
Towards Property Profiling: SYNTHESIS and SAR Probing of New Tetracyclic Diazaphenothiazine Analogues

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
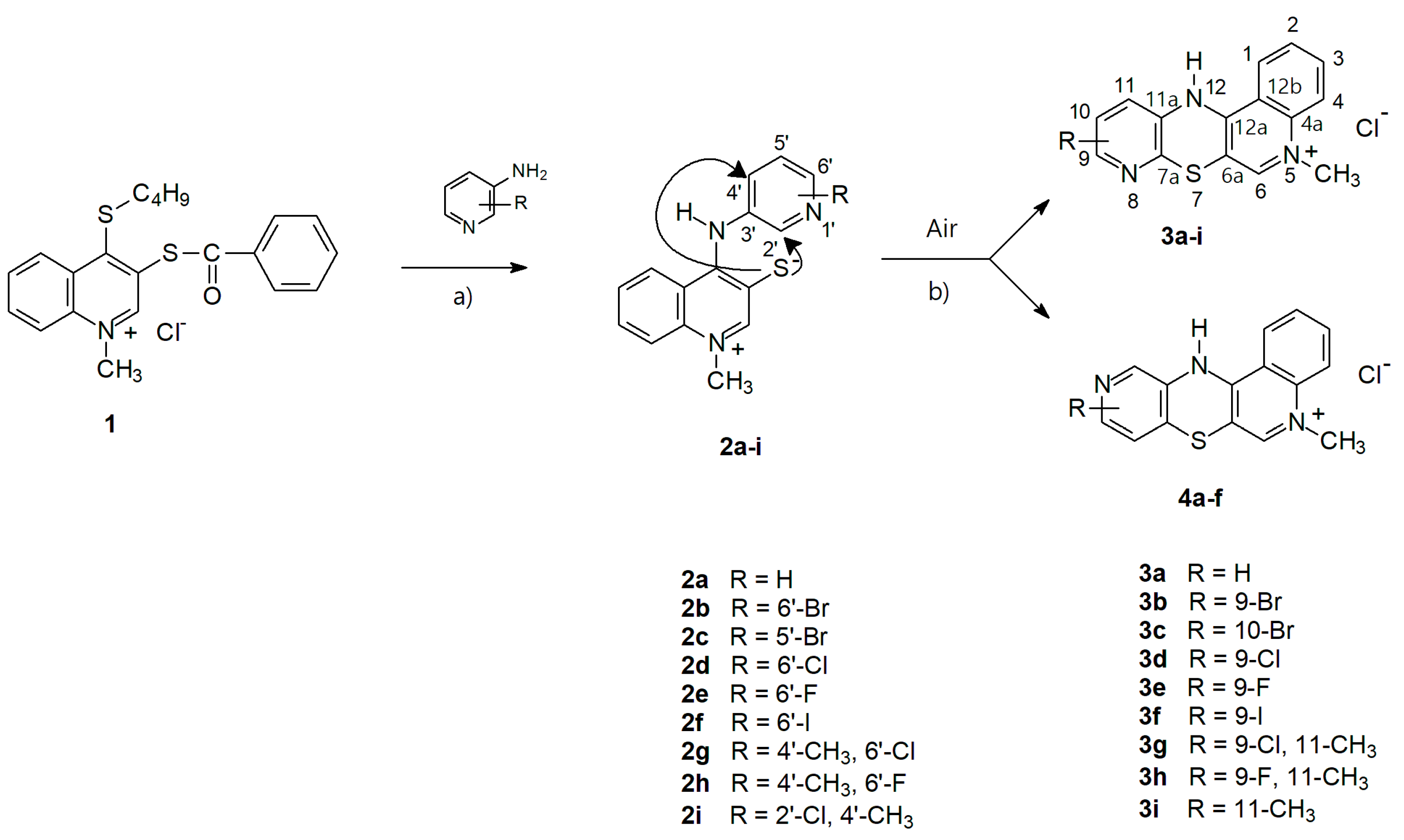
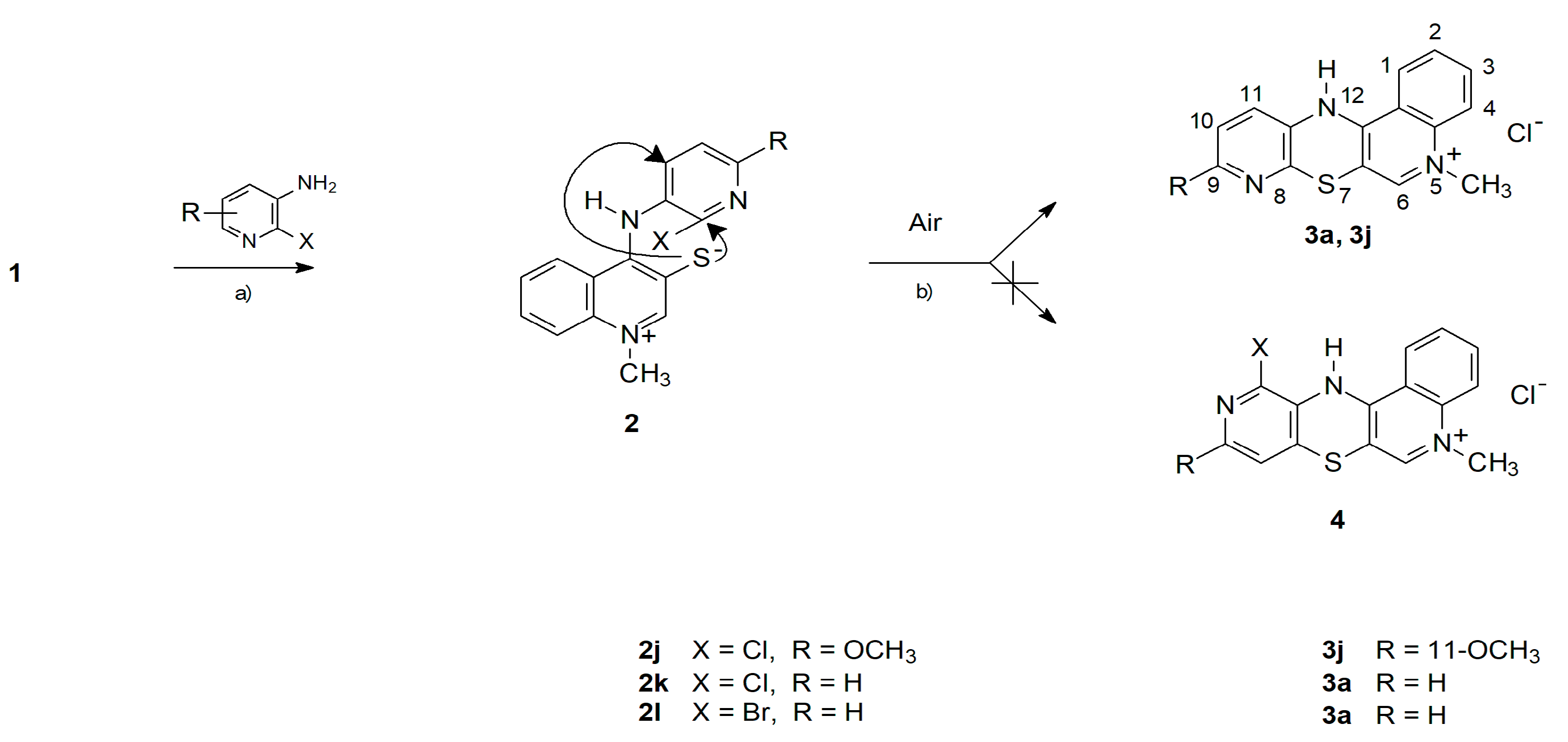
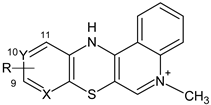
2.1. Chemistry—Design and Synthesis
- -
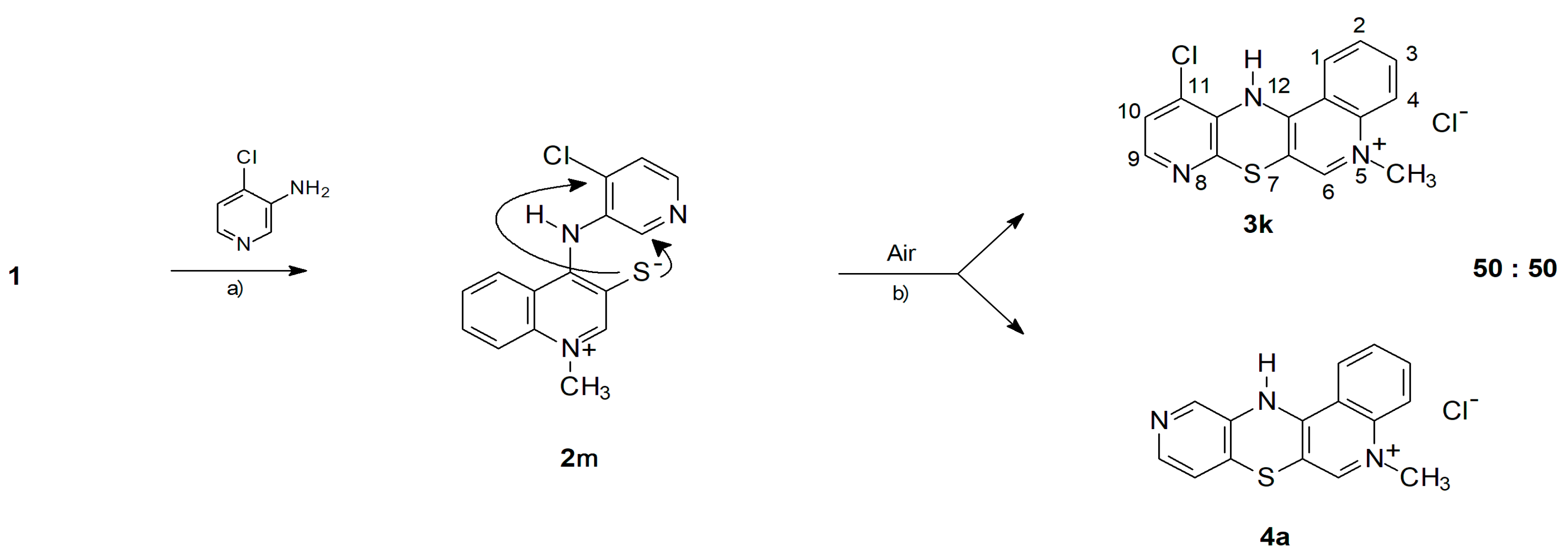
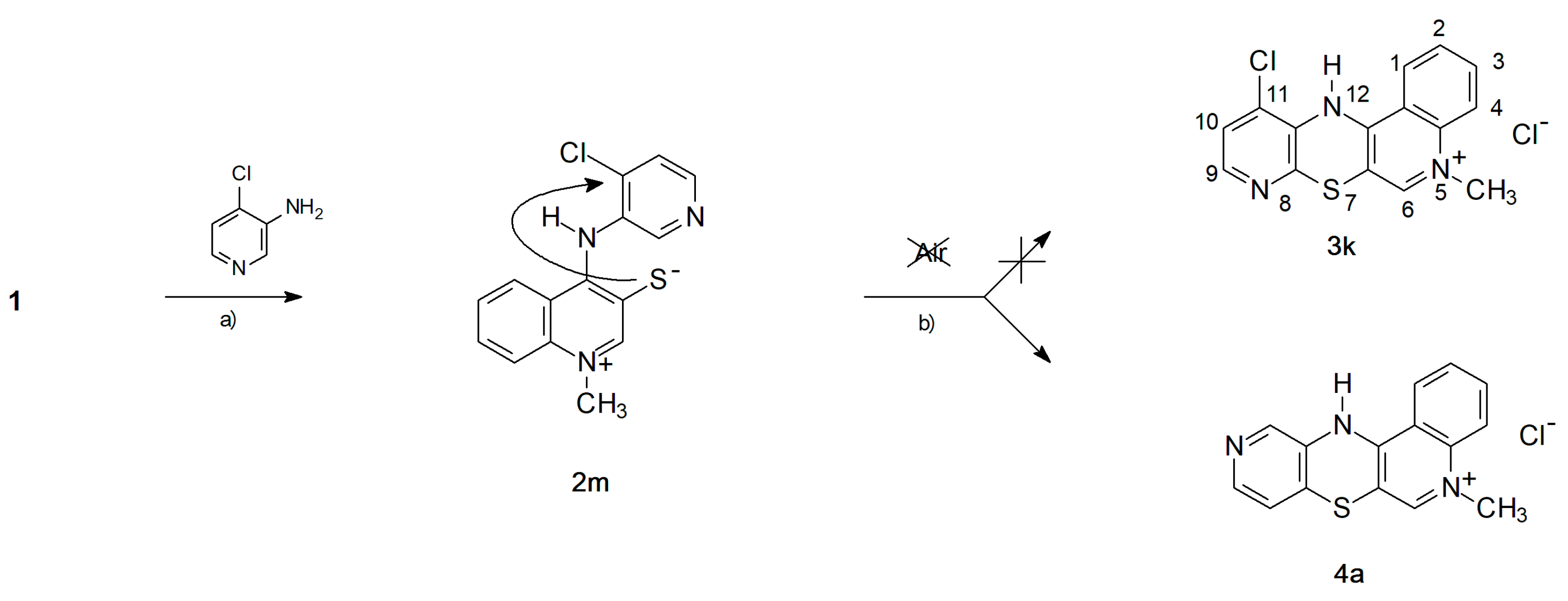
- The cyclization to 3k takes place as a nucleophilic substitution of a hydrogen atom. The hydride anion is a very difficult leaving group and its substitution in nucleophilic substitution reactions requires the presence of an oxidizing agent. In this case, it is atmospheric oxygen;
- -
- The cyclization to 4a takes place as a nucleophilic substitution of a halogen atom by a thiolate sulfur atom. It is a direct process and does not require the presence of an oxidant in the reaction medium.
2.2. Spectroscopic Structural Analysis
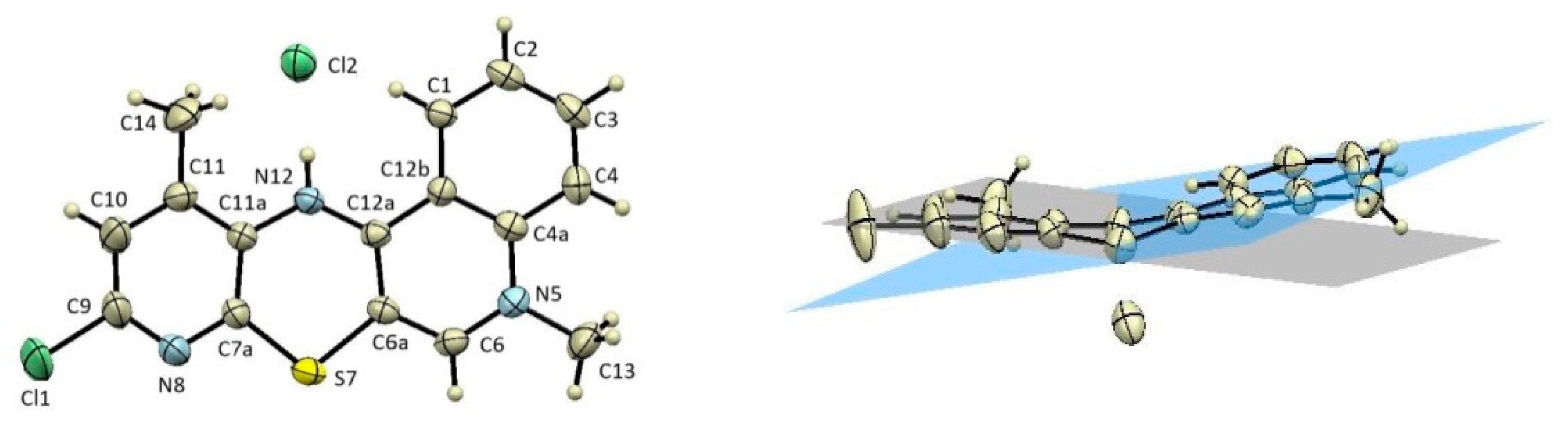
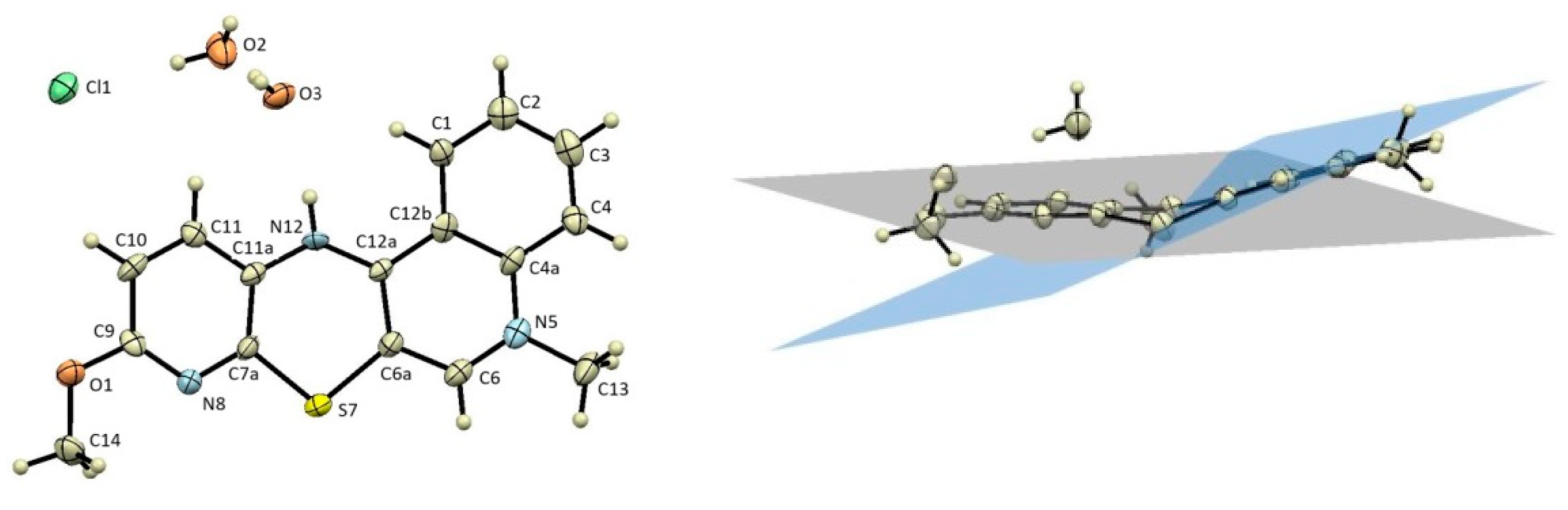
2.3. X-ray Structural Analysis
2.4. In Vitro Cytotoxic Activity
2.5. In Vitro Antimicrobial Activity
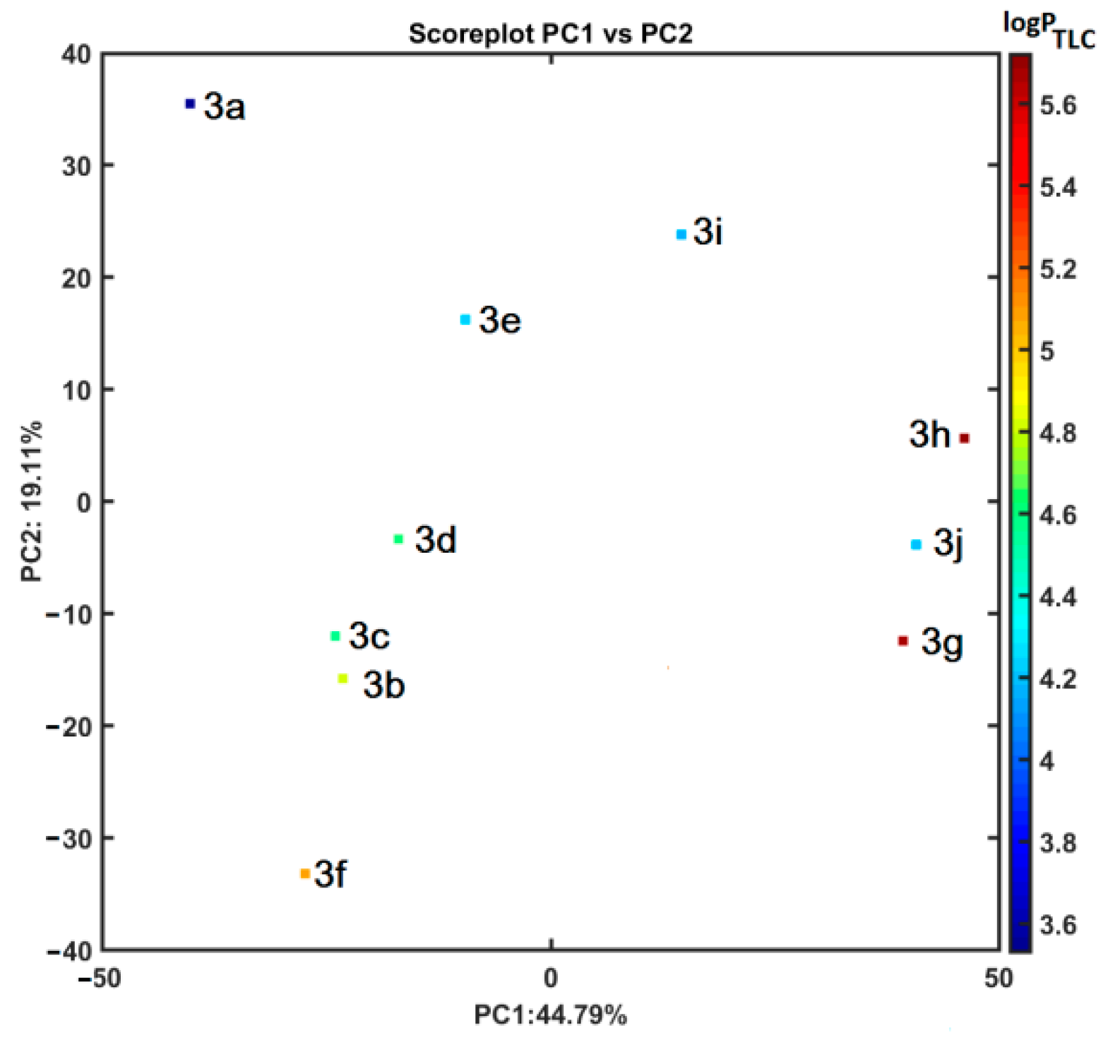
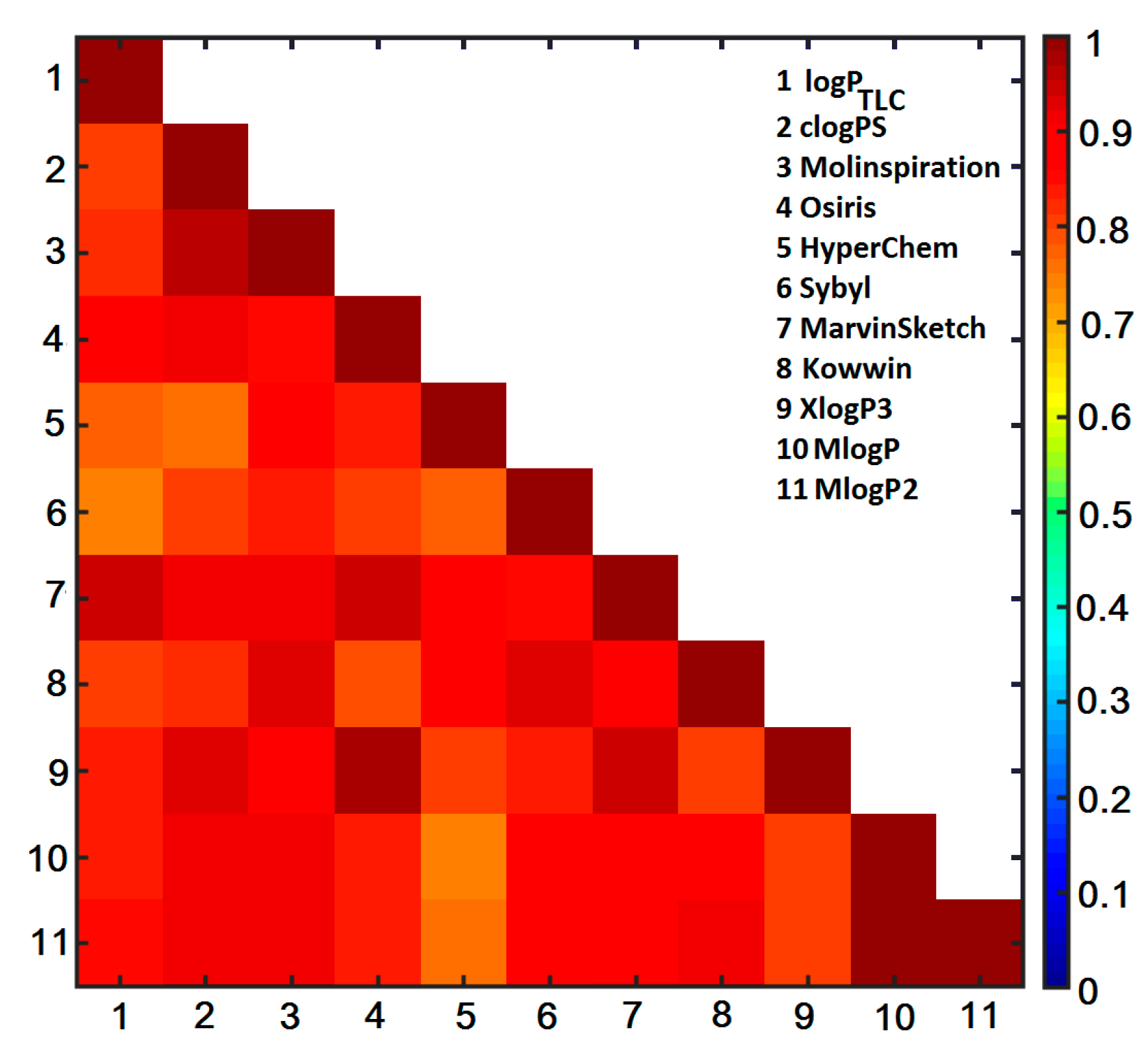
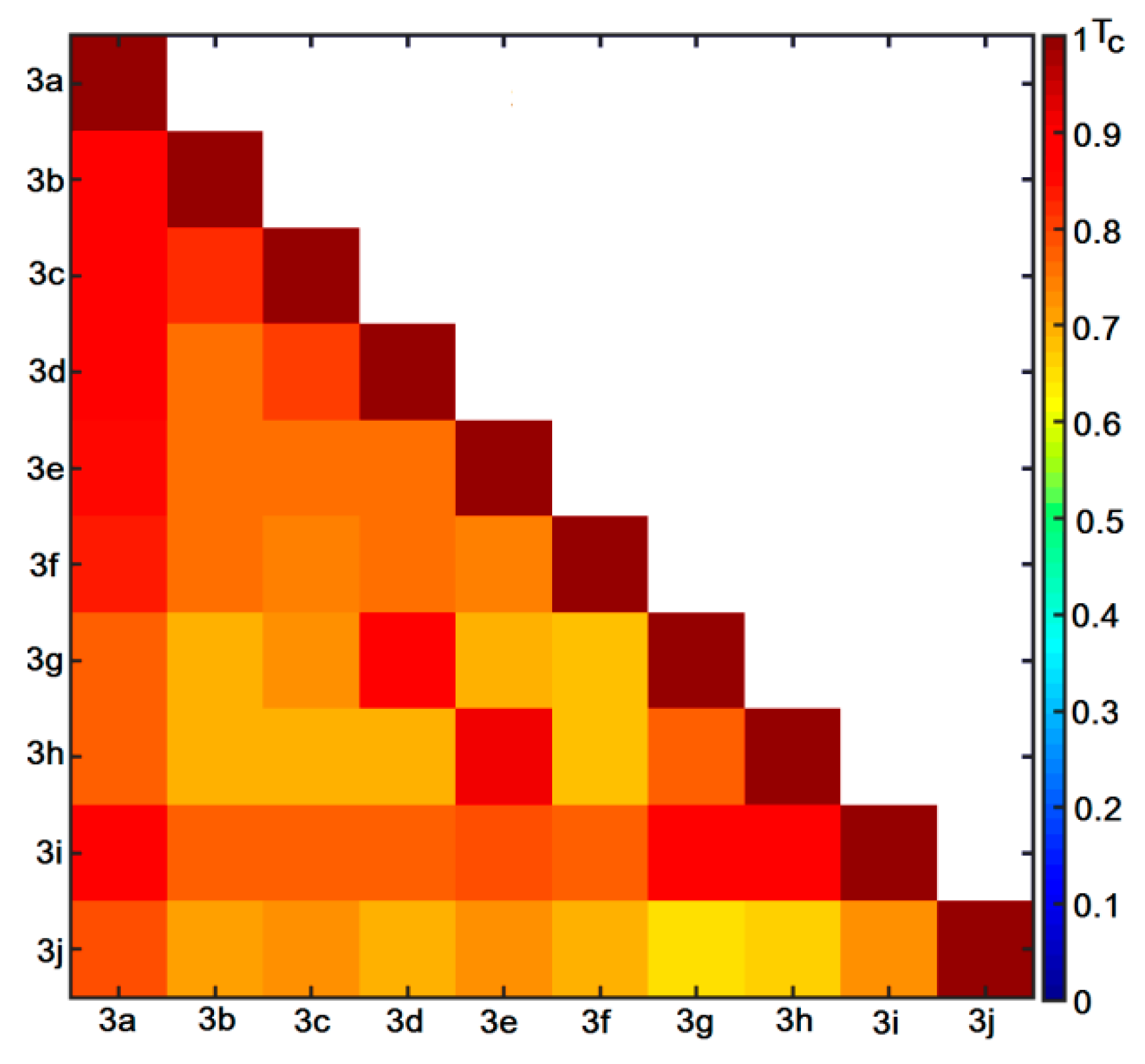
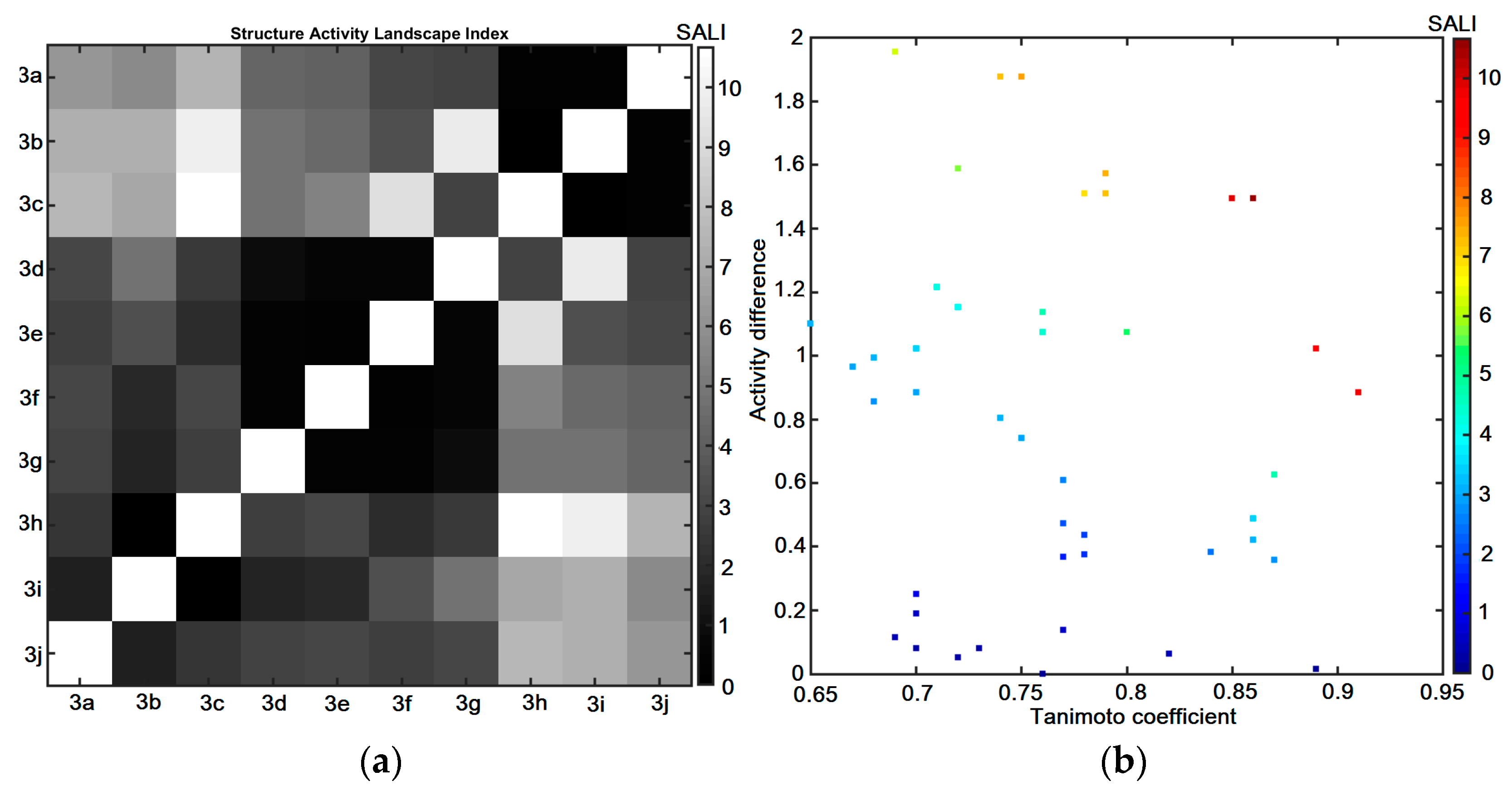
2.6. Similarity-Oriented Property Mapping
2.7. Probability-Driven Pharmacophore Probing
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 5-Methyl-12H-quino [3,4-b]pyrido[5,6-e][1,4]thiazinium Chloride 3a–j
- 5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3a): Yield: 80%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.10 (s, 3H, NCH3), 7.00–6.99 (d.d, 3J = 4.8 Hz, 3J = 7.8 Hz, 1H, H10), 7.33–7.34 (d.d, 3J = 7.8 Hz, 4J = 1.2 Hz, 1H, H11), 7.74–7.75 (m, 1H, H2), 7.91–7.92 (d.d, 3J = 4.8 Hz, 4J = 1.2 Hz, 1H, H9), 7.96–7.98 (m, 2H, H3, H4), 8.36 (s, 1H, H6), 8.40–8.41 (m, 1H, H1); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 42.29 (NCH3), 108.46 (C4a), 115.86 (C12b), 118.33 (C4), 122.79 (C1), 123.14 (C10), 124.26 (C11), 128.35 (C2), 133.44 (C11a), 134.77 (C3), 139.24 (C12a), 141.41 (C7a), 143.04 (C6), 146.42 (C9), 150.96 (C6a); ESI-HRMS Calcd for C15H12N3S ([M]+): 266.0746, Found: 266.0749.
- 9-bromo-5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3b): Yield: 75%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.22 (s, 3H, NCH3), 7.10–7.16 (d, 3J = 8.1 Hz 1H, H10), 7.37–7.42 (d, 3J = 8.1 Hz, 1H, H11), 7.80–7.90 (m, 1H, Harom), 8.06–8.13 (m, 2H, Harom), 8.42–8.50 (m, 2H, Harom); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 42.47 (NCH3), 108.64 (C), 116.01 (C), 118.40 (C), 118.63 (C), 122.79 (C), 126.12 (C), 128.55 (C), 134.31 (C), 134.91 (C), 139.19 (C), 140.44 (C), 143.31 (C), 146.70 (C), 148.17 (C); ESI-HRMS Calcd for C15H11Br[79]N3S ([M]+): 343.9852, Found: 343.9849; ESI-HRMS Calcd for C15H11Br[81]N3S ([M]+): 345.9831, Found: 345.9828.
- 10-bromo-5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3c): Yield: 72%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.22 (s, 3H, NCH3), 7.25–7.32 (m, 2H, Harom), 7.83–7.92 (m, 1H, Harom), 8.05–8.14 (m, 2H, Harom), 8.42–8.50 (m, 2H, Harom); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 42.37 (NCH3), 108.01 (C), 115.91 (C), 118.37 (C), 122.75 (C), 126.39 (C), 126.93 (C), 128.48 (C), 133.08 (C), 134.93 (C), 136.36 (C), 139.31 (C), 142.06 (C), 143.13 (C), 150.86 (C); ESI-HRMS Calcd for C15H11Br[79]N3S ([M]+): 343.9852, Found: 343.9849; ESI-HRMS Calcd for C15H11Br[81]N3S ([M]+): 345.9831, Found: 345.9828.
- 9-chloro-5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3d): Yield: 85%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.23 (s, 3H, NCH3), 7.15–7.18 (d, J = 8.4Hz, 1H, H10), 7.38–7.41 (d, J = 8.4 Hz, 1H, H11), 7.85–7.90 (m, 1H, H2), 8.06–8.14 (m, 2H, H3, H4), 8.44–8.47 (m, 1H, H1), 8.48 (s, 1H, H6); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 42.35 (NCH3), 107.90 (C4a), 115.89 (C12b), 118.36 (C4), 122.76 (C1), 123.04 (C10), 126.72 (C11), 128.46 (C2), 132.67 (C11a), 134.93 (C3), 139.31 (C12a), 141.66 (C), 143.12 (C6), 146.86 (C), 150.91 (C6a); ESI-HRMS Calcd for C15H11ClN3S ([M]+): 300.0357, Found: 300.0352.
- 9-fluoro-5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3e): Yield: 69%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.21 (s, 3H, NCH3), 6.70–6.82 (m, 1H, Harom), 7.54–7.60 (m, 1H, Harom), 7.84–7.89 (m, 1H, Harom), 8.05–8.12 (m, 2H, Harom), 8.44 (s, 1H, H6), 8.48–8.50 (m, 1H, Harom); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 42.25 (NCH3), 107.41 (C, JC–F = 36.2 Hz), 107.78 (C), 115.77 (C), 118.31 (C), 122.79 (C), 128.34 (C), 129.53 (C, JC–F = 7.5 Hz), 131.28 (C, JC–F = 4.5 Hz), 134.90 (C), 139.09 (C, JC–F = 15.1 Hz), 139.35 (C), 142.99 (C), 151.29 (C), 160.85 (C, JC–F = 241.4 Hz); ESI-HRMS Calcd for C15H11FN3S ([M]+): 284.0652, Found: 284.0649.
- 9-iodo-5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3f): Yield: 78%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.22 (s, 3H, NCH3), 7.04–7.09 (d, 3J = 8.4 Hz, 1H, H11), 7.49–7.53 (d, 3J = 8.4 Hz, 1H, H10), 7.83–7.90 (m, 1H, Harom), 8.05–8.13 (m, 2H, Harom), 8.44–8.46 (m, 1H, H1), 8.48 (s, 1H, H6); 13C NMR (DMSOd-6, 150.9 MHz) δ (ppm): 43.34 (NCH3), 107.24 (C), 111.99 (C) 116.24 (C), 119.35 (C), 124.45 (C), 125.98 (C), 126.85 (C), 128.64 (C), 134.27 (C), 135.30 (C), 139.23 (C), 142.55 (C), 143.95 (C), 150.33 (C); ESI-HRMS Calcd for C15H11IN3S ([M]+): 391.9713, Found: 391.9713.
- 9-chloro-5,11-dimethyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3g): Yield: 61%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 2.45 (s, 3H, 11CH3), 4.28 (s, 3H, NCH3), 7.14 (s, 1H, H10), 7.85–7.95 (m, 1H, Harom), 8.10–8.21 (m, 2H, Harom), 8.40–8.45 (m, 1H, Harom), 8.55–8.66 (m, 1H, Harom); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 15.53 (CH3), 42.60 (NCH3), 110.05 (C), 116.73 (C), 118.36 (C), 123.05 (C), 124.81 (C), 128.52 (C), 131.48 (C), 134.97 (C), 138.73 (C), 139.26 (C), 142.55 (C), 143.65 (C), 146.69 (C); 151.94 (C); ESI-HRMS Calcd for C16H13ClN3S ([M]+): 314.0513, Found: 314.0513.
- 9-fluoro-5,11-dimethyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3h): Yield: 63%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 2.50 (s, 3H, CH3), 4.28 (s, 3H, NCH3), 6.78 (s, 1H, H10), 7.88–7.95 (m, 1H, Harom), 8.11–8.20 (m, 2H, Harom), 8.40–8.45 (m, 1H, Harom), 8.61 (s, 1H, H6); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 16.015 (CH3, JC–F = 1.5 Hz), 42.57 (C), 109.28 (C, JC–F = 37.7 Hz), 116.63 (C), 118.35 (C), 123.08 (C), 128.46 (C), 130.08 (C), 134.97 (C), 139.27 (C), 140.08 (C, JC–F = 16.6 Hz), 141.73 (C, JC–F = 7.5 Hz), 143. 56 (C), 152.35 (C), 160.74 (C, JC–F = 239.9 Hz); ESI-HRMS Calcd for C16H13FN3S ([M]+): 298.0809, Found: 298.0814.
- 5,11-dimethyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3i): Yield: 65%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 2.57 (s, 3H, 11-CH3), 4.33 (s, 3H, NCH3), 7.27–7.31 (d,3J = 5.4 Hz, 1H, Hpirydyl), 7.91–7.00 (m, 1H, Harom), 7.08–7.13 (d, 3J = 5.4 Hz, 1H, Hpirydyl), 7.14–7.20 (m, 1H, Harom), 7.2–7.25 (m, 1H, Harom), 7.47–7.52 (m, 1H, Harom), 8.74 (s, 1H, H6); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 16.02 (CH3), 42.86 (NCH3), 109.44 (C), 116.89 (C), 118.51 (C), 123.14 (C), 126.21 (C), 128.75 (C), 133.61 (C), 135.16 (C), 138.48 (C), 139.30 (C), 141.47 (C), 143.50 (C), 144.11 (C), 152.16 (C); ESI-HRMS Calcd for C16H14N3S ([M]+): 280.0903, Found: 280.0907.
- 9-methoxy-5-methyl-12H-quino[3,4-b]pyrido[5,6-e][1,4]thiazinium chloride (3j): Yield: 86%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 3.83 (s, 3H, OCH3), 4.15 (s, 3H, NCH3), 6.40-6.52 (d, 3J = 8.4 Hz, 1H, H10), 7.38–7.42 (d, 3J = 8.4 Hz, 1H, H11), 7.80–7.83 (m, 1H, H2), 8.00–8.05 (m, 2H, H3, H4), 8.32 (s, 1H, H6), 8.43–8.46 (m, 1H, H1); 13C NMR (DMSOd-6, 150.9 MHz) δ (ppm): 42.90 (NCH3), 54.40 (OCH3), 106.25 (C4a), 109.22 (C10), 115.86 (C12b), 119.06 (C4), 124.86 (C1), 127.75 (C2), 128.14 (C11), 129.77 (C3), 135.08 (C), 137.25 (C12a), 139.26 (C11a), 143.0 (C6), 150.74 (C6a), 161.77 (C); ESI-HRMS Calcd for C16H14N3OS ([M]+): 296.0852, Found: 296.0861.
3.1.2. Synthesis of 5-Methyl-12H-quino[3,4-b]pyrido[3,4-e][1,4]thiazinium Chloride 4a
- 5-methyl-5H-qujino[3,4-b]pyrido[3,4-e][1,4]thiazinium chloride (4a): Yield: 80%; 1H NMR (CD3OD, 600 MHz) δ (ppm): 4.11 (s, 3H, NCH3), 6.96–6.97 (d, 3J = 4.8 Hz, 1H, H8), 7.75–7.77 (m, 1H, H2), 7.99–8.00 (m, 3H, H3, H4, H9), 8.15 (s, 1H, H11), 8.36 (s, 1H, H6), 8.43–8.45 (m, 1H, H1); 13C NMR (CD3OD, 150.9 MHz) δ (ppm): 42.34 (NCH3), 105.91 (C6a), 116.15 (C12b), 118.36 (C4), 121.25 (C8), 122.77 (C1), 128.39 (C2), 129.17 (C7a), 133.50 (C11a), 134.92 (C3), 137.12 (C11), 139.33 (C4a), 143.08 (C6), 146.99 (C9), 152.25 (C12a); ESI-HRMS Calcd for C15H12N3OS ([M]+): 266.0751, Found: 266.0749.
- 1H NMR and 13C NMR spectrum data are reported in Supplementary Materials (Figures S1–S22).
3.2. X-ray Structural Analysis
3.3. Biological Evaluation
3.3.1. Cell Culture
3.3.2. Proliferation Assay
3.3.3. In Vitro Antistaphylococcal Evaluation
3.4. Molecular Modeling
3.4.1. Model Building
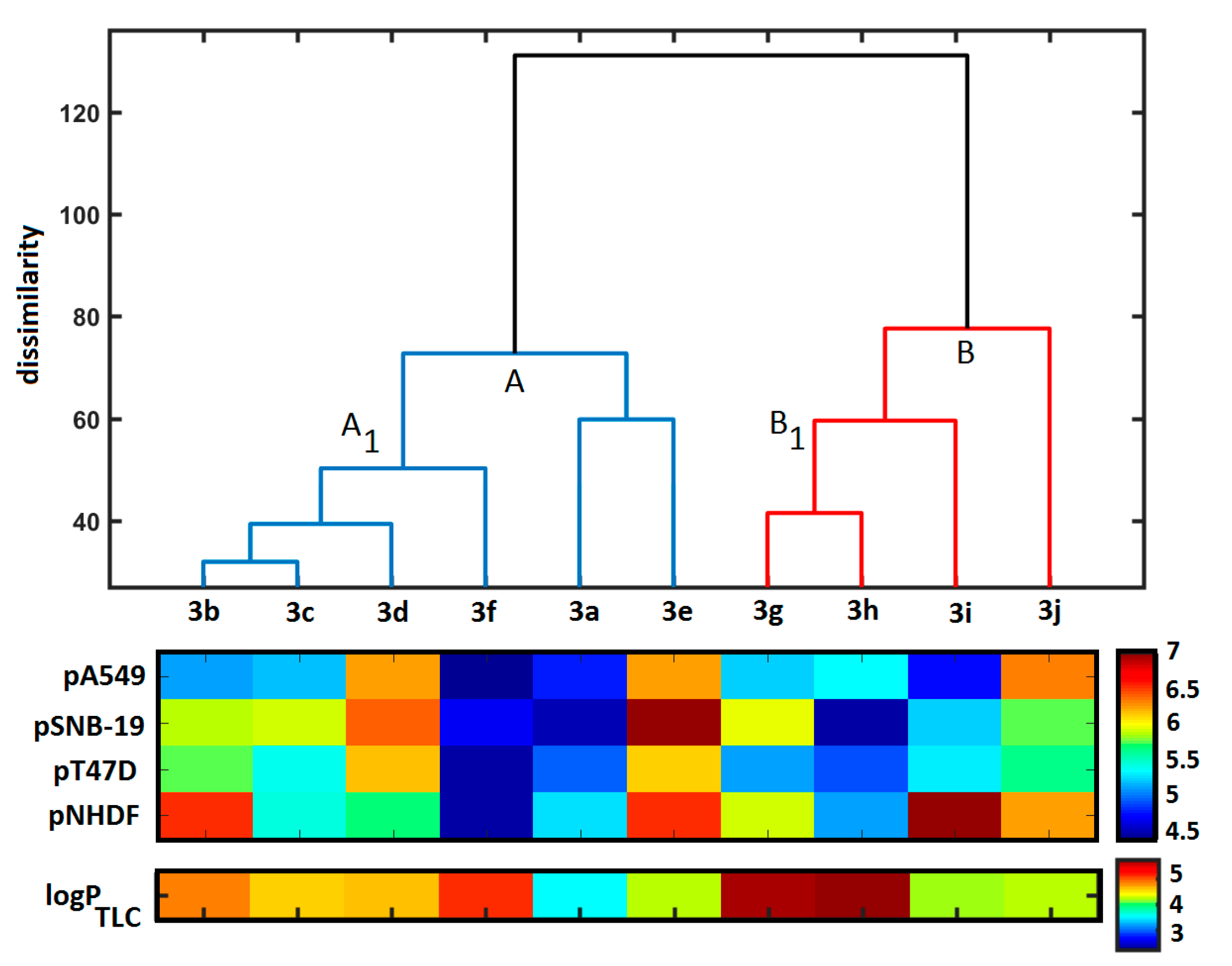
3.4.2. Principal Component and Hierarchical Clustering Analysis
3.4.3. Theoretical Lipophilicity Evaluation
3.4.4. Similarity-Related Activity Landscape Index
3.4.5. Ligand-Based SAR Probing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sneader, W. Chronology of drug introduction. In Comprehensive Medicinal Chemistry. The Rational Design, Mechanistic Study and Therapeutic Application of Chemical Compounds; Hansch, C., Sammes, P.G., Taylor, J.B., Eds.; Pergamon Press: Oxford, UK, 1990; Volume 1, pp. 7–80. [Google Scholar]
- Li, J.J. Heterocyclic Chemistry in Drug Discovery; Wiley: Hoboken, NJ, USA, 2013; pp. 1–16. [Google Scholar]
- Gupta, R.R.; Kumar, M. Synthesis properties and reactions of phenothiazines. In Phenothiazine and 1,4-Benzothiazines–Chemical and Biological Aspect; Elsevier: Amsterdam, The Netherlands, 1988; pp. 1–161. [Google Scholar]
- Mosnaim, A.D.; Ranade, V.V.; Wolf, M.E.; Puente, J.; Valenzuela, M.A. Phenothiazine molecule provides the basic chemical structure for various classes of pharmacotherapeutic agents. Am. J. Therapeut. 2006, 13, 261–273. [Google Scholar] [CrossRef]
- Zięba, A.; Czuba, Z.; Król, W. In vitro antimicrobial activity of novel azapheno thiazine derivatives. Acta Pol. Pharm. Drug Res. 2012, 69, 1149–1152. [Google Scholar]
- Jeleń, M.; Pluta, K.; Zimecki, M.; Morak-Młodawska, B.; Artym, J.; Kocięba, M. 6-Substituted 9-fluoroquino[3,2-b]benzo[1,4]thiazines display strong antiproliferative and antitumor properties. Eur. J. Med. Chem. 2015, 89, 411–420. [Google Scholar] [CrossRef]
- Otręba, M.; Kośmider, L.; Rzepecka-Stojko, A. Antiviral activity of chlorpromazine, fluphenazine, perphenazine, prochlorperazine, and thioridazine towards RNA-viruses. A review. Eur. J. Pharmacol. 2020, 887, 173553. [Google Scholar] [CrossRef]
- Warman, A.J.; Rito, T.S.; Fisher, N.E.; Moss, D.M.; Berry, N.G.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antitubercular pharmacodynamics of phenothiazines. J. Antimicrob. Chemother. 2013, 68, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Amaral, L.; Viveiros, M.; Kristiansen, J.E. Phenothiazines: Potential alternatives for the management of antibiotic resistant infections of tuberculosis and malaria in developing countries. Trop. Int. Health 2001, 6, 1016–1022. [Google Scholar] [CrossRef] [Green Version]
- Jeleń, M.; Bavavea, E.; Pappa, M.; Kourounakis, A.P.; Morak-Młodawska, B.; Pluta, K. Synthesis of quinoline/naphthalene-containing azaphenothiazines and their potent in vitro antioxidant properties. Med. Chem. Res. 2015, 24, 1725–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zięba, A.; Maślankiewicz, A.; Suwińska, K. 1-Alkyl-4-(arylamino)quinolinium-3-thiolate and 7-alkyl-12(H)-quino[3,4-b]1,4-benzothiazinium salts. Eur. J. Org. Chem. 2000, 16, 2947–2953. [Google Scholar] [CrossRef]
- Zięba, A.; Sochanik, A.; Szurko, A.; Rams, M.; Mrozek, A.; Cmoch, P. Synthesis and in vitro antiproliferative activity of 5-alkyl-12(H)-quino[3,4-b][1,4]benzothiazinium salts. Eur. J. Med. Chem. 2010, 45, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Zięba, A.; Latocha, M.; Sochanik, A. Synthesis and in vitro antiproliferative activity of novel 12(H)-quino[3,4-b][1,4]benzothiazine derivatives. Med. Chem. Res. 2013, 22, 4158–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zięba, A.; Latocha, M.; Sochanik, A.; Nycz, A.; Kuśmierz, D. Synthesis and in vitro antiproliferative activity of novel phenyl ring-substituted 5-alkyl-12(H)-quino[3,4-b][1,4]benzothiazine derivatives. Molecules 2016, 21, 1455. [Google Scholar] [CrossRef] [Green Version]
- Zięba, A.; Suwińska, K. 1-Alkyl-4-(3-pyridinylamino)quinolinium-3-thiolates and their transformation into new diazaphenothiazine derivatives. Heterocycles 2006, 68, 495–503. [Google Scholar] [CrossRef]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355–365. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Walczak, M.; Fraczyk, J.; Kaminski, Z.; Kolesinska, B.; Smolinski, A.; Jampilek, J. Towards intelligent drug design system: Application of artificial dipeptide receptor library in QSAR-oriented studies. Molecules 2018, 23, 1964. [Google Scholar] [CrossRef] [Green Version]
- Peltason, L.; Bajorath, J. Systematic computational analysis of structure-activity relationships: Concepts, challenges and recent advances. Future Med. Chem. 2009, 1, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Terfloth, L. Calculation of structure descriptors. In Chemoinformatics: A Textbook; Gasteiger, J., Engel, T., Eds.; Wiley VCH: Viernheim, Germany, 2003; p. 401. [Google Scholar]
- De Almeida, A.F.; Moreira, R.; Rodrigues, T. Synthetic organic chemistry driven by artificial intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Devillers, J. Methods for building QSARs. Methods Mol. Biol. 2013, 930, 3–27. [Google Scholar] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Kozakiewicz, D.; Gajcy, K.; Strub, D.J.; Swietlicka, A.; Stepankova, S.; Imramovsky, A.; Polanski, J.; Smolinski, A.; et al. Novel benzene-based carbamates for AChE/BChE inhibition: Synthesis and ligand/structure-oriented SAR study. Int. J. Mol. Sci. 2019, 20, 1524. [Google Scholar] [CrossRef] [Green Version]
- Bak, A.; Pizova, H.; Kozik, V.; Vorcakova, K.; Kos, J.; Treml, J.; Odehnalova, K.; Oravec, M.; Imramovsky, A.; Bobal, P.; et al. SAR-mediated similarity assessment of the property profile for new, silicon-based AChE/BChE inhibitors. Int. J. Mol. Sci. 2019, 20, 5385. [Google Scholar] [CrossRef] [Green Version]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef]
- Kos, J.; Kozik, V.; Pindjakova, D.; Jankech, T.; Smolinski, A.; Stepankova, S.; Hosek, J.; Oravec, M.; Jampilek, J.; Bak, A. Synthesis and hybrid SAR property modeling of novel cholinesterase inhibitors. Int. J. Mol. Sci. 2021, 22, 3444. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Van Drie, J.H. Structure–activity landscape index: Identifying and quantifying activity cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Peltason, L.; Wawer, M.; Guha, R.; Lajiness, M.S.; Van Drie, J.H. Navigating structure–activity landscapes. Drug Discov. Today 2009, 14, 698–705. [Google Scholar] [CrossRef]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Ferriz, J.M.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Nubel, U.; Dordel, J.; Kurt, K.; Strommenger, B.; Westh, H.; Shukla, S.K.; Zemlickova, H.; Leblois, R.; Wirth, T.; Jombart, T.; et al. A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog. 2010, 6, e1000855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolinski, A.; Drobek, L.; Dombek, V.; Bak, A. Modeling of experimental data on trace elements and organic content in industrial waste dumps. Chemosphere 2016, 162, 189–198. [Google Scholar] [CrossRef]
- Smolinski, A.; Howaniec, N.; Bak, A. Utilization of energy crops and sewage sludge in the process of co-gasificiation for sustainable hydrogen production. Energies 2018, 11, 809. [Google Scholar] [CrossRef] [Green Version]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Assessing how well a modeling protocol captures a structure—Activity landscape. J. Chem. Inf. Modeling 2008, 48, 1716–1728. [Google Scholar] [CrossRef]
- Lopez-Lopez, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colquhoun, D. The quantitative analysis of drug–receptor interactions: A short history. Trends Pharmacol. Sci. 2006, 27, 149–157. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Environ. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [PubMed] [Green Version]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Modeling 2003, 46, 2310–2318. [Google Scholar] [CrossRef]
- Pauk, K.; Zadrazilova, I.; Imramovsky, A.; Vinsova, J.; Pokorna, M.; Masarikova, M.; Cizek, A.; Jampilek, J. New derivatives of salicylamides: Preparation and antimicrobial activity against various bacterial species. Bioorg. Med. Chem. 2013, 21, 6574–6581. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; M07; NCCLS: Wayne, PA, USA, 2018. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | X | Y | R | logP | IC50 [μM] | |||
| A549 (std) | SNB-19 (std) | T47D (std) | NHDF (std) | |||||
| 3a | N | CH | H | 3.53 | 70.3 ± 9.8 | 132.3 ± 11.6 | 46.2 ± 7.1 | 21.1 ± 1.5 |
| 3b | N | CH | 9-Br | 4.80 | 23.8 ± 1.7 | 4.1 ± 0.3 | 5.2 ± 0.4 | 0.8 ± 0.05 |
| 3c | N | C | 10-Br | 4.57 | 20.6 ± 1.2 | 3.4 ± 0.2 | 12.2 ± 1.7 | 11.0 ± 1.4 |
| 3d | N | CH | 9-Cl | 4.62 | 2.0 ± 0.3 | 1.3 ± 0.1 | 2.3 ± 0.3 | 7.7 ± 0.5 |
| 3e | N | CH | 9-F | 4.24 | 2.1 ± 0.35 | 0.4 ± 0.08 | 2.8 ± 0.35 | 1.1 ± 0.14 |
| 3f | N | CH | 9-I | 5.09 | 115.3 ± 10.7 | 62.5 ± 6.6 | 128.4 ± 18.2 | 97.4 ± 6.2 |
| 3g | N | CH | 9-Cl, 11-CH3 | 5.66 | 20 ± 1.6 | 3.5 ± 0.2 | 26.7 ± 1.6 | 3.8 ± 0.3 |
| 3h | N | CH | 9-F, 11-CH3 | 5.72 | 15.4 ± 1.0 | 125.5 ± 14.8 | 47.7 ± 2.8 | 28.5 ± 4.0 |
| 3i | N | CH | 11-CH3 | 4.17 | 69.3 ± 5.3 | 22.1 ± 1.5 | 18.9 ± 1.4 | 0.4 ± 0.07 |
| 3j | N | CH | 9-OCH3 | 4.20 | 1.7 ± 0.1 | 6.1 ± 0.4 | 8.1 ± 1.0 | 2.0 ± 0.3 |
| 4a | CH | N | H | 2.93 | 30.1 ± 3.4 | 18.7 ± 1.2 | 36.8 ± 2.1 | 7.5 ± 0.2 |
| cisplatinum | - | - | - | - | 3.0 ± 0.2 | 16.7 ± 1.3 | 9.0 ± 0.7 | 29.9 ± 3.3 |
| Comp. | MIC (μM) | |||
|---|---|---|---|---|
| SA | MRSA 63718 | MRSA SA 630 | MRSA SA 3202 | |
| 3a | 212 | 212 | 106 | 212 |
| 3b | 337 | 337 | 337 | 337 |
| 3c | 42.1 | 42.1 | 21.1 | 42.1 |
| 3d | 381 | 381 | 381 | 381 |
| 3e | 50.2 | 25.1 | 12.5 | 25.1 |
| 3f | 299 | 299 | 299 | 299 |
| 3g | >731 | >731 | >731 | >731 |
| 3h | 383 | 383 | 383 | 383 |
| 3i | 203 | 203 | 101 | 203 |
| 3j | 193 | 193 | 96 | 193 |
| 4a | 424 | 424 | 424 | 424 |
| AMP | 5.72 | 45.8 | 45.8 | 45.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Empel, A.; Bak, A.; Kozik, V.; Latocha, M.; Cizek, A.; Jampilek, J.; Suwinska, K.; Sochanik, A.; Zieba, A. Towards Property Profiling: SYNTHESIS and SAR Probing of New Tetracyclic Diazaphenothiazine Analogues. Int. J. Mol. Sci. 2021, 22, 12826. https://doi.org/10.3390/ijms222312826
Empel A, Bak A, Kozik V, Latocha M, Cizek A, Jampilek J, Suwinska K, Sochanik A, Zieba A. Towards Property Profiling: SYNTHESIS and SAR Probing of New Tetracyclic Diazaphenothiazine Analogues. International Journal of Molecular Sciences. 2021; 22(23):12826. https://doi.org/10.3390/ijms222312826
Chicago/Turabian StyleEmpel, Anna, Andrzej Bak, Violetta Kozik, Malgorzata Latocha, Alois Cizek, Josef Jampilek, Kinga Suwinska, Aleksander Sochanik, and Andrzej Zieba. 2021. "Towards Property Profiling: SYNTHESIS and SAR Probing of New Tetracyclic Diazaphenothiazine Analogues" International Journal of Molecular Sciences 22, no. 23: 12826. https://doi.org/10.3390/ijms222312826
APA StyleEmpel, A., Bak, A., Kozik, V., Latocha, M., Cizek, A., Jampilek, J., Suwinska, K., Sochanik, A., & Zieba, A. (2021). Towards Property Profiling: SYNTHESIS and SAR Probing of New Tetracyclic Diazaphenothiazine Analogues. International Journal of Molecular Sciences, 22(23), 12826. https://doi.org/10.3390/ijms222312826