
DIA-Based Quantitative Proteomics Reveals the Protein Regulatory Networks of Floral Thermogenesis in Nelumbo nucifera

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
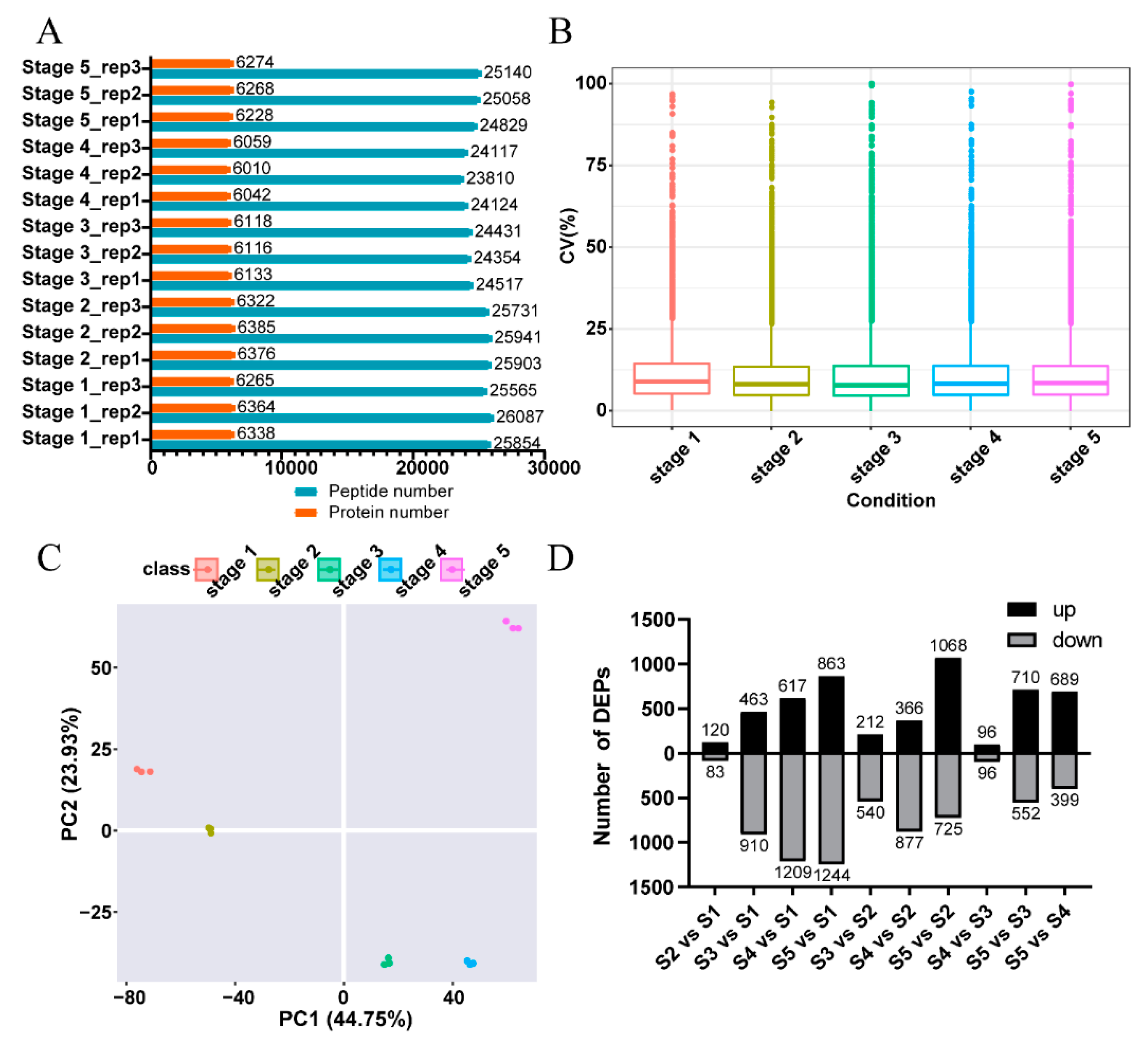
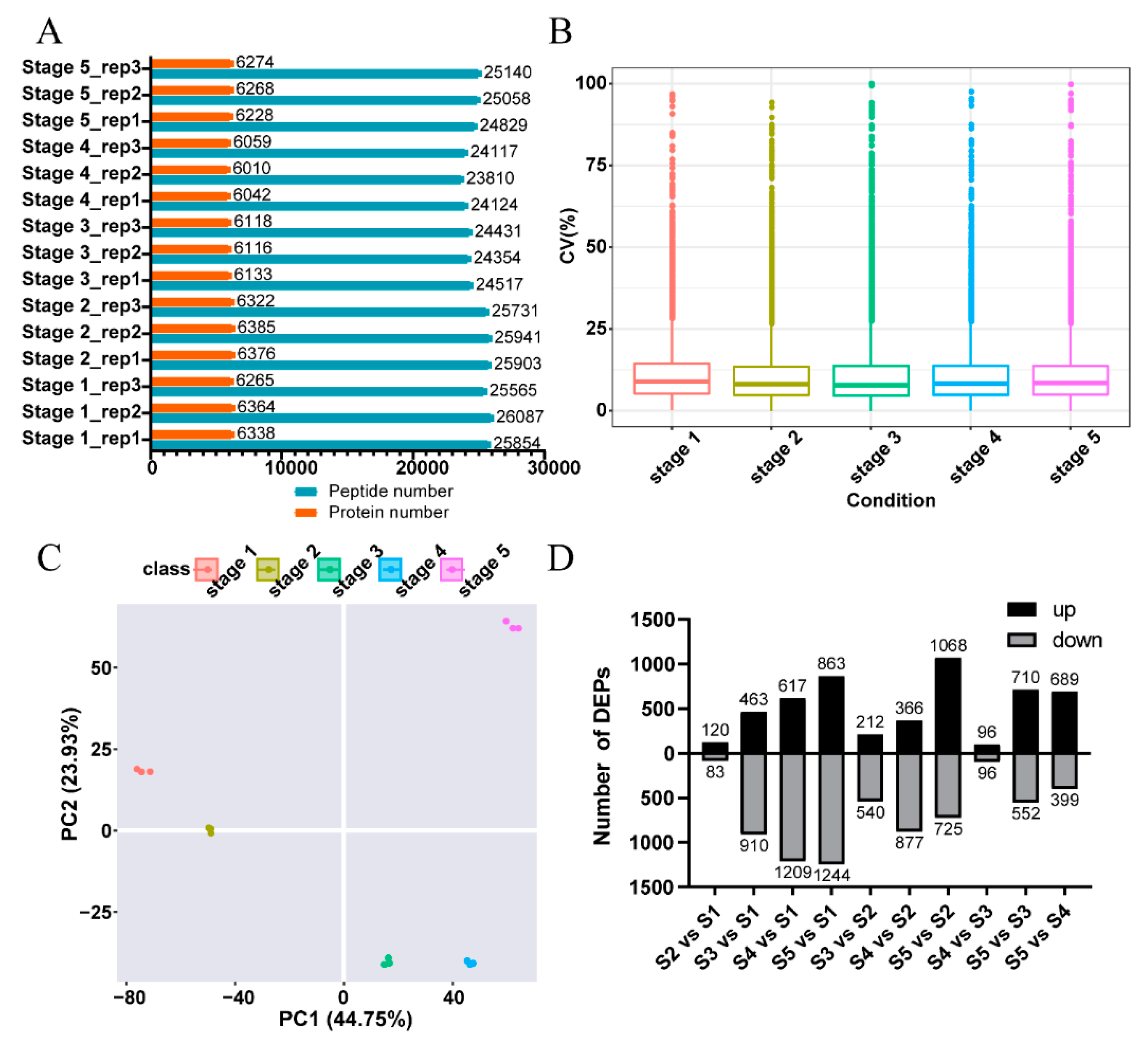
2.1. An Overview of the Quantitative Proteomics Analysis
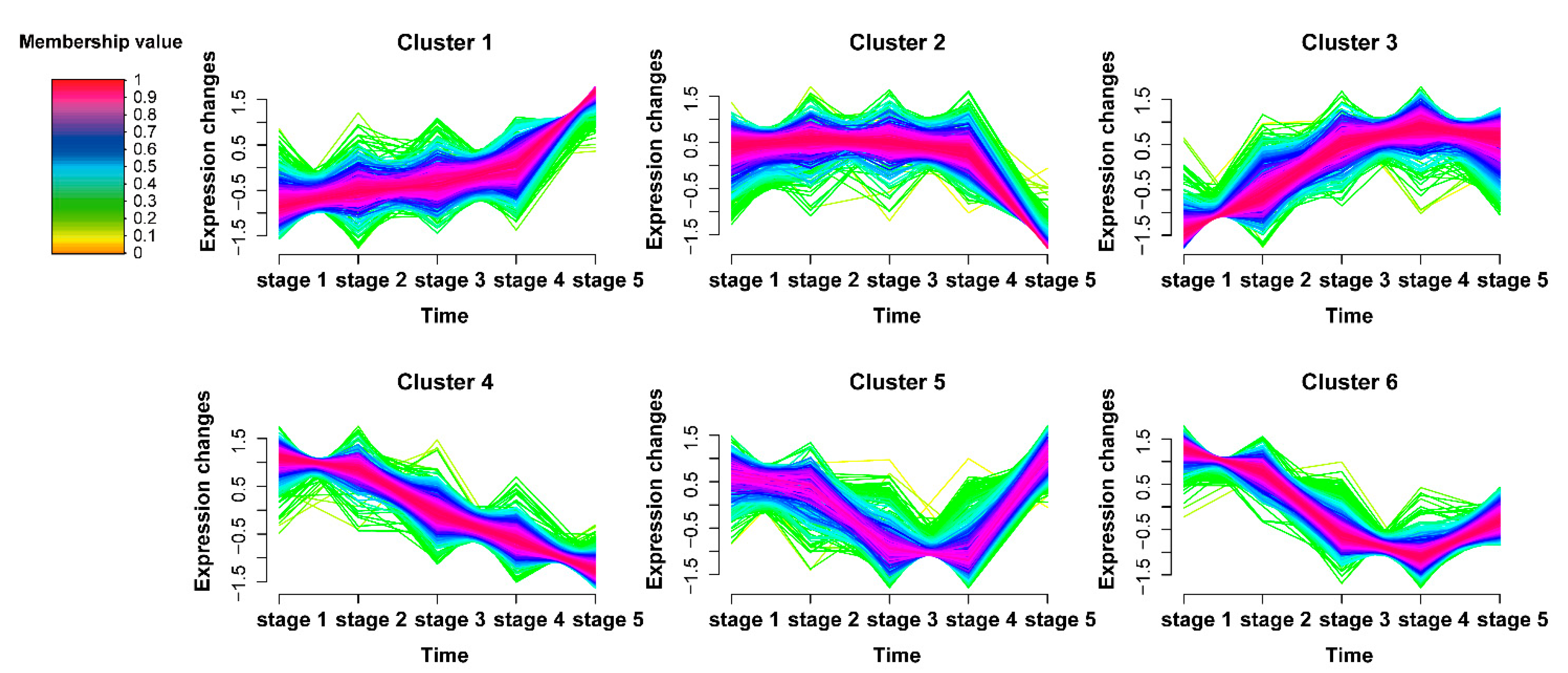
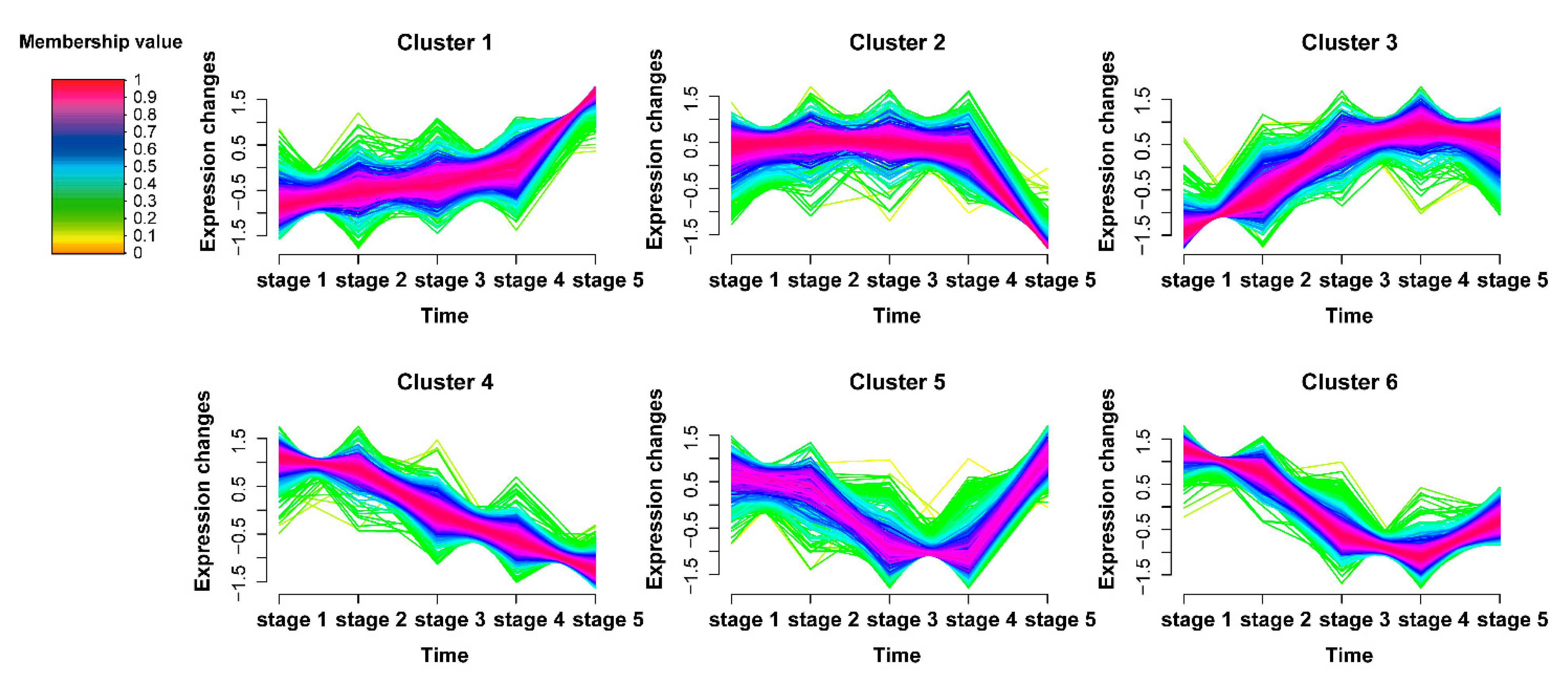
2.2. Time-Series Analysis of DAPs
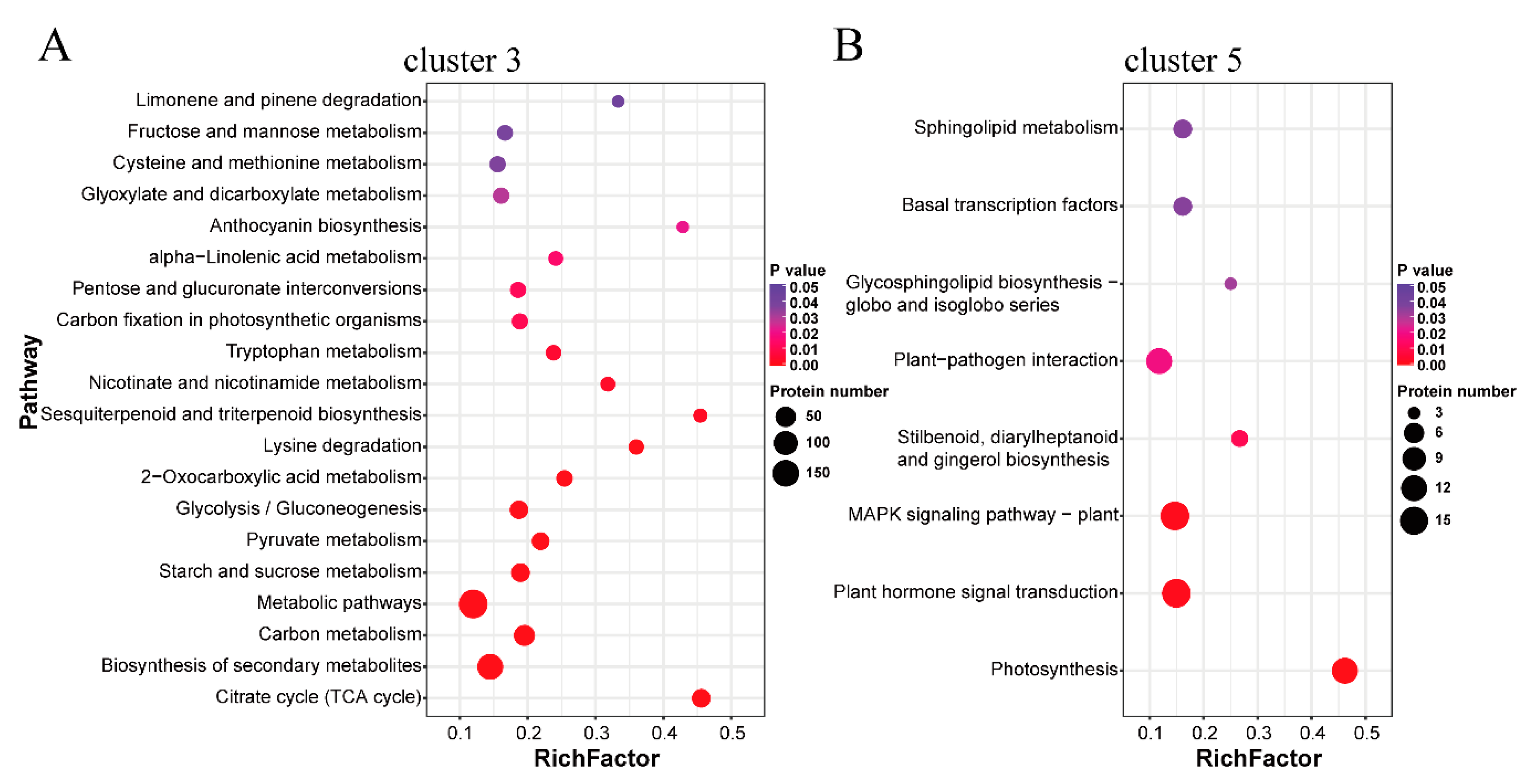
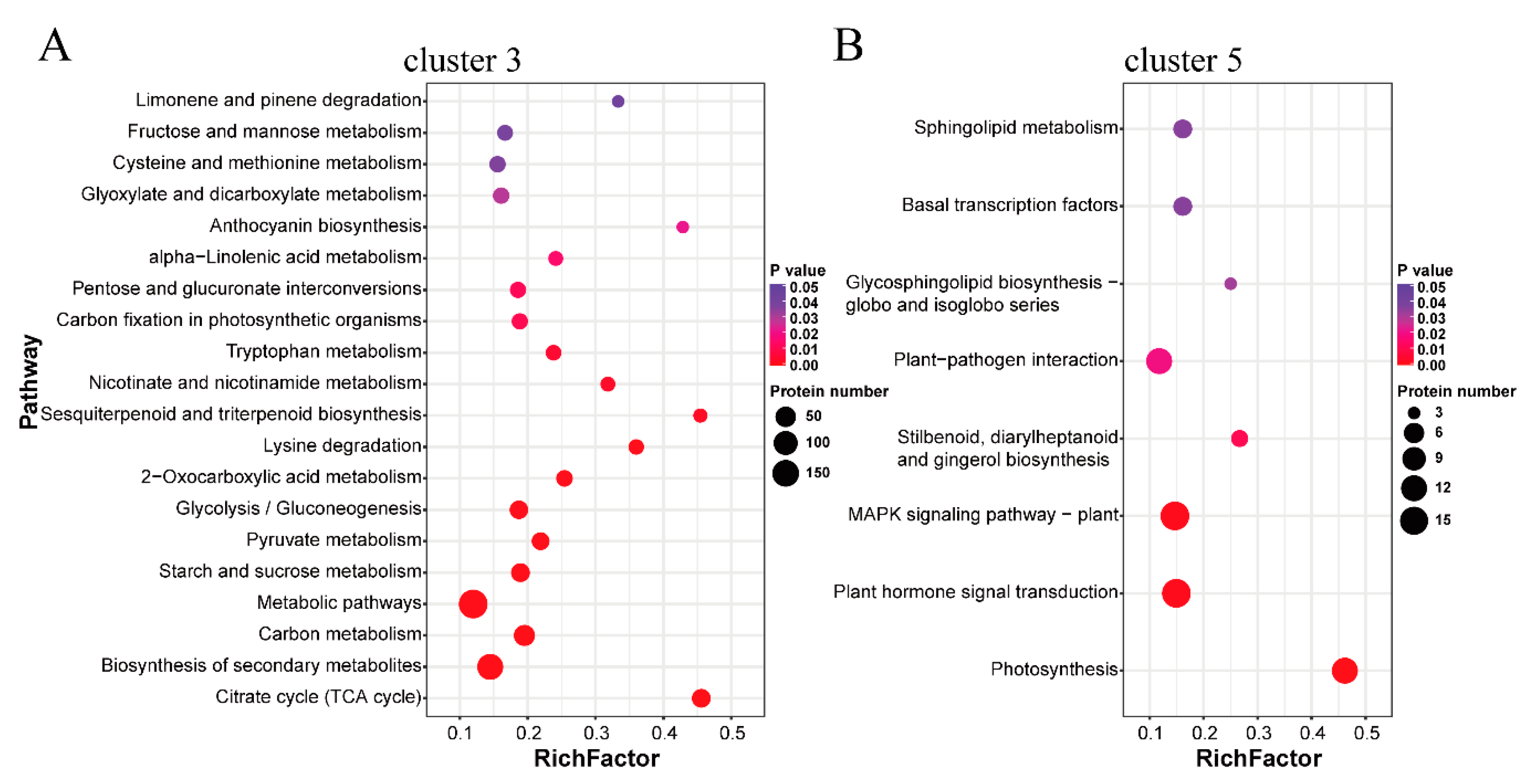
2.3. Functional Annotation Analysis of the Protein Clusters Related to the Thermogenic Process
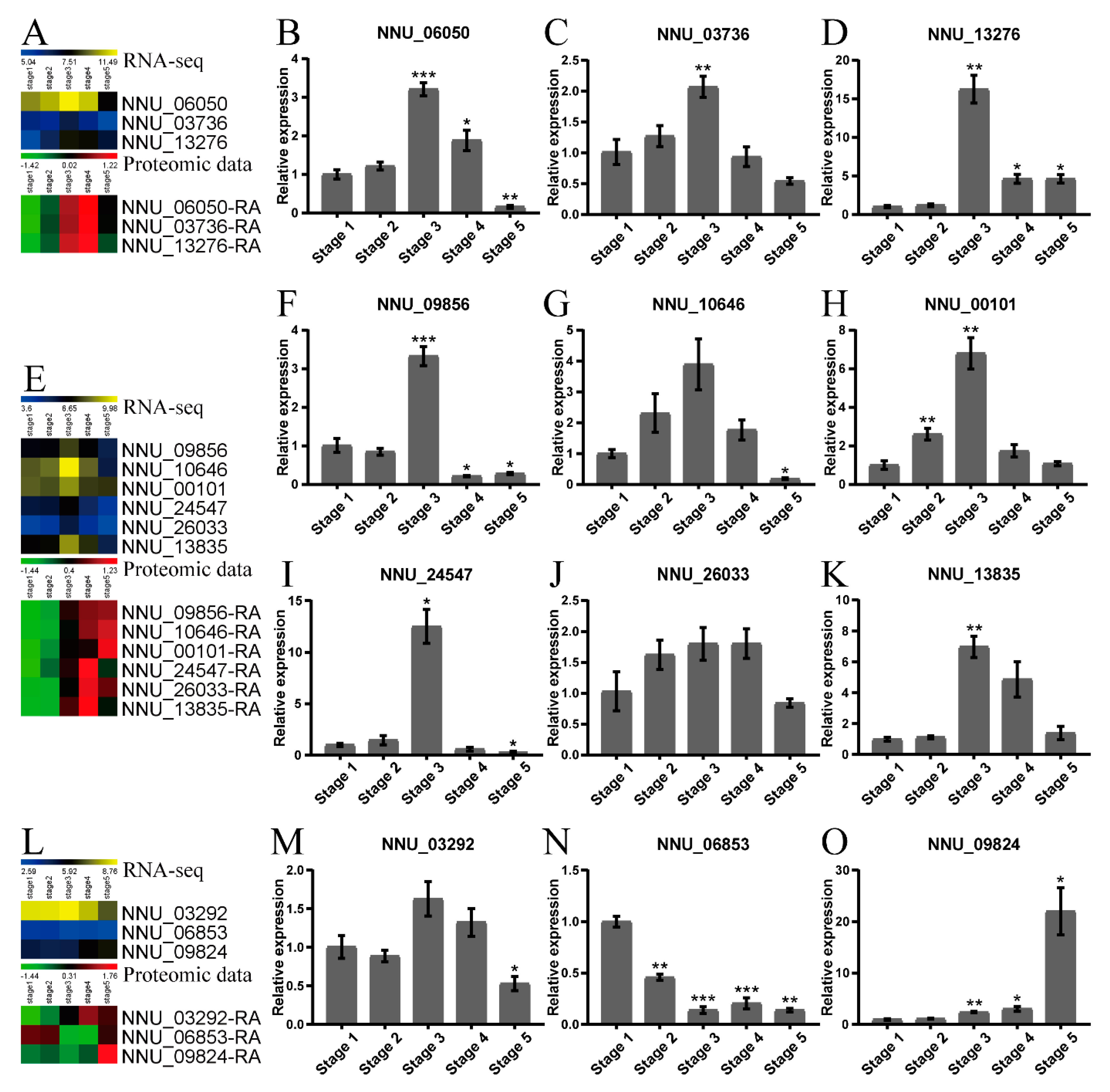
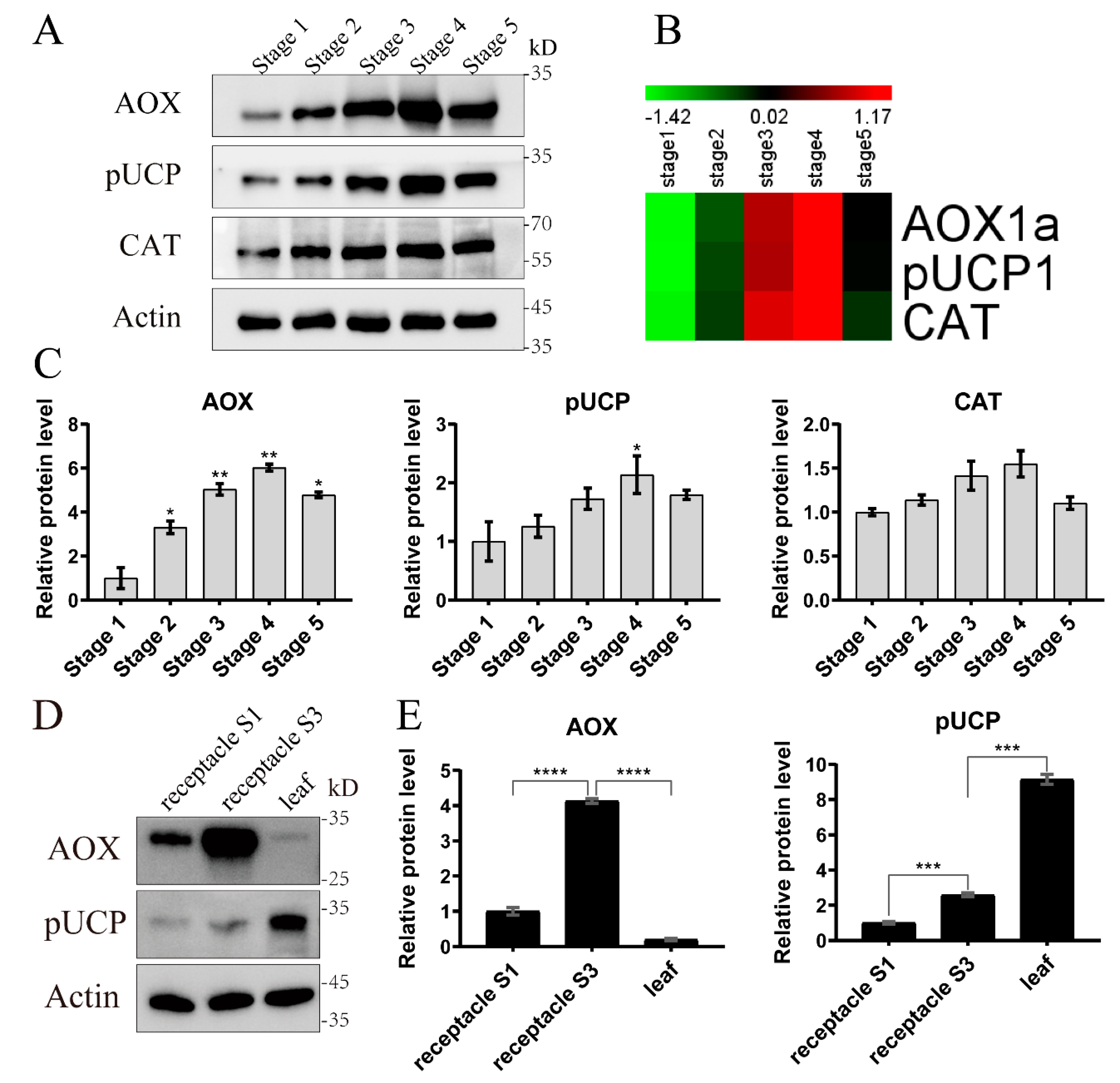
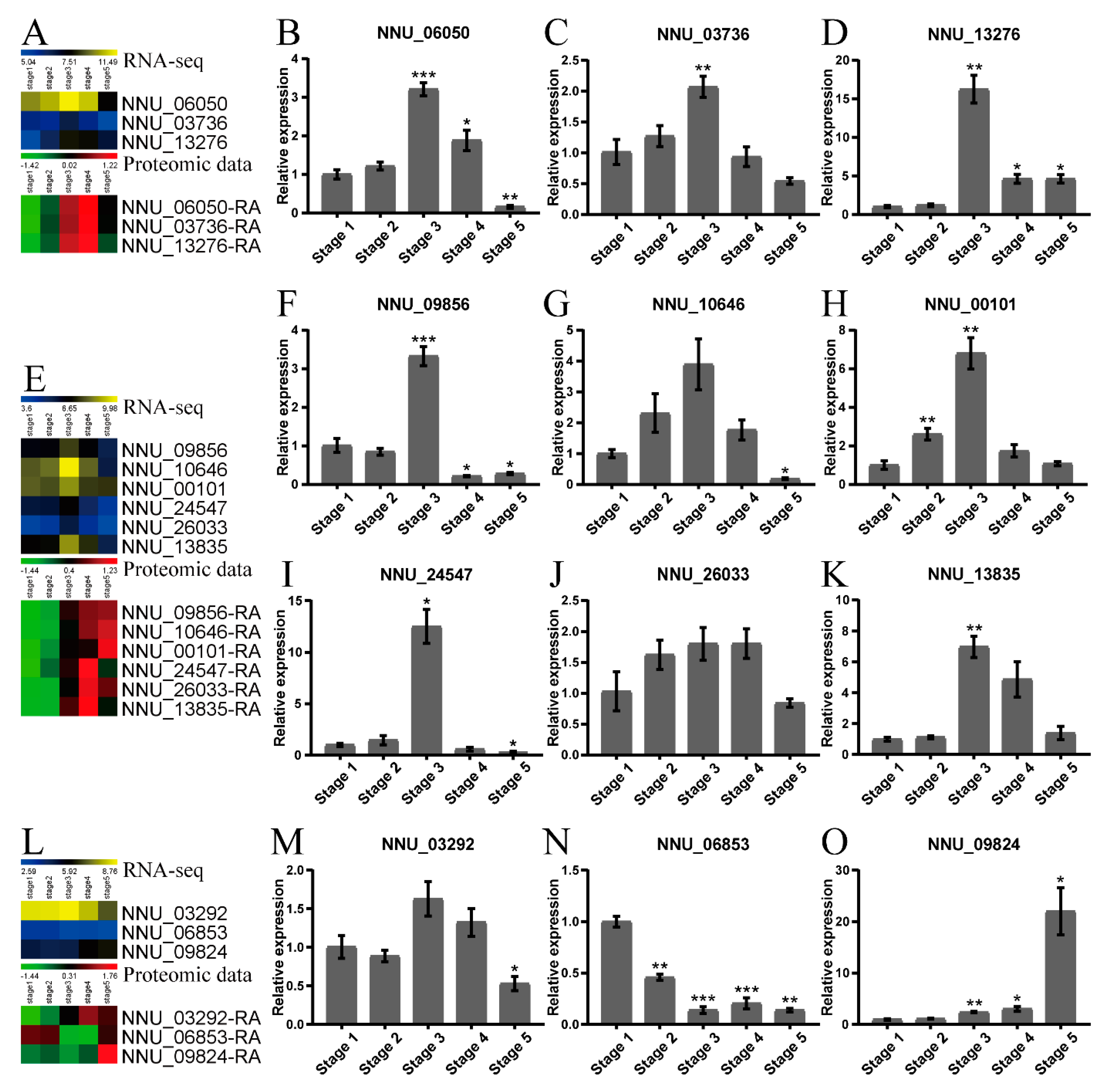
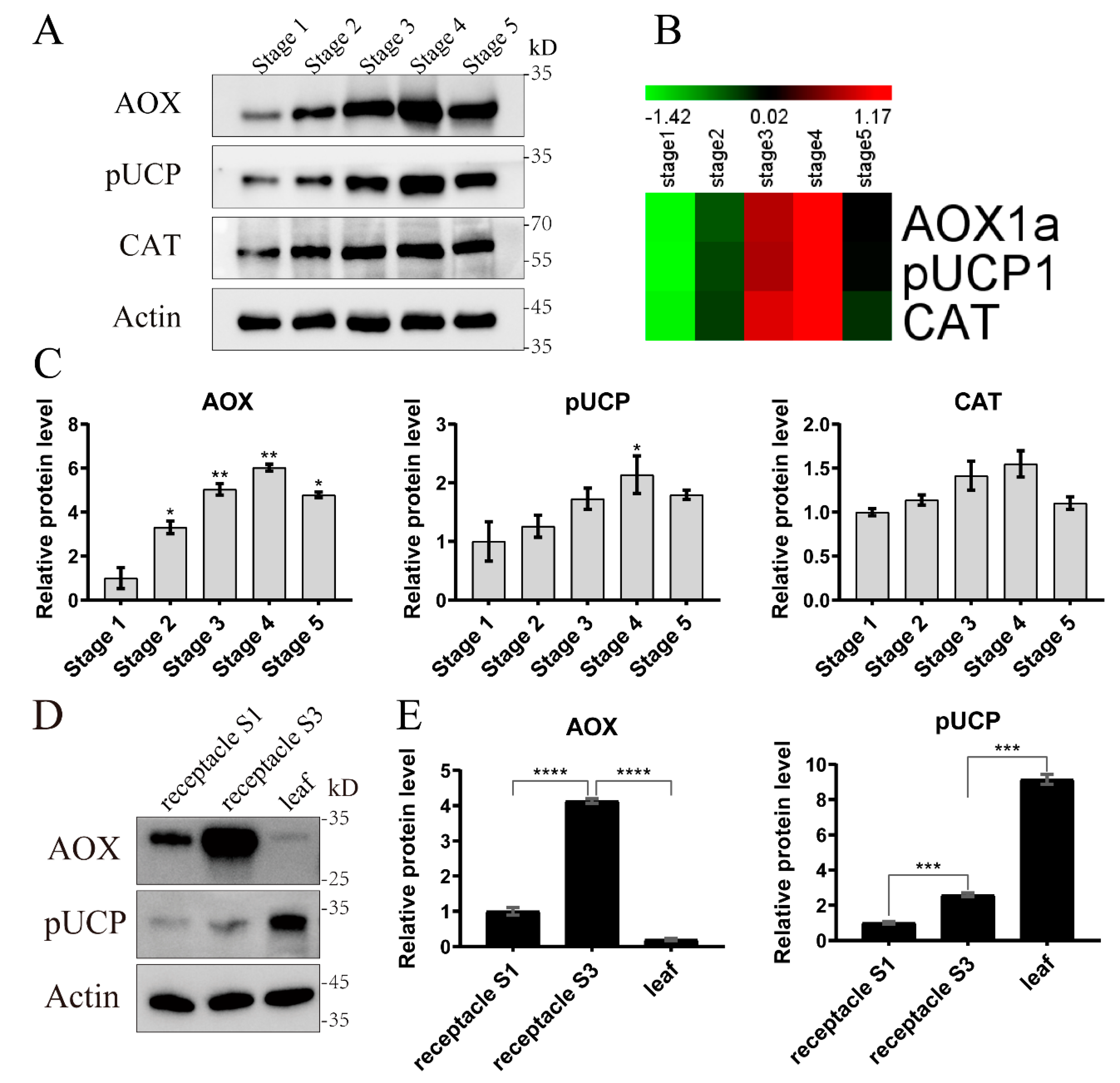
2.4. Validation of the Proteomic Data by qRT-PCR and Western Blot
2.5. Abundance of the Proteins AOX and pUCP between Thermogenic and non-Thermogenic Tissues
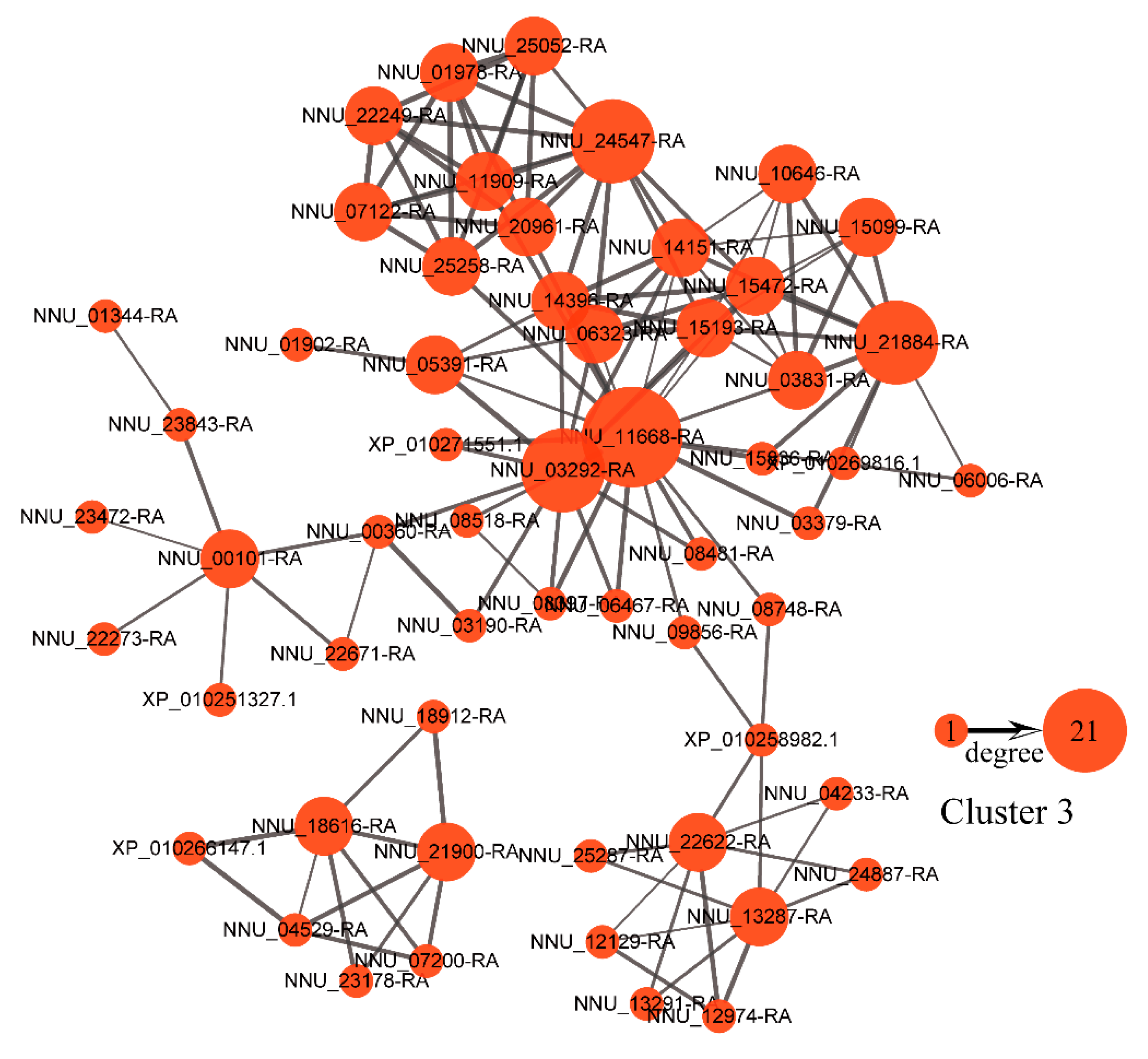
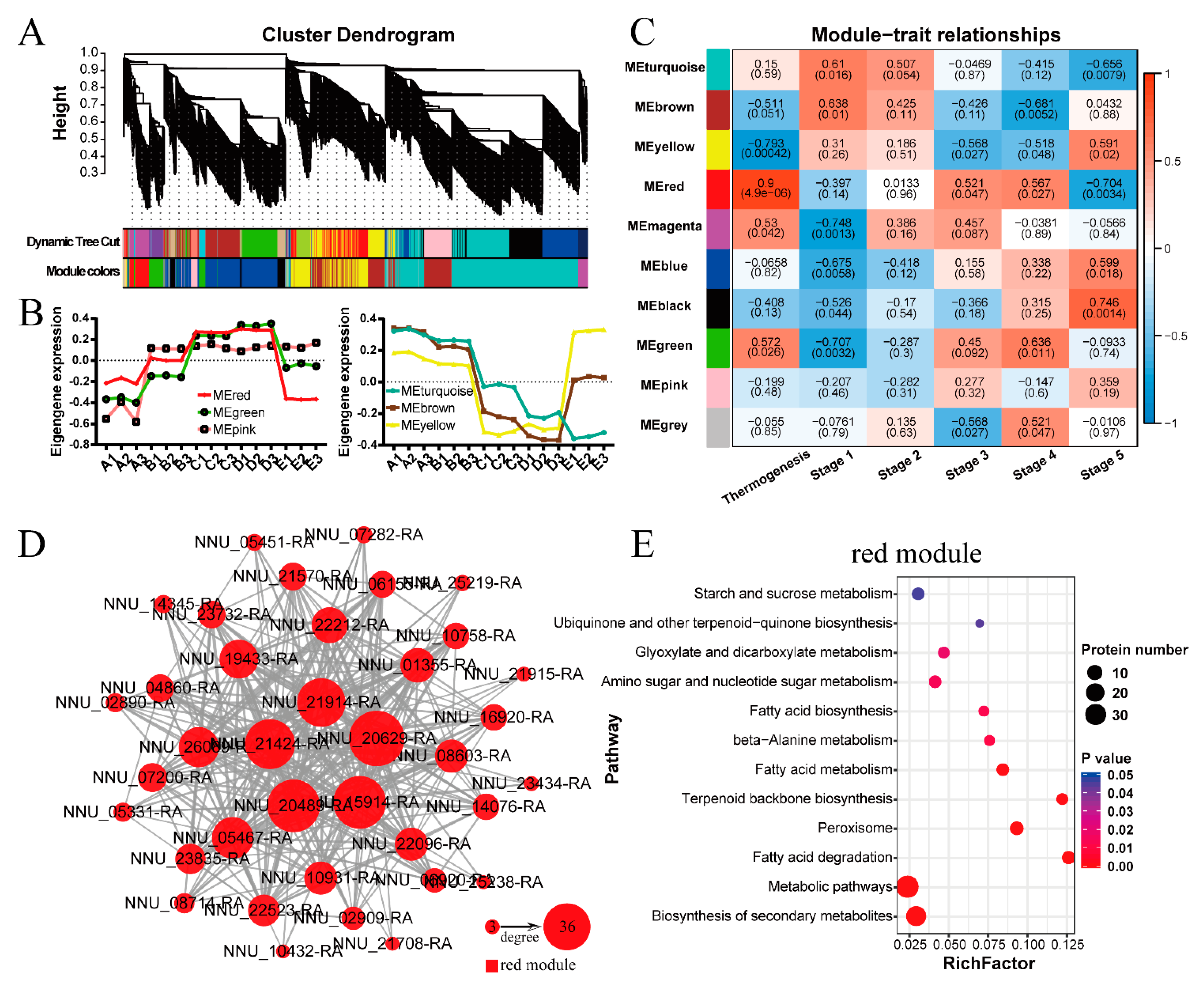
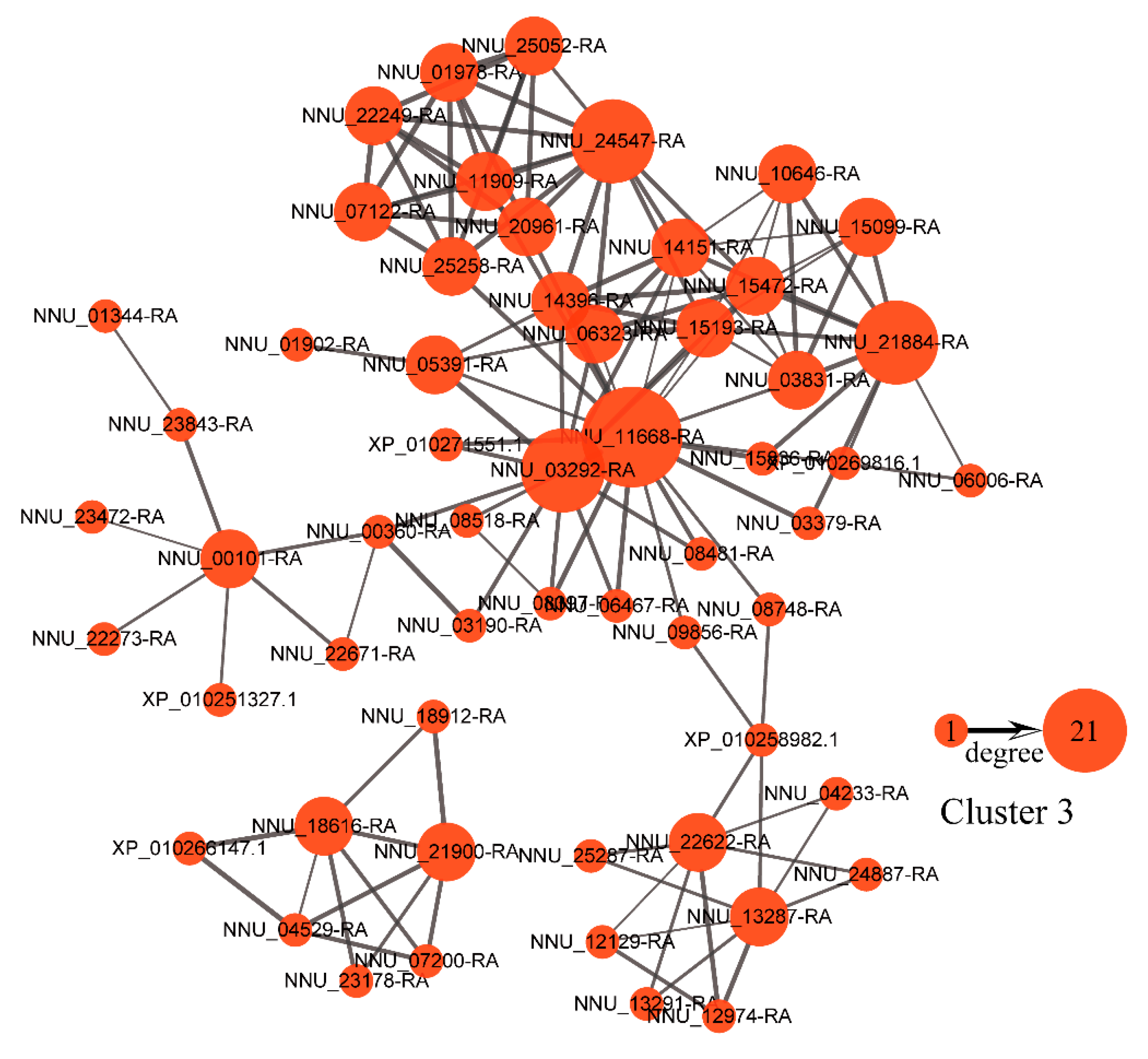
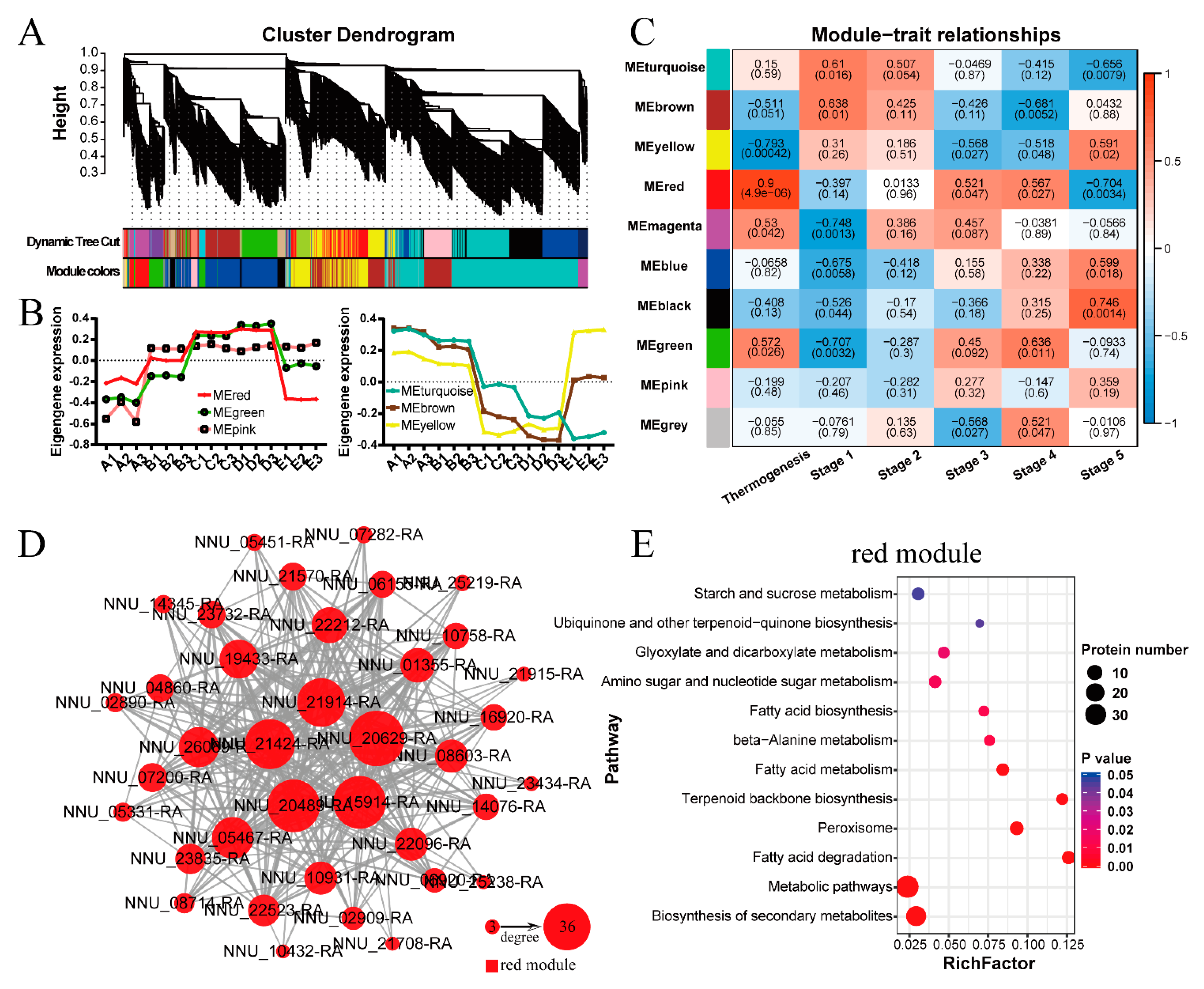
2.6. Protein-Protein Interaction (PPI) Network and Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Discussion
3.1. The Low-Correlation Omics Data Showed Similar Functions That Related to Thermogenesis
3.2. The Protein Expression Patterns of AOX and pUCP between Thermogenic and non-Thermogenic Tissues were Completely Different
3.3. TCA Cycle Metabolism, Fatty Acids Degradation, and Ubiquinone Biosynthesis Participate in Regulating Floral Thermogenesis of N. nucifera
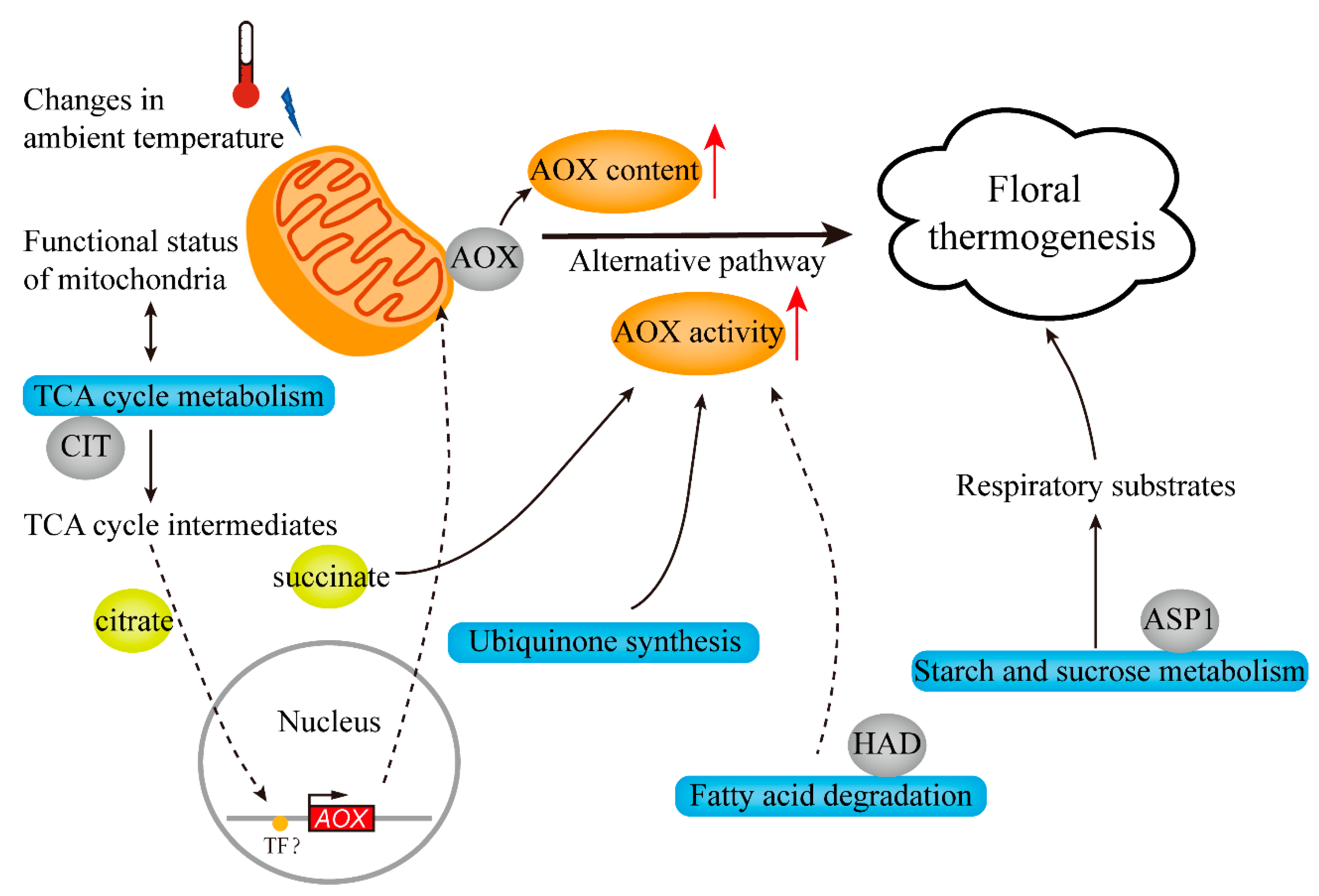
3.4. Putative Model for the Regulatory Networks Involved in the Regulation Mechanism of Floral Thermogenesis in N. nucifera
4. Materials and Methods
4.1. Plant Materials
4.2. Protein Preparation and Digestion
4.3. High pH Reverse Phase Fractionation
4.4. High-Performance Liquid Chromatography (HPLC)
4.5. Data-Dependent Acquisition Mass Spectrometry
4.6. Data-Independent Acquisition Mass Spectrometry
4.7. Data Analysis
4.8. Bioinformatic Analysis
4.9. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.10. Western Blot Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaffee, R.R.; Roberts, J.C. Temperature acclimation in birds and mammals. Annu. Rev. Physiol. 1971, 33, 155–202. [Google Scholar] [CrossRef]
- Nagy, K.A.; Odell, D.K.; Seymour, R.S. Temperature regulation by the inflorescence of philodendron. Science 1972, 178, 1195–1197. [Google Scholar] [CrossRef] [PubMed]
- Knutson, R.M. Heat production and temperature regulation in eastern skunk cabbage. Science 1974, 186, 746–747. [Google Scholar] [CrossRef]
- Raskin, I.; Ehmann, A.; Melander, W.R.; Meeuse, B.J. Salicylic acid: A natural inducer of heat production in Arum lilies. Science 1987, 237, 1601–1602. [Google Scholar] [CrossRef]
- Wagner, A.M.; Krab, K.; Wagner, M.J.; Moore, A.L. Regulation of thermogenesis in flowering Araceae: The role of the alternative oxidase. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Seymour, R.S.; Schultze-Motel, P. Thermoregulating lotus flowers. Nature 1996, 383, 305. [Google Scholar] [CrossRef]
- Dieringer, G.; Leticia Cabrera, R.; Mottaleb, M. Ecological relationship between floral thermogenesis and pollination in Nelumbo lutea (Nelumbonaceae). Am. J. Bot. 2014, 101, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Liu, X.; Mou, S.; Xu, S.; Zhang, Z. Temperature regulation of floral buds and floral thermogenicity in Magnolia denudata (Magnoliaceae). Trees 2013, 27, 1755–1762. [Google Scholar] [CrossRef]
- Ito-Inaba, Y.; Sato, M.; Sato, M.P.; Kurayama, Y.; Yamamoto, H.; Ohata, M.; Ogura, Y.; Hayashi, T.; Toyooka, K.; Inaba, T. Alternative oxidase capacity of mitochondria in microsporophylls may function in cycad thermogenesis. Plant Physiol. 2019, 180, 743–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.K.; Huang, S.Q. Flower thermoregulation facilitates fertilization in Asian sacred lotus. Ann. Bot. 2009, 103, 1159–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, R.S.; Ito, Y.; Onda, Y.; Ito, K. Effects of floral thermogenesis on pollen function in Asian skunk cabbage Symplocarpus renifolius. Biol. Lett. 2009, 5, 568–570. [Google Scholar] [CrossRef] [Green Version]
- Seymour, R.S.; Schultze-Motel, P. Heat-producing flowers. Endeavour 1997, 21, 125–129. [Google Scholar] [CrossRef]
- Seymour, R.S.; White, C.R.; Gibernau, M. Heat reward for insect pollinators. Nature 2003, 426, 243–244. [Google Scholar] [CrossRef]
- Jarmuszkiewicz, W.; Sluse-Goffart, C.M.; Vercesi, A.E.; Sluse, F.E. Alternative oxidase and uncoupling protein: Thermogenesis versus cell energy balance. Biosci. Rep. 2001, 21, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watling, J.R.; Grant, N.M.; Miller, R.E.; Robinson, S.A. Mechanisms of thermoregulation in plants. Plant Signal. Behav. 2008, 3, 595–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.L.; Siedow, J.N. The regulation and nature of the cyanide-resistant alternative oxidase of plant mitochondria. Biochim. Biophys. Acta Bioenerg. 1991, 1059, 121–140. [Google Scholar] [CrossRef]
- Vanlerberghe, G.C.; McLntosh, L. Signals regulating the expression of the nuclear gene encoding alternative oxidase of plant mitochondria. Plant Physiol. 1996, 111, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, D.M.; Mcintosh, L. Salicylic acid regulation of respiration in higher plants: Alternative oxidase expression. Plant Cell 1992, 4, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Chivasa, S.; Berry, J.O.; Rees, T.A.; Carr, J.P. Changes in gene expression during development and thermogenesis in Arum. Aust. J. Plant Physiol. 1999, 26, 391–399. [Google Scholar] [CrossRef]
- Grant, N.M.; Miller, R.E.; Watling, J.R.; Robinson, S.A. Synchronicity of thermogenic activity, alternative pathway respiratory flux, AOX protein content, and carbohydrates in receptacle tissues of sacred lotus during floral development. J. Exp. Bot. 2008, 59, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef]
- Hourton-Cabassa, U.; Matos, A.R.; Zachowski, A.; Moreau, F. The plant uncoupling protein homologues: A new family of energy-dissipating proteins in plant mitochondria. Plant Physiol. Biochem. 2004, 42, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Vercesi, A.E.; Borecky, J.; Godoy Maia, I.D.; Arruda, P.; Cuccovia, I.M.; Chaimovich, H. Plant uncoupling mitochondrial proteins. Annu. Rev. Plant Biol. 2006, 57, 383–404. [Google Scholar] [CrossRef]
- Ito-Inaba, Y.; Hida, Y.; Ichikawa, M.; Kato, Y.; Yamashita, T. Characterization of the plant uncoupling protein, SrUCPA, expressed in spadix mitochondria of the thermogenic skunk cabbage. J. Exp. Bot. 2008, 59, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Seymour, R.S.; Schultze-Motel, P. Physiological temperature regulation by flowers of the sacred lotus. Philos. Trans. R. Soc. Lond. B 1998, 353, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Watling, J.R.; Robinson, S.A.; Seymour, R.S. Contribution of the alternative pathway to respiration during thermogenesis in flowers of the sacred lotus. Plant Physiol. 2006, 140, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Grant, N.; Onda, Y.; Kakizaki, Y.; Ito, K.; Watling, J.; Robinson, S. Two cys or not two cys? That is the question; alternative oxidase in the thermogenic plant sacred lotus. Plant Physiol. 2009, 150, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Raskin, I.; Turner, I.M.; Melander, W.R. Regulation of heat production in the inflorescences of an Arum lily by endogenous salicylic acid. Proc. Natl. Acad. Sci. USA 1989, 86, 2214–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito-Inaba, Y.; Hida, Y.; Inaba, T. What is critical for plant thermogenesis? Differences in mitochondrial activity and protein expression between thermogenic and non-thermogenic skunk cabbages. Planta 2009, 231, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Ito-Inaba, Y.; Hida, Y.; Matsumura, H.; Masuko, H.; Yazu, F.; Terauchi, R.; Watanabe, M.; Inaba, T. The gene expression landscape of thermogenic skunk cabbage suggests critical roles for mitochondrial and vacuolar metabolic pathways in the regulation of thermogenesis. Plant Cell Environ. 2012, 35, 554–566. [Google Scholar] [CrossRef]
- Onda, Y.; Mochida, K.; Yoshida, T.; Sakurai, T.; Seymour, R.S.; Umekawa, Y.; Pirintsos, S.A.; Shinozaki, K.; Ito, K. Transcriptome analysis of thermogenic Arum concinnatum reveals the molecular components of floral scent production. Sci. Rep. 2015, 5, 8753. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Chen, G.; Jin, J.; Wang, Y.; Xu, M.; Peng, J.; Ding, Y. Small RNA and transcriptome sequencing reveals miRNA regulation of floral thermogenesis in Nelumbo nucifera. Int. J. Mol. Sci. 2020, 21, 3324. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Quan, J.; Kang, Y.; Li, L.; Zhao, G.; Sun, J.; Liu, Z. Proteome analysis of rainbow trout (Oncorhynchus mykiss) liver responses to chronic heat stress using DIA/SWATH. J. Proteomics 2021, 233, 104079. [Google Scholar] [CrossRef]
- Meng, K.; Lu, S.; Yan, X.; Sun, Y.; Gao, J.; Wang, Y.; Yin, X.; Sun, Z.; He, Q.Y. Quantitative mitochondrial proteomics reveals ANXA7 as a crucial factor in mitophagy. J. Proteome Res. 2020, 19, 1275–1284. [Google Scholar] [CrossRef]
- Miller, R.E.; Watling, J.R.; Robinson, S.A. Functional transition in the floral receptacle of the sacred lotus (Nelumbo nucifera): From thermogenesis to photosynthesis. Funct. Plant Biol. 2009, 36, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Vanlerberghe, G.C.; Day, D.A.; Wiskich, J.T.; Vanlerberghe, A.E.; McIntosh, L. Alternative oxidase activity in tobacco leaf mitochondria (dependence on tricarboxylic acid cycle-mediated redox regulation and pyruvate activation). Plant Physiol. 1995, 109, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, G.R.; Maxwell, D.P.; Villarimo, A.R.; McIntosh, L. Mitochondria/nuclear signaling of alternative oxidase gene expression occurs through distinct pathways involving organic acids and reactive oxygen species. Plant Cell Rep. 2004, 23, 497–503. [Google Scholar] [CrossRef]
- Finkemeier, I.; König, A.C.; Heard, W.; Nunes-Nesi, A.; Pham, P.A.; Leister, D.; Fernie, A.R.; Sweetlove, L.J. Transcriptomic analysis of the role of carboxylic acids in metabolite signaling in Arabidopsis leaves. Plant Physiol. 2013, 162, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Bhagavan, N.V.; Ha, C.-E. Essentials of Medical Biochemistry, 2nd ed.; Academic Press: San Diego, CA, USA, 2015; pp. 165–185. [Google Scholar]
- Weitzman, P.D.; Danson, M.J. Citrate synthase. Curr. Top. Cell. Regul. 1976, 10, 161–204. [Google Scholar]
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Vanlerberghe, G.C.; McIntosh, L. Alternative oxidase: From gene to function. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 703–734. [Google Scholar] [CrossRef]
- Vanlerberghe, G.C.; Dahal, K.; Alber, N.A.; Chadee, A. Photosynthesis, respiration and growth: A carbon and energy balancing act for alternative oxidase. Mitochondrion 2020, 52, 197–211. [Google Scholar] [CrossRef]
- Sluse, F.E.; Almeida, A.M.; Jarmuszkiewicz, W.; Vercesi, A.E. Free fatty acids regulate the uncoupling protein and alternative oxidase activities in plant mitochondria. FEBS Lett. 1998, 433, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Dry, I.B.; Moore, A.L.; Day, D.A.; Wiskich, J.T. Regulation of alternative pathway activity in plant mitochondria: Nonlinear relationship between electron flux and the redox poise of the quinone pool. Arch. Biochem. Biophys. 1989, 273, 148–157. [Google Scholar] [CrossRef]
- Ribas-Carbo, M.; Wiskich, J.T.; Berry, J.A.; Siedow, J.N. Ubiquinone redox behavior in plant mitochondria during electron transport. Arch. Biochem. Biophys. 1995, 317, 156–160. [Google Scholar] [CrossRef]
- Ng, S.; De Clercq, I.; Van Aken, O.; Law, S.R.; Ivanova, A.; Willems, P.; Giraud, E.; Van Breusegem, F.; Whelan, J. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol Plant 2014, 7, 1075–1093. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Wang, K.; Deng, J.; Damaris, R.N.; Yang, M.; Xu, L.; Yang, P. LOTUS-DB: An integrative and interactive database for Nelumbo nucifera study. Database 2015, 2015, bav023. [Google Scholar] [CrossRef] [Green Version]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, L.; Rinner, O.; Picotti, P.; Hüttenhain, R.; Beck, M.; Brusniak, M.Y.; Hengartner, M.O.; Aebersold, R. mProphet: Automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef]
- Choi, M.; Chang, C.Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, L.; Futschik, M. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhu, Z.; Gui, S.; Jin, J.; Yi, R.; Wu, Z.; Qian, Q.; Ding, Y. The NnCenH3 protein and centromeric DNA sequence profiles of Nelumbo nucifera Gaertn. (sacred lotus) reveal the DNA structures and dynamics of centromeres in basal eudicots. Plant J. 2016, 87, 568–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic. Acids. Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Zou, Y.; Jin, J.; Chen, H.; Liu, Z.; Zi, Q.; Xiong, Z.; Wang, Y.; Li, Q.; Peng, J.; et al. DIA-Based Quantitative Proteomics Reveals the Protein Regulatory Networks of Floral Thermogenesis in Nelumbo nucifera. Int. J. Mol. Sci. 2021, 22, 8251. https://doi.org/10.3390/ijms22158251
Sun Y, Zou Y, Jin J, Chen H, Liu Z, Zi Q, Xiong Z, Wang Y, Li Q, Peng J, et al. DIA-Based Quantitative Proteomics Reveals the Protein Regulatory Networks of Floral Thermogenesis in Nelumbo nucifera. International Journal of Molecular Sciences. 2021; 22(15):8251. https://doi.org/10.3390/ijms22158251
Chicago/Turabian StyleSun, Yueyang, Yu Zou, Jing Jin, Hao Chen, Zhiying Liu, Qinru Zi, Zeyang Xiong, Ying Wang, Qian Li, Jing Peng, and et al. 2021. "DIA-Based Quantitative Proteomics Reveals the Protein Regulatory Networks of Floral Thermogenesis in Nelumbo nucifera" International Journal of Molecular Sciences 22, no. 15: 8251. https://doi.org/10.3390/ijms22158251
APA StyleSun, Y., Zou, Y., Jin, J., Chen, H., Liu, Z., Zi, Q., Xiong, Z., Wang, Y., Li, Q., Peng, J., & Ding, Y. (2021). DIA-Based Quantitative Proteomics Reveals the Protein Regulatory Networks of Floral Thermogenesis in Nelumbo nucifera. International Journal of Molecular Sciences, 22(15), 8251. https://doi.org/10.3390/ijms22158251