) (biglycan, decorin) | 1. regulation of cell–matrix crosstalk and anti-cancer effect (decorin) 2. pro-angiogenic and pro-inflammatory factor (biglycan) |
| Matrix Metalloproteinases (MMPs) | 1. collagenases, gelatinases, membrane-type, stromelysins, and matrilysins 2. tissue inhibitors of metalloproteinases (TIMPs) | 1. tissue degradation and remodeling; cell proliferation, migration, differentiation, or apoptosis 2. regulation of the activity of MMPs in tissues |
| Cytokines and growth factors | 1. transforming growth factor beta (TGF-β) 2. tumor necrosis factor-alpha (TNF-α) 3. vascular endothelial growth factor (VEGF) | 1. binding to leucine-rich repeat structures and fibrillar proteins 2. binding to fibronectin and laminin 3. interacting with fibronectin and tenascin-C, resulting in the promotion of cell proliferation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kołakowski, A.; Dziemitko, S.; Chmielecka, A.; Żywno, H.; Bzdęga, W.; Charytoniuk, T.; Chabowski, A.; Konstantynowicz-Nowicka, K. Molecular Advances in MAFLD—A Link between Sphingolipids and Extracellular Matrix in Development and Progression to Fibrosis. Int. J. Mol. Sci. 2022, 23, 11380. https://doi.org/10.3390/ijms231911380
Kołakowski A, Dziemitko S, Chmielecka A, Żywno H, Bzdęga W, Charytoniuk T, Chabowski A, Konstantynowicz-Nowicka K. Molecular Advances in MAFLD—A Link between Sphingolipids and Extracellular Matrix in Development and Progression to Fibrosis. International Journal of Molecular Sciences. 2022; 23(19):11380. https://doi.org/10.3390/ijms231911380
Chicago/Turabian StyleKołakowski, Adrian, Sylwia Dziemitko, Aleksandra Chmielecka, Hubert Żywno, Wiktor Bzdęga, Tomasz Charytoniuk, Adrian Chabowski, and Karolina Konstantynowicz-Nowicka. 2022. "Molecular Advances in MAFLD—A Link between Sphingolipids and Extracellular Matrix in Development and Progression to Fibrosis" International Journal of Molecular Sciences 23, no. 19: 11380. https://doi.org/10.3390/ijms231911380
APA StyleKołakowski, A., Dziemitko, S., Chmielecka, A., Żywno, H., Bzdęga, W., Charytoniuk, T., Chabowski, A., & Konstantynowicz-Nowicka, K. (2022). Molecular Advances in MAFLD—A Link between Sphingolipids and Extracellular Matrix in Development and Progression to Fibrosis. International Journal of Molecular Sciences, 23(19), 11380. https://doi.org/10.3390/ijms231911380