
Promyelocytic Leukemia Proteins Regulate Fanconi Anemia Gene Expression

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
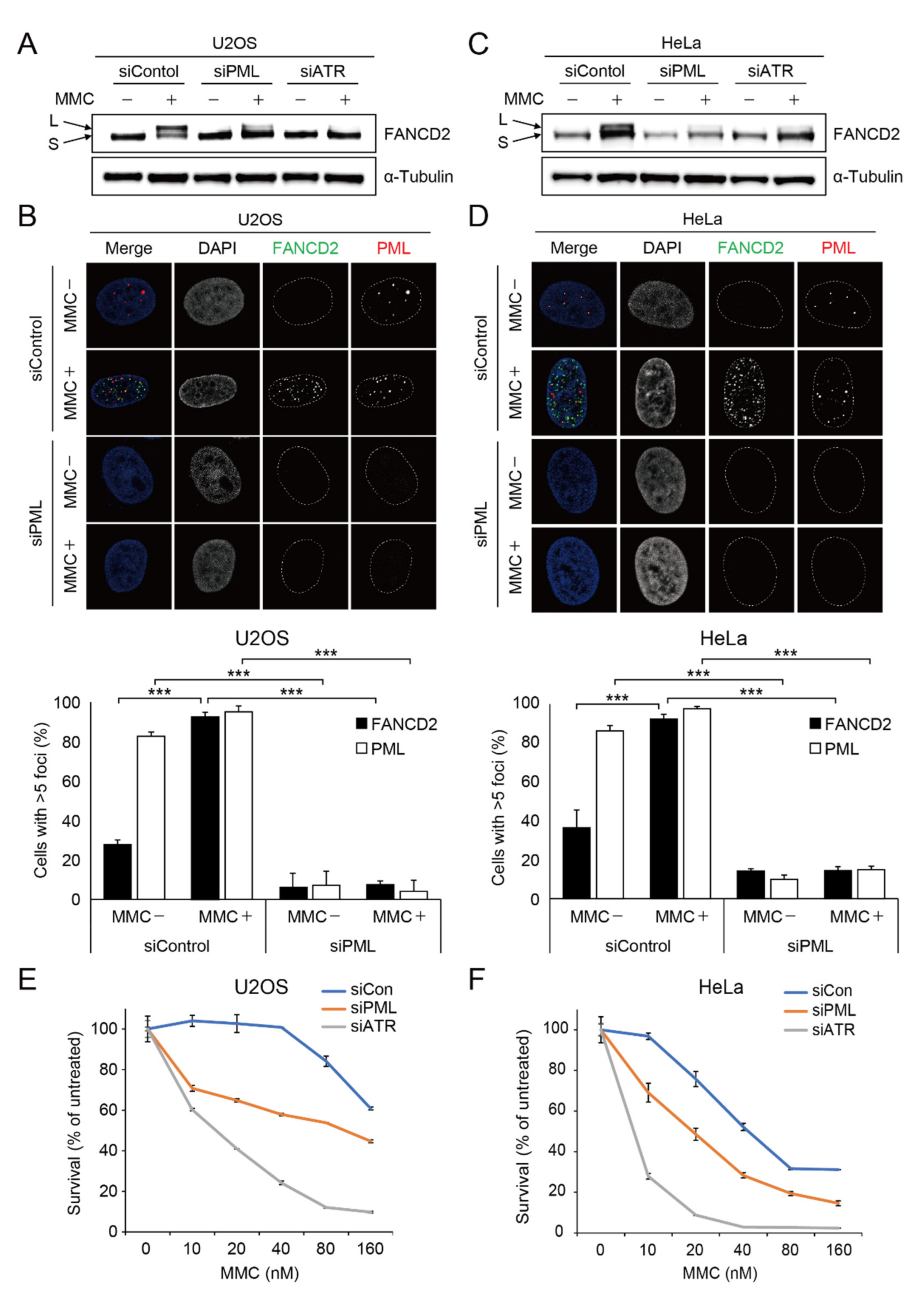
2.1. Depletion of PML Causes Impairment of Damage-Induced FANCD2 Mono-Ubiquitination and FANCD2 Foci Formation
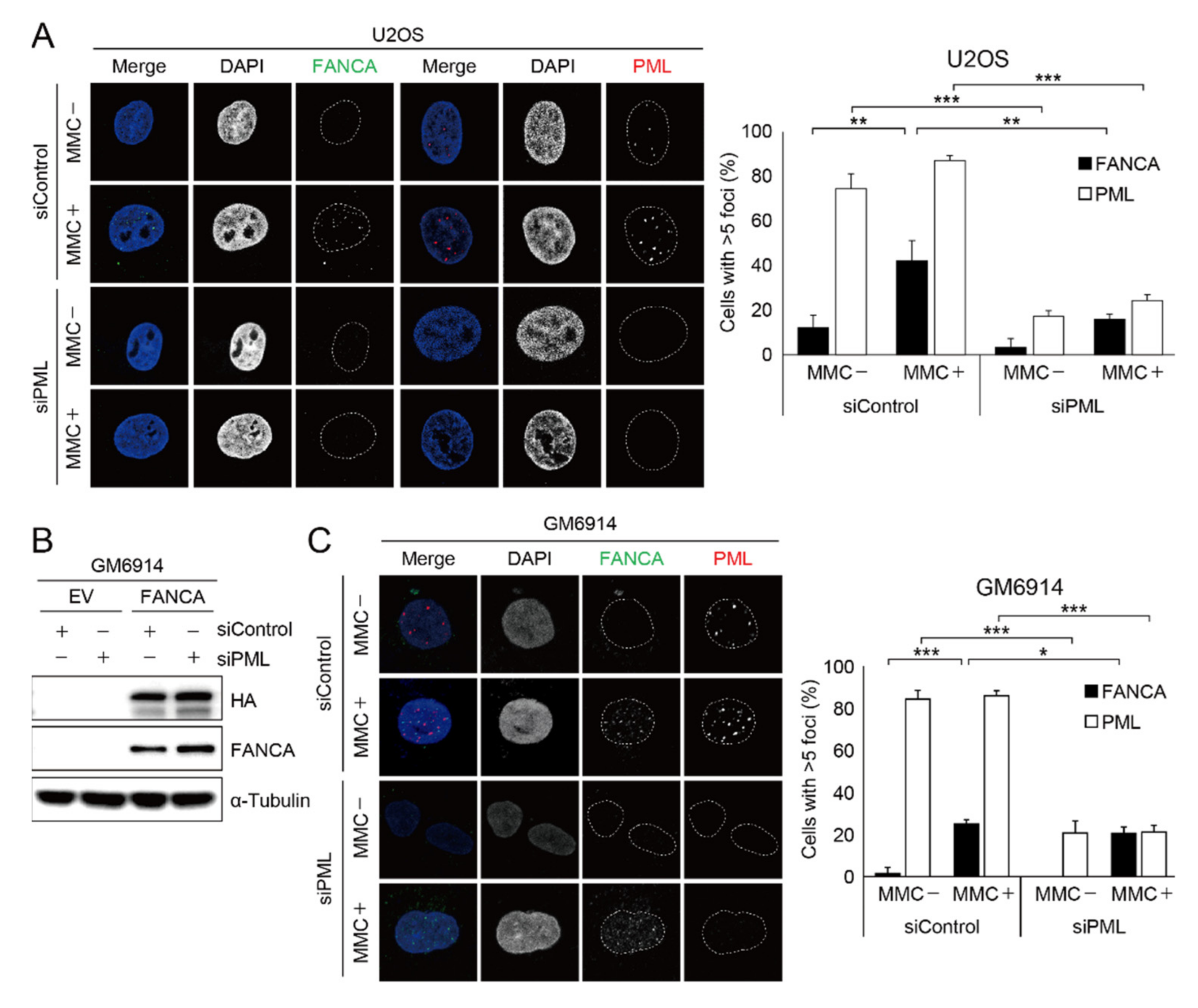
2.2. Damage-Induced FANCA Foci Formation Is Impaired in Cells with PML Depletion
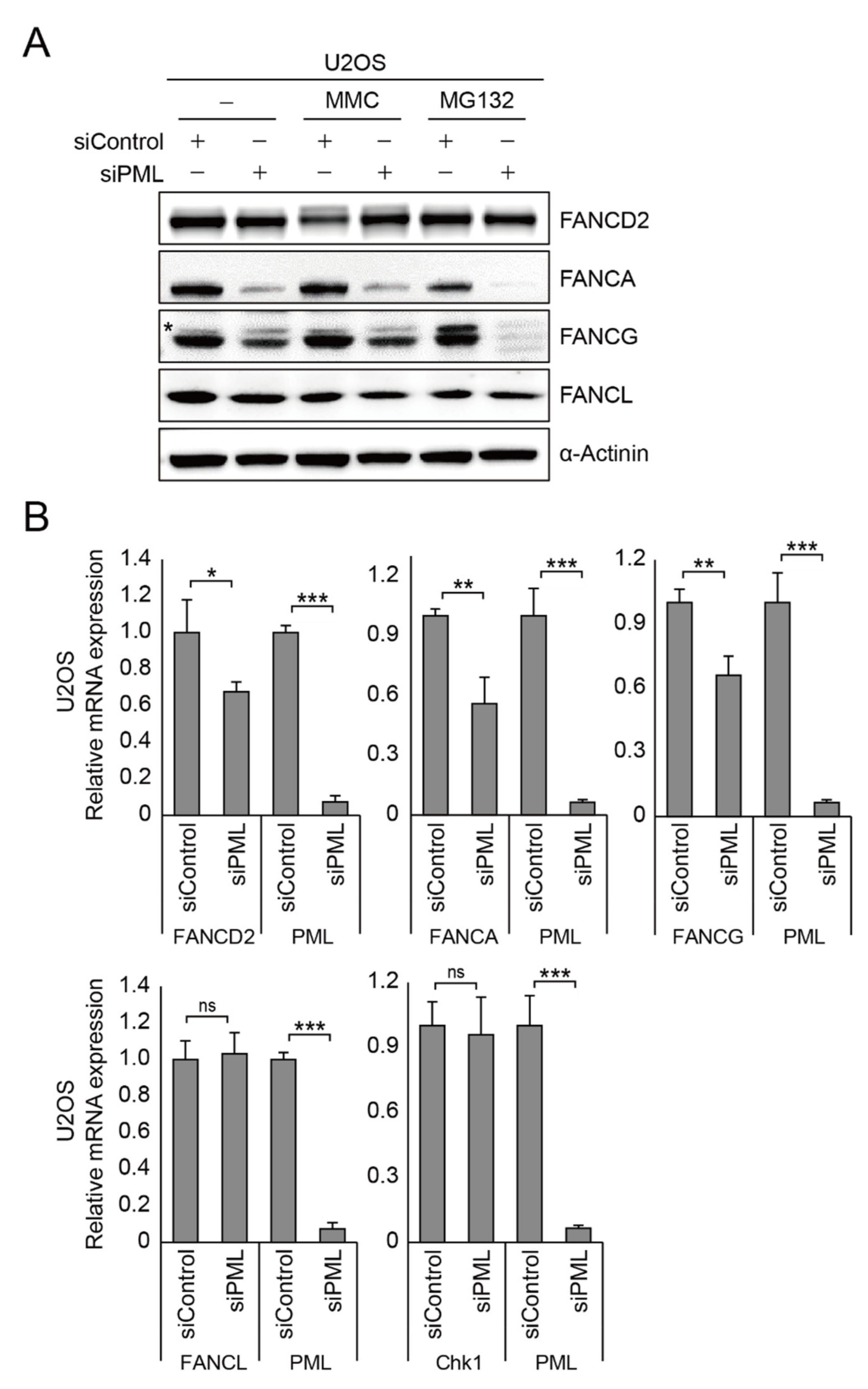
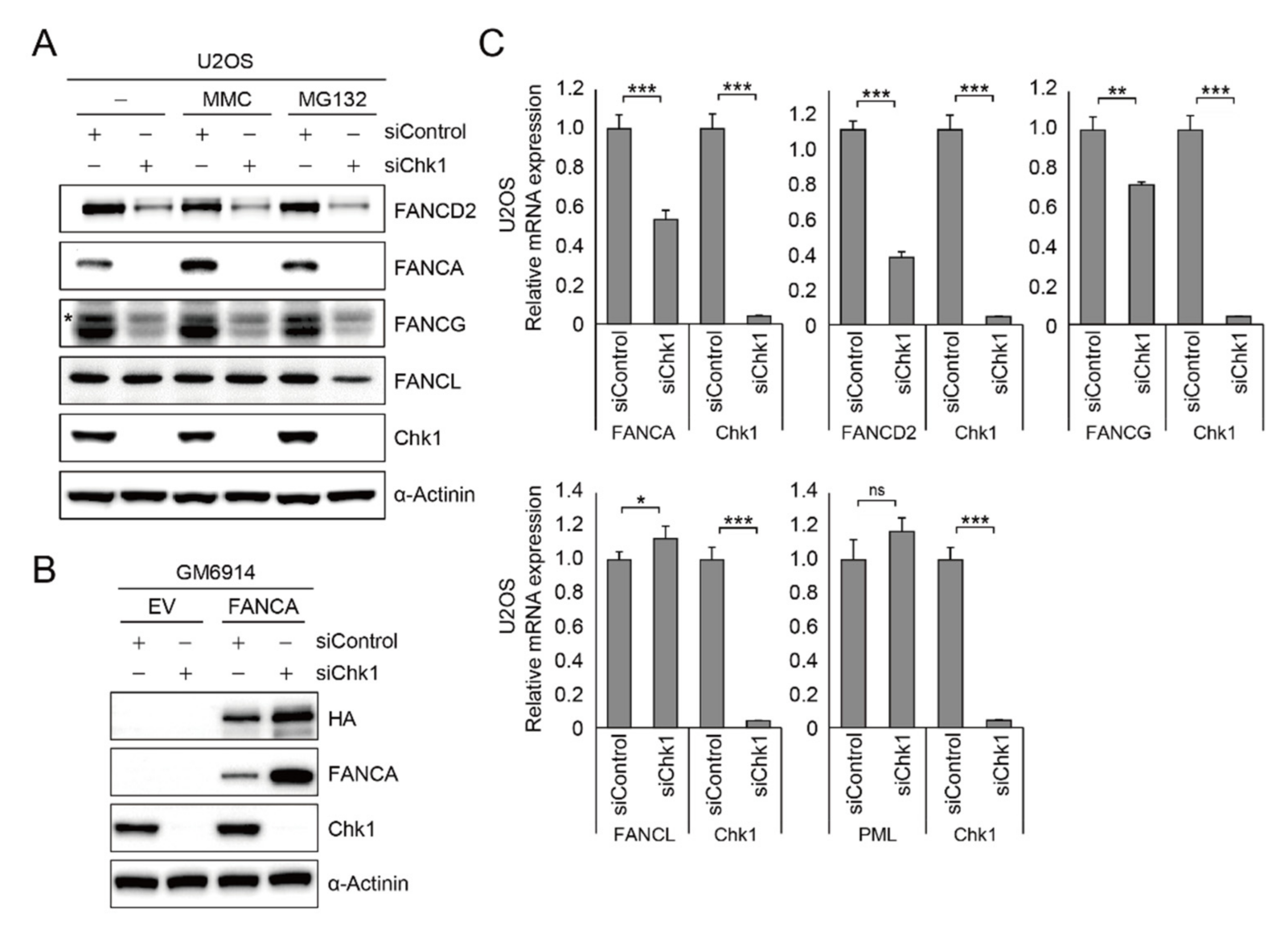
2.3. PML Regulates FANCA, FANCG, and FANCD2 Gene Expression
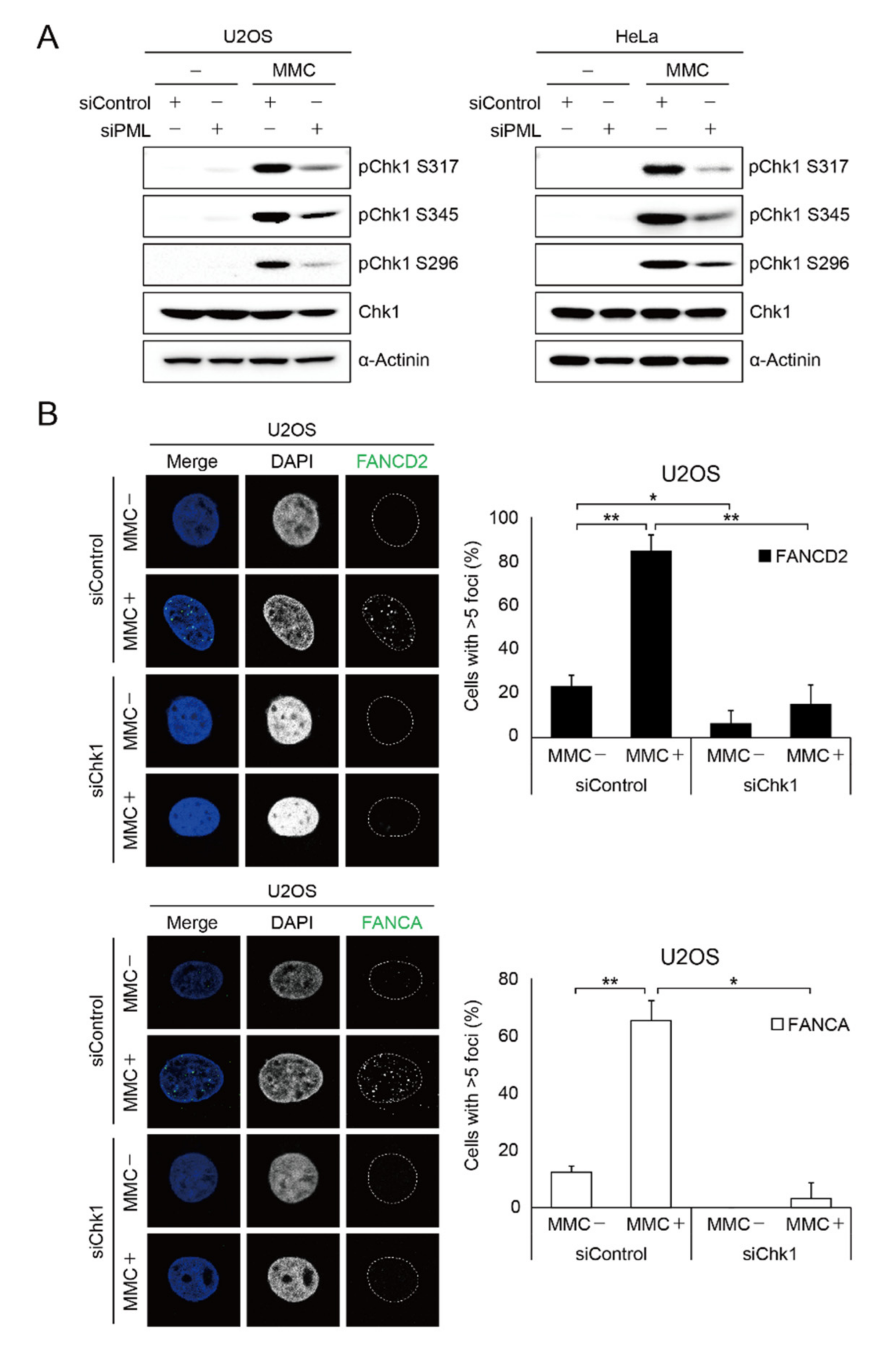
2.4. PML Promotes CHK1 Phosphorylation in Response to DNA Damage
2.5. CHK1 Is Implicated in FA Gene Expression
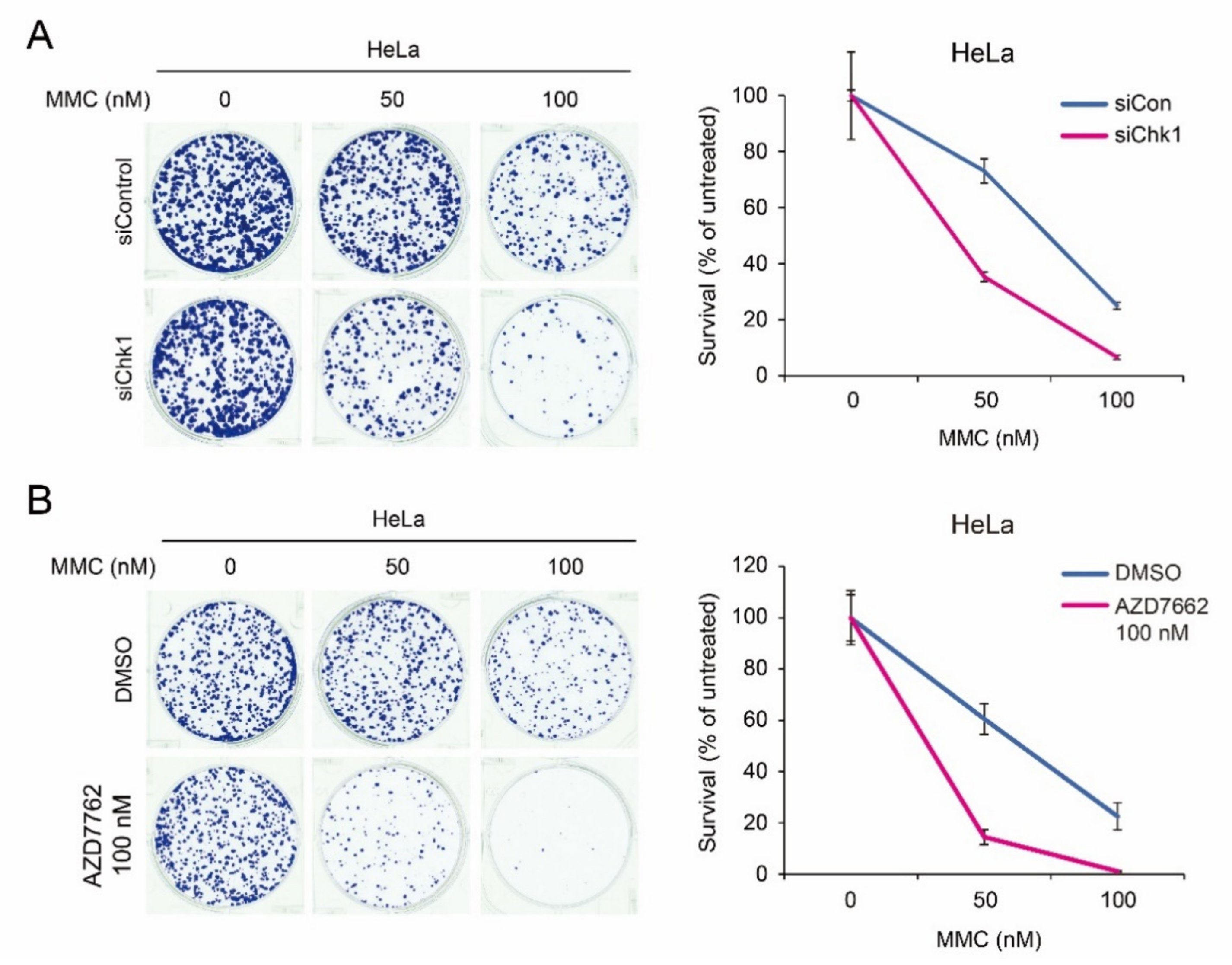
2.6. Synergistic Chemotherapeutic Effects of CHK1 Inhibitors and Crossliking Agents
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Small Interfering RNA (siRNA)
4.3. Western Blotting
4.4. Immunofluorescence
4.5. Mitomycin C Sensitivity Assay
4.6. RNA Isolation and Quantitative Real-Time PCR
4.7. Clonogenic Assay
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De The, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Kao, H.Y. The function, regulation and therapeutic implications of the tumor suppressor protein, PML. Cell Biosci. 2015, 5, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomoni, P.; Pandolfi, P.P. The role of PML in tumor suppression. Cell 2002, 108, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7, e2308. [Google Scholar] [CrossRef]
- Chang, H.R.; Munkhjargal, A.; Kim, M.J.; Park, S.Y.; Jung, E.; Ryu, J.H.; Yang, Y.; Lim, J.S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat Res. 2018, 809, 99–107. [Google Scholar] [CrossRef]
- Sahin, U.; de The, H.; Lallemand-Breitenbach, V. PML nuclear bodies: Assembly and oxidative stress-sensitive sumoylation. Nucleus 2014, 5, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.; Shiels, C.; Freemont, P.S. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001, 20, 7223–7233. [Google Scholar] [CrossRef] [Green Version]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Nisole, S.; Maroui, M.A.; Mascle, X.H.; Aubry, M.; Chelbi-Alix, M.K. Differential Roles of PML Isoforms. Front. Oncol. 2013, 3, 125. [Google Scholar] [CrossRef] [Green Version]
- Fogal, V.; Gostissa, M.; Sandy, P.; Zacchi, P.; Sternsdorf, T.; Jensen, K.; Pandolfi, P.P.; Will, H.; Schneider, C.; Del Sal, G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000, 19, 6185–6195. [Google Scholar] [CrossRef] [Green Version]
- Boichuk, S.; Hu, L.; Makielski, K.; Pandolfi, P.P.; Gjoerup, O.V. Functional connection between Rad51 and PML in homology-directed repair. PLoS ONE 2011, 6, e25814. [Google Scholar] [CrossRef] [Green Version]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009, 668, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagby, G.C., Jr. Genetic basis of Fanconi anemia. Curr. Opin. Hematol. 2003, 10, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Lalai, A.S.; Hoeijmakers, J.H. Fanconi anemia (cross) linked to DNA repair. Cell 2005, 123, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Castella, M.; Pujol, R.; Callen, E.; Trujillo, J.P.; Casado, J.A.; Gille, H.; Lach, F.P.; Auerbach, A.D.; Schindler, D.; Benitez, J.; et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood 2011, 117, 3759–3769. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Castella, M.; Jacquemont, C.; Thompson, E.L.; Yeo, J.E.; Cheung, R.S.; Huang, J.W.; Sobeck, A.; Hendrickson, E.A.; Taniguchi, T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet. 2015, 11, e1005563. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [Green Version]
- Longerich, S.; Kwon, Y.; Tsai, M.S.; Hlaing, A.S.; Kupfer, G.M.; Sung, P. Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic Acids Res. 2014, 42, 5657–5670. [Google Scholar] [CrossRef] [Green Version]
- Walworth, N.; Davey, S.; Beach, D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 1993, 363, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Kumagai, A.; Guo, Z.; Emami, K.H.; Wang, S.X.; Dunphy, W.G. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol. 1998, 142, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, C.; Bartkova, J.; Latella, L.; Falck, J.; Mailand, N.; Schroeder, T.; Sehested, M.; Lukas, J.; Bartek, J. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001, 61, 4990–4993. [Google Scholar] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.H.; Zhao, X.; Zhu, J.; Kim, I.K.; Rao, G.; McCutcheon, J.; Hsu, S.T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Checkpoint Kinase 1 Inhibition Enhances Cisplatin Cytotoxicity and Overcomes Cisplatin Resistance in SCLC by Promoting Mitotic Cell Death. J. Thorac. Oncol. 2019, 14, 1032–1045. [Google Scholar] [CrossRef]
- Verlinden, L.; Vanden Bempt, I.; Eelen, G.; Drijkoningen, M.; Verlinden, I.; Marchal, K.; De Wolf-Peeters, C.; Christiaens, M.R.; Michiels, L.; Bouillon, R.; et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor /progesterone receptor /HER-2 breast carcinomas. Cancer Res. 2007, 67, 6574–6581. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Hu, K.; Yuan, Y.; Sang, Y.; Bu, Q.; Chen, G.; Yang, L.; Li, B.; Huang, P.; Chen, D.; et al. CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J. Clin. Investig. 2012, 122, 2165–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [Green Version]
- Grobelny, J.V.; Godwin, A.K.; Broccoli, D. ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J. Cell Sci. 2000, 113 Pt 24, 4577–4585. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Wu, W.; Chen, Z.; Meng, G. PML Nuclear Body Biogenesis, Carcinogenesis, and Targeted Therapy. Trends Cancer 2020, 6, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; Kim, J.M.; D’Andrea, A.D. Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev. 2009, 23, 555–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Zhang, F.; Barrett, B.; Ren, K.; Andreassen, P.R. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009, 37, 1740–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskins, E.E.; Gunawardena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Wells, S.I. Coordinate regulation of Fanconi anemia gene expression occurs through the Rb/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munkhjargal, A.; Kim, M.-J.; Kim, D.-Y.; Jeon, Y.-J.; Kee, Y.-H.; Kim, L.-K.; Kim, Y.-H. Promyelocytic Leukemia Proteins Regulate Fanconi Anemia Gene Expression. Int. J. Mol. Sci. 2021, 22, 7782. https://doi.org/10.3390/ijms22157782
Munkhjargal A, Kim M-J, Kim D-Y, Jeon Y-J, Kee Y-H, Kim L-K, Kim Y-H. Promyelocytic Leukemia Proteins Regulate Fanconi Anemia Gene Expression. International Journal of Molecular Sciences. 2021; 22(15):7782. https://doi.org/10.3390/ijms22157782
Chicago/Turabian StyleMunkhjargal, Anudari, Myung-Jin Kim, Da-Yeon Kim, Young-Jun Jeon, Young-Hoon Kee, Lark-Kyun Kim, and Yong-Hwan Kim. 2021. "Promyelocytic Leukemia Proteins Regulate Fanconi Anemia Gene Expression" International Journal of Molecular Sciences 22, no. 15: 7782. https://doi.org/10.3390/ijms22157782
APA StyleMunkhjargal, A., Kim, M.-J., Kim, D.-Y., Jeon, Y.-J., Kee, Y.-H., Kim, L.-K., & Kim, Y.-H. (2021). Promyelocytic Leukemia Proteins Regulate Fanconi Anemia Gene Expression. International Journal of Molecular Sciences, 22(15), 7782. https://doi.org/10.3390/ijms22157782