
The Biology of Classic Hairy Cell Leukemia

 , , , and
, , , and 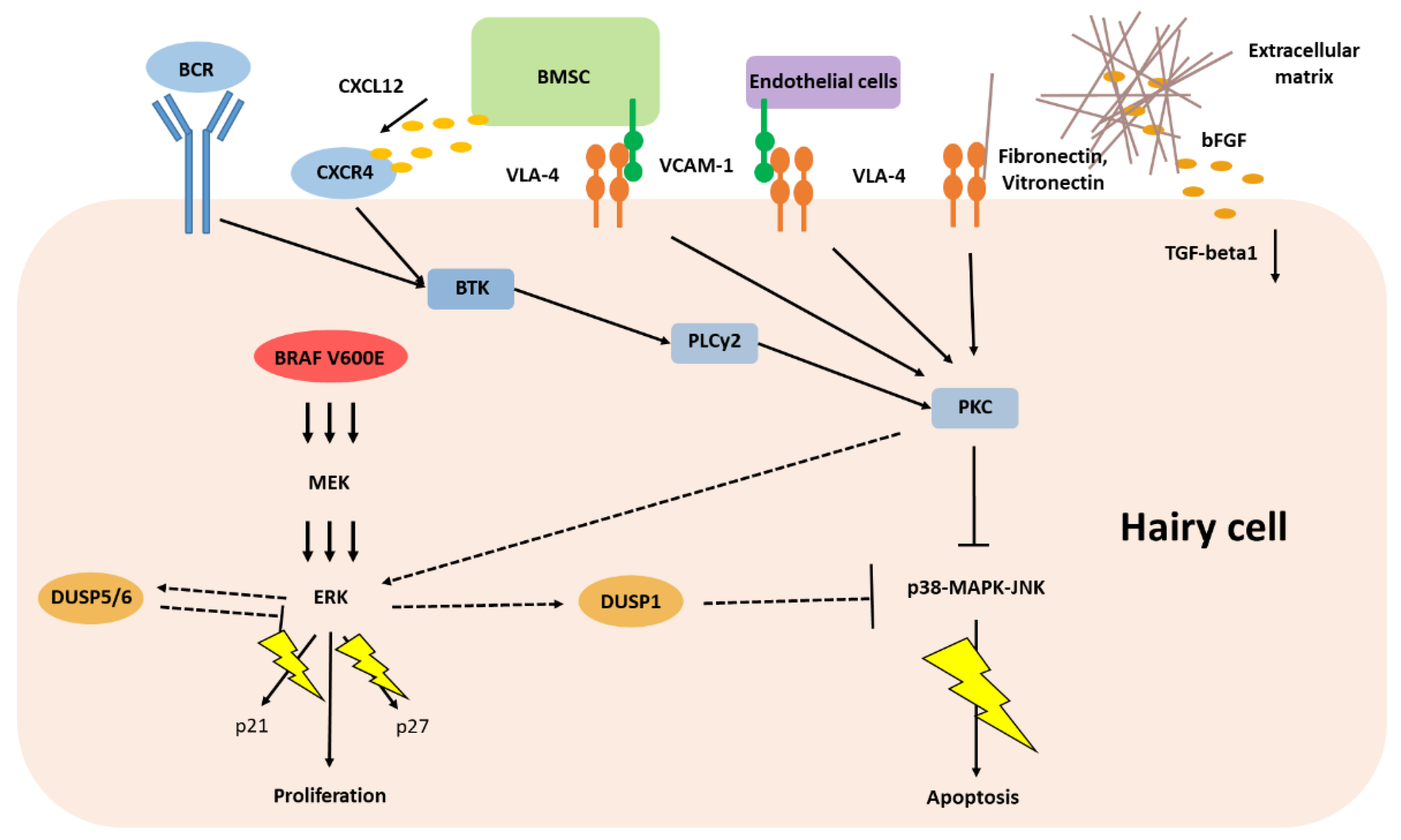{kind=link}
Abstract
1. Introduction
2. Mature Clonal B Cells with ‘Hairy’ Cytoplasmatic Projections
3. Malignant Cell Inherent Features Driving Classic Hairy Cell Leukemia
4. The Epigenetic Landscape of Classic Hairy Cell Leukemia
5. Classic Hairy Cell Leukemia Depends on Extracellular Stimuli
6. Immune Deficiency in Classic Hairy Cell Leukemia
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef]
- Bouroncle, B.A.; Wiseman, B.K.; Doan, C.A. Leukemic reticuloendotheliosis. Blood 1958, 13, 609–630. [Google Scholar] [CrossRef] [PubMed]
- Else, M.; Dearden, C.E.; Catovsky, D. Long-term follow-up after purine analogue therapy in hairy cell leukaemia. Best Pract. Res. Clin. Haematol. 2015, 28, 217–229. [Google Scholar] [CrossRef]
- Dinmohamed, A.G.; Posthuma, E.F.M.; Visser, O.; Kater, A.P.; Raymakers, R.A.P.; Doorduijn, J.K. Relative survival reaches a plateau in hairy cell leukemia: A population-based analysis in The Netherlands. Blood 2018, 131, 1380–1383. [Google Scholar] [CrossRef]
- Schrek, R.; Donnelly, W.J. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966, 27, 199–211. [Google Scholar] [CrossRef]
- Scheinberg, M.; Brenner, A.I.; Sullivan, A.L.; Cathcart, E.S.; Katayama, I. The heterogeneity of leukemic reticuloendotheliosis, “hairy cell leukemia”. Evidence for its monocytic origin. Cancer 1976, 37, 1302–1307. [Google Scholar] [CrossRef]
- Rosner, M.C.; Golomb, H.M. Phagocytic capacity of hairy cells from seventeen patients. Virchows Arch. BCell Pathol. Incl. Mol. Pathol. 1982, 40, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, E.K.; Stewart, P.L.; Kincade, P.W.; Lee, M.B.; Thompson, A.A.; Wall, R. Aberrant expression and localization of the cytoskeleton-binding pp52 (LSP1) protein in hairy cell leukemia. Leuk. Res. 2001, 25, 57–67. [Google Scholar] [CrossRef]
- Zhang, X.; Machii, T.; Matsumura, I.; Ezoe, S.; Kawasaki, A.; Tanaka, H.; Ueda, S.; Sugahara, H.; Shibayama, H.; Mizuki, M.; et al. Constitutively activated Rho guanosine triphosphatases regulate the growth and morphology of hairy cell leukemia cells. Int. J. Hematol. 2003, 77, 263–273. [Google Scholar] [CrossRef]
- Catovsky, D.; Pettit, J.E.; Galetto, J.; Okos, A.; Galton, D.A. The B-lymphocyte nature of the hairy cell of leukaemic reticuloendotheliosis. Br. J. Haematol 1974, 26, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Matutes, E.; Morilla, R.; Owusu-Ankomah, K.; Houliham, A.; Meeus, P.; Catovsky, D. The immunophenotype of hairy cell leukemia (HCL). Proposal for a scoring system to distinguish HCL from B-cell disorders with hairy or villous lymphocytes. Leuk. Lymphoma 1994, 14 (Suppl. 1), 57–61. [Google Scholar]
- Forconi, F.; Sozzi, E.; Rossi, D.; Sahota, S.S.; Amato, T.; Raspadori, D.; Trentin, L.; Leoncini, L.; Gaidano, G.; Lauria, F. Selective influences in the expressed immunoglobulin heavy and light chain gene repertoire in hairy cell leukemia. Haematologica 2008, 93, 697–705. [Google Scholar] [CrossRef]
- Forconi, F.; Sahota, S.S.; Raspadori, D.; Ippoliti, M.; Babbage, G.; Lauria, F.; Stevenson, F.K. Hairy cell leukemia: At the crossroad of somatic mutation and isotype switch. Blood 2004, 104, 3312–3317. [Google Scholar] [CrossRef]
- Basso, K.; Liso, A.; Tiacci, E.; Benedetti, R.; Pulsoni, A.; Foa, R.; Di Raimondo, F.; Ambrosetti, A.; Califano, A.; Klein, U.; et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J. Exp. Med. 2004, 199, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Arribas, A.J.; Rinaldi, A.; Chiodin, G.; Kwee, I.; Mensah, A.A.; Cascione, L.; Rossi, D.; Kanduri, M.; Rosenquist, R.; Zucca, E.; et al. Genome-wide promoter methylation of hairy cell leukemia. Blood Adv. 2019, 3, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Vanhentenrijk, V.; De Wolf-Peeters, C.; Wlodarska, I. Comparative expressed sequence hybridization studies of hairy cell leukemia show uniform expression profile and imprint of spleen signature. Blood 2004, 104, 250–255. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef]
- Pettirossi, V.; Santi, A.; Imperi, E.; Russo, G.; Pucciarini, A.; Bigerna, B.; Schiavoni, G.; Fortini, E.; Spanhol-Rosseto, A.; Sportoletti, P.; et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood 2015, 125, 1207–1216. [Google Scholar] [CrossRef]
- Tiacci, E.; Park, J.H.; De Carolis, L.; Chung, S.S.; Broccoli, A.; Scott, S.; Zaja, F.; Devlin, S.; Pulsoni, A.; Chung, Y.R.; et al. Targeting Mutant BRAF in Relapsed or Refractory Hairy-Cell Leukemia. N. Engl. J. Med. 2015, 373, 1733–1747. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Capponi, M.; Ambrosetti, A.; Lucia, E.; Antolino, A.; Pulsoni, A.; Ferrari, S.; Zinzani, P.L.; et al. Vemurafenib plus Rituximab in Refractory or Relapsed Hairy-Cell Leukemia. N. Engl. J. Med. 2021, 384, 1810–1823. [Google Scholar] [CrossRef]
- Bohn, J.P.; Pircher, A.; Wanner, D.; Vill, D.; Foeger, B.; Wolf, D.; Steurer, M. Low-dose vemurafenib in hairy cell leukemia patients with active infection. Am. J. Hematol. 2019, 94, E180–E182. [Google Scholar] [CrossRef]
- Chung, S.S.; Kim, E.; Park, J.H.; Chung, Y.R.; Lito, P.; Teruya-Feldstein, J.; Hu, W.; Beguelin, W.; Monette, S.; Duy, C.; et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci. Transl. Med. 2014, 6, 238ra271. [Google Scholar] [CrossRef] [PubMed]
- Durham, B.H.; Getta, B.; Dietrich, S.; Taylor, J.; Won, H.; Bogenberger, J.M.; Scott, S.; Kim, E.; Chung, Y.R.; Chung, S.S.; et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood 2017, 130, 1644–1648. [Google Scholar] [CrossRef]
- Dietrich, S.; Hüllein, J.; Lee, S.C.; Hutter, B.; Gonzalez, D.; Jayne, S.; Dyer, M.J.; Oleś, M.; Else, M.; Liu, X.; et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 2015, 126, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.M.; Gysin, S.; Malek, N.; Nakayama, K.; Roberts, J.M.; McMahon, M. Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Mol. Cell. Biol. 2004, 24, 10868–10881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maitre, E.; Bertrand, P.; Maingonnat, C.; Viailly, P.J.; Wiber, M.; Naguib, D.; Salaün, V.; Cornet, E.; Damaj, G.; Sola, B.; et al. New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget 2018, 9, 28866–28876. [Google Scholar] [CrossRef]
- Hart, G.T.; Wang, X.; Hogquist, K.A.; Jameson, S.C. Krüppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc. Natl. Acad. Sci. USA 2011, 108, 716–721. [Google Scholar] [CrossRef]
- Forconi, F.; Sozzi, E.; Cencini, E.; Zaja, F.; Intermesoli, T.; Stelitano, C.; Rigacci, L.; Gherlinzoni, F.; Cantaffa, R.; Baraldi, A.; et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 2009, 114, 4696–4702. [Google Scholar] [CrossRef]
- Hockley, S.L.; Else, M.; Morilla, A.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; Matutes, E. The prognostic impact of clinical and molecular features in hairy cell leukaemia variant and splenic marginal zone lymphoma. Br. J. Haematol. 2012, 158, 347–354. [Google Scholar] [CrossRef]
- Grever, M.R.; Abdel-Wahab, O.; Andritsos, L.A.; Banerji, V.; Barrientos, J.; Blachly, J.S.; Call, T.G.; Catovsky, D.; Dearden, C.; Demeter, J.; et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017, 129, 553–560. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.; Romá-Mateo, C.; Ríos, P.; Tárrega, C.; Cejudo-Marín, R.; Tabernero, L.; Pulido, R. Dual-specificity MAP kinase phosphatases as targets of cancer treatment. Anticancer Agents Med. Chem. 2011, 11, 109–132. [Google Scholar] [CrossRef]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, J.Y.; Han, Z.D.; He, H.C.; Chen, J.H.; Chen, Y.R.; Yang, S.B.; Wu, Y.D.; Zeng, Y.R.; Zou, J.; et al. Down-regulation of dual-specificity phosphatase 5 predicts poor prognosis of patients with prostate cancer. Int. J. Clin. Exp. Med. 2015, 8, 4186–4194. [Google Scholar] [PubMed]
- Rushworth, L.K.; Kidger, A.M.; Delavaine, L.; Stewart, G.; van Schelven, S.; Davidson, J.; Bryant, C.J.; Caddye, E.; East, P.; Caunt, C.J.; et al. Dual-specificity phosphatase 5 regulates nuclear ERK activity and suppresses skin cancer by inhibiting mutant Harvey-Ras (HRasQ61L)-driven SerpinB2 expression. Proc. Natl. Acad. Sci. USA 2014, 111, 18267–18272. [Google Scholar] [CrossRef]
- Kidger, A.M.; Rushworth, L.K.; Stellzig, J.; Davidson, J.; Bryant, C.J.; Bayley, C.; Caddye, E.; Rogers, T.; Keyse, S.M.; Caunt, C.J. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E317–E326. [Google Scholar] [CrossRef]
- Van Herpen, C.M.L.; Agarwala, S.S.; Hauschild, A.; Berking, C.; Beck, J.T.; Schadendorf, D.; Jansen, R.; Queirolo, P.; Ascierto, P.A.; Blank, C.U.; et al. Biomarker results from a phase II study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS- or BRAF-mutated melanoma. Oncotarget 2019, 10, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Shojaee, S.; Caeser, R.; Buchner, M.; Park, E.; Swaminathan, S.; Hurtz, C.; Geng, H.; Chan, L.N.; Klemm, L.; Hofmann, W.-K.; et al. Erk Negative Feedback Control Enables Pre-B Cell Transformation and Represents a Therapeutic Target in Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Scott, D.A.; Hatch, E.; Tian, X.; Mansour, S.L. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 2007, 134, 167–176. [Google Scholar] [CrossRef]
- Tashiro, E.; Maruki, H.; Minato, Y.; Doki, Y.; Weinstein, I.B.; Imoto, M. Overexpression of cyclin D1 contributes to malignancy by up-regulation of fibroblast growth factor receptor 1 via the pRB/E2F pathway. Cancer Res. 2003, 63, 424–431. [Google Scholar] [PubMed]
- Slack, D.N.; Seternes, O.-M.; Gabrielsen, M.; Keyse, S.M. Distinct Binding Determinants for ERK2/p38α and JNK MAP Kinases Mediate Catalytic Activation and Substrate Selectivity of MAP Kinase Phosphatase-1*210. J. Biol. Chem. 2001, 276, 16491–16500. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef]
- Kamiguti, A.S.; Harris, R.J.; Slupsky, J.R.; Baker, P.K.; Cawley, J.C.; Zuzel, M. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003, 22, 2272–2284. [Google Scholar] [CrossRef]
- Ramkissoon, A.; Chaney, K.E.; Milewski, D.; Williams, K.B.; Williams, R.L.; Choi, K.; Miller, A.; Kalin, T.V.; Pressey, J.G.; Szabo, S.; et al. Targeted Inhibition of the Dual Specificity Phosphatases DUSP1 and DUSP6 Suppress MPNST Growth via JNK. Clin. Cancer Res. 2019, 25, 4117–4127. [Google Scholar] [CrossRef]
- Rinaldi, A.; Kwee, I.; Young, K.H.; Zucca, E.; Gaidano, G.; Forconi, F.; Bertoni, F. Genome-wide high resolution DNA profiling of hairy cell leukaemia. Br. J. Haematol. 2013, 162, 566–569. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Oakes, C.C.; Martin-Subero, J.I. Insight into origins, mechanisms, and utility of DNA methylation in B-cell malignancies. Blood 2018, 132, 999–1006. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. Tig. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Baer, C.; Claus, R.; Frenzel, L.P.; Zucknick, M.; Park, Y.J.; Gu, L.; Weichenhan, D.; Fischer, M.; Pallasch, C.P.; Herpel, E.; et al. Extensive promoter DNA hypermethylation and hypomethylation is associated with aberrant microRNA expression in chronic lymphocytic leukemia. Cancer Res. 2012, 72, 3775–3785. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Sumazin, P.; Morozov, P.; Schneider, C.; Maute, R.L.; Kitagawa, Y.; Mandelbaum, J.; Haddad, J., Jr.; Chen, C.Z.; Califano, A.; et al. Identification of the human mature B cell miRNome. Immunity 2009, 30, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Brahmachary, M.; Tiacci, E.; Dalla-Favera, R.; Falini, B.; Basso, K. A microRNA signature specific for hairy cell leukemia and associated with modulation of the MAPK-JNK pathways. Leukemia 2012, 26, 2564–2567. [Google Scholar] [CrossRef]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafrè, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef]
- Chilosi, M.; Chiarle, R.; Lestani, M.; Menestrina, F.; Montagna, L.; Ambrosetti, A.; Prolla, G.; Pizzolo, G.; Doglioni, C.; Piva, R.; et al. Low expression of p27 and low proliferation index do not correlate in hairy cell leukaemia. Br. J. Haematol. 2000, 111, 263–271. [Google Scholar] [CrossRef]
- Lal, A.; Kim, H.H.; Abdelmohsen, K.; Kuwano, Y.; Pullmann, R., Jr.; Srikantan, S.; Subrahmanyam, R.; Martindale, J.L.; Yang, X.; Ahmed, F.; et al. p16(INK4a) translation suppressed by miR-24. PLoS ONE 2008, 3, e1864. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Izumiya, M.; Ogata-Kawata, H.; Okamoto, K.; Fujiwara, Y.; Nakai, M.; Okabe, A.; Schetter, A.J.; Bowman, E.D.; Midorikawa, Y.; et al. Tumor suppressor miR-22 determines p53-dependent cellular fate through post-transcriptional regulation of p21. Cancer Res. 2011, 71, 4628–4639. [Google Scholar] [CrossRef]
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef]
- Sivina, M.; Kreitman, R.J.; Peled, A.; Ravandi, F.; Burger, J.A. Adhesion of Hairy Cells Leukemia (HCL) Cells to Stromal Cells Can Be Inhibited by Blocking VLA-4 Integrins and CXCR4 Chemokine Receptors. Blood 2011, 118, 1760. [Google Scholar] [CrossRef]
- Iwasaki, H.; Suda, T. Cancer stem cells and their niche. Cancer Sci. 2009, 100, 1166–1172. [Google Scholar] [CrossRef]
- Baker, P.K.; Pettitt, A.R.; Slupsky, J.R.; Chen, H.J.; Glenn, M.A.; Zuzel, M.; Cawley, J.C. Response of hairy cells to IFN-α involves induction of apoptosis through autocrine TNF-α and protection by adhesion. Blood 2002, 100, 647–653. [Google Scholar] [CrossRef]
- Sivina, M.; Kreitman, R.J.; Arons, E.; Ravandi, F.; Burger, J.A. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: A new therapeutic approach. Br. J. Haematol. 2014, 166, 177–188. [Google Scholar] [CrossRef]
- Barak, V.; Nisman, B.; Polliack, A.; Vannier, E.; Dinarello, C.A. Correlation of serum levels of interleukin-1 family members with disease activity and response to treatment in hairy cell leukemia. Eur. Cytokine Netw. 1998, 9, 33–39. [Google Scholar] [PubMed]
- Semenzato, G.; Trentin, L.; Zambello, R.; Agostini, C.; Bulian, P.; Siviero, F.; Ambrosetti, A.; Vinante, F.; Prior, M.; Chilosi, M.; et al. Origin of the soluble interleukin-2 receptor in the serum of patients with hairy cell leukemia. Leukemia 1988, 2, 788–792. [Google Scholar] [PubMed]
- Aziz, K.A.; Till, K.J.; Zuzel, M.; Cawley, J.C. Involvement of CD44-hyaluronan interaction in malignant cell homing and fibronectin synthesis in hairy cell leukemia. Blood 2000, 96, 3161–3167. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.A.; Till, K.J.; Chen, H.; Slupsky, J.R.; Campbell, F.; Cawley, J.C.; Zuzel, M. The role of autocrine FGF-2 in the distinctive bone marrow fibrosis of hairy-cell leukemia (HCL). Blood 2003, 102, 1051–1056. [Google Scholar] [CrossRef]
- Shehata, M.; Schwarzmeier, J.D.; Hilgarth, M.; Hubmann, R.; Duechler, M.; Gisslinger, H. TGF-beta1 induces bone marrow reticulin fibrosis in hairy cell leukemia. J. Clin. Investig. 2004, 113, 676–685. [Google Scholar] [CrossRef]
- Burthem, J.; Baker, P.K.; Hunt, J.A.; Cawley, J.C. Hairy cell interactions with extracellular matrix: Expression of specific integrin receptors and their role in the cell’s response to specific adhesive proteins. Blood 1994, 84, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, R.W.; Yuvaraj, S.; Kil, L.P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. [Google Scholar] [CrossRef]
- Bohn, J.P.; Wanner, D.; Steurer, M. Ibrutinib for relapsed refractory hairy cell leukemia variant. Leuk. Lymphoma 2017, 58, 1224–1226. [Google Scholar] [CrossRef]
- Rogers, K.A.; Andritsos, L.A.; Wei, L.; McLaughlin, E.M.; Ruppert, A.S.; Anghelina, M.; Blachly, J.S.; Call, T.; Chihara, D.; Dauki, A.; et al. Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood 2021, 137, 3473–3483. [Google Scholar] [CrossRef]
- Arnaout, M.A. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 1990, 75, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, F.; Teodoridis, J.M.; Park, H.; Georgakis, A.; Farokhzad, O.C.; Böttinger, E.P.; Da Silva, N.; Rousselot, P.; Chomienne, C.; Ferenczi, K.; et al. CD11c gene expression in hairy cell leukemia is dependent upon activation of the proto-oncogenes ras and junD. Blood 2003, 101, 4033–4041. [Google Scholar] [CrossRef]
- Gazon, H.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M., Jr. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front. Microbiol. 2017, 8, 2686. [Google Scholar] [CrossRef] [PubMed]
- Gasché, C.; Reinisch, W.; Schwarzmeier, J.D. Evidence of colony suppressor activity and deficiency of hematopoietic growth factors in hairy cell leukemia. Hematol. Oncol. 1993, 11, 97–104. [Google Scholar] [CrossRef]
- Van de Corput, L.; Falkenburg, J.H.; Kester, M.G.; Willemze, R.; Kluin-Nelemans, J.C. Impaired expression of CD28 on T cells in hairy cell leukemia. Clin. Immunol. (OrlandoFla.) 1999, 93, 256–262. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohn, J.-P.; Salcher, S.; Pircher, A.; Untergasser, G.; Wolf, D. The Biology of Classic Hairy Cell Leukemia. Int. J. Mol. Sci. 2021, 22, 7780. https://doi.org/10.3390/ijms22157780
Bohn J-P, Salcher S, Pircher A, Untergasser G, Wolf D. The Biology of Classic Hairy Cell Leukemia. International Journal of Molecular Sciences. 2021; 22(15):7780. https://doi.org/10.3390/ijms22157780
Chicago/Turabian StyleBohn, Jan-Paul, Stefan Salcher, Andreas Pircher, Gerold Untergasser, and Dominik Wolf. 2021. "The Biology of Classic Hairy Cell Leukemia" International Journal of Molecular Sciences 22, no. 15: 7780. https://doi.org/10.3390/ijms22157780
APA StyleBohn, J.-P., Salcher, S., Pircher, A., Untergasser, G., & Wolf, D. (2021). The Biology of Classic Hairy Cell Leukemia. International Journal of Molecular Sciences, 22(15), 7780. https://doi.org/10.3390/ijms22157780