The Heat Shock Protein HSP70 Promotes Th17 Genes’ Expression via Specific Regulation of microRNA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
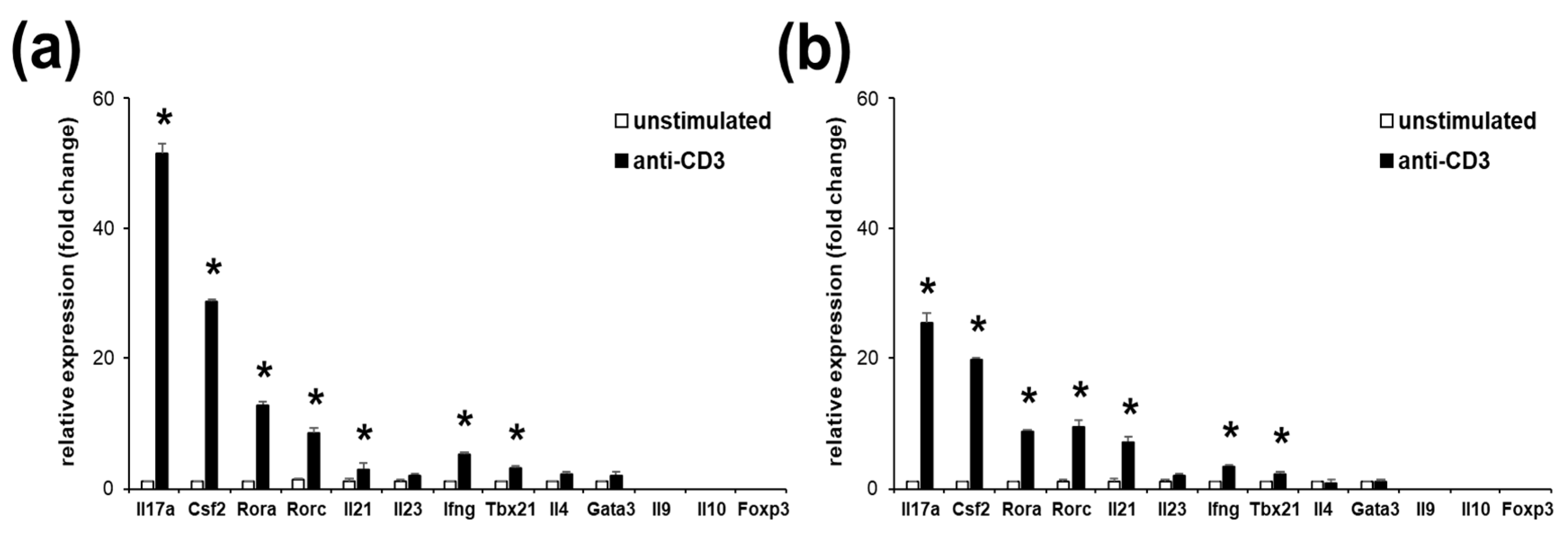
2.1. EL4 TCR-Positive Cells Stimulation Induces Expression of Th17 Marker Genes
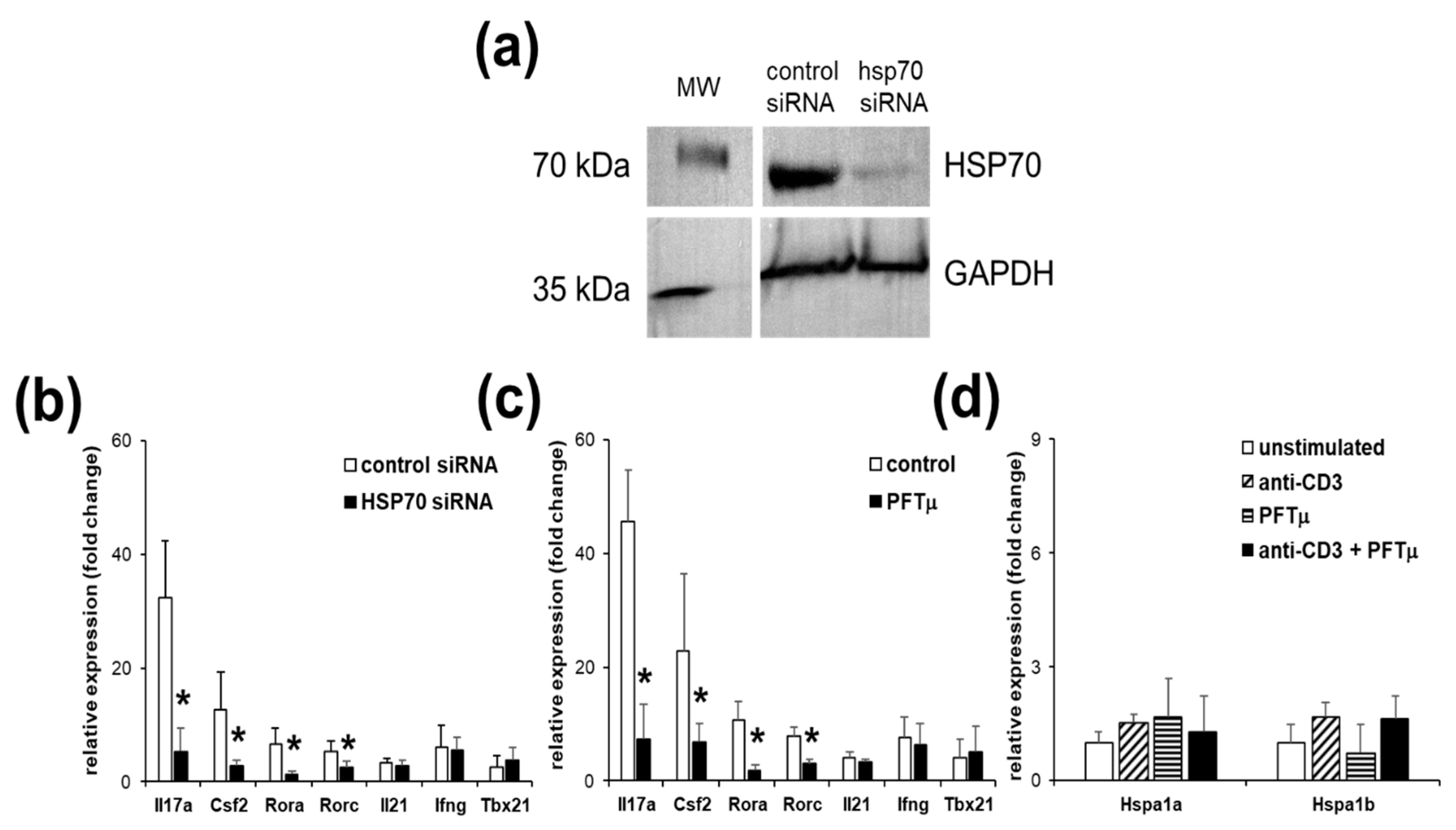
2.2. HSP70 Inhibition Reduces Marker Gene Expressions of Th17 Cells
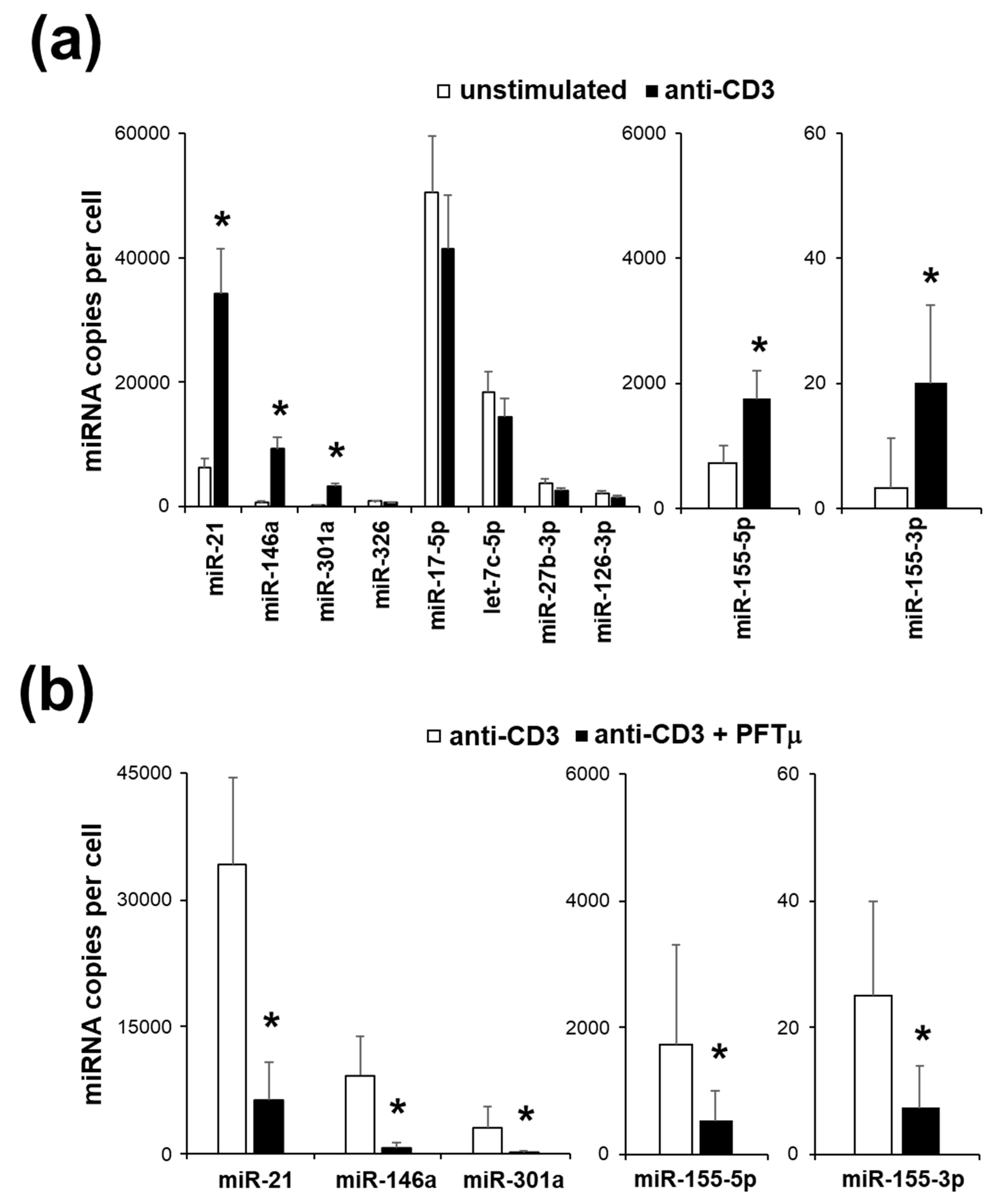
2.3. miRNA Expression Profile of EL4 TCR+ Cells
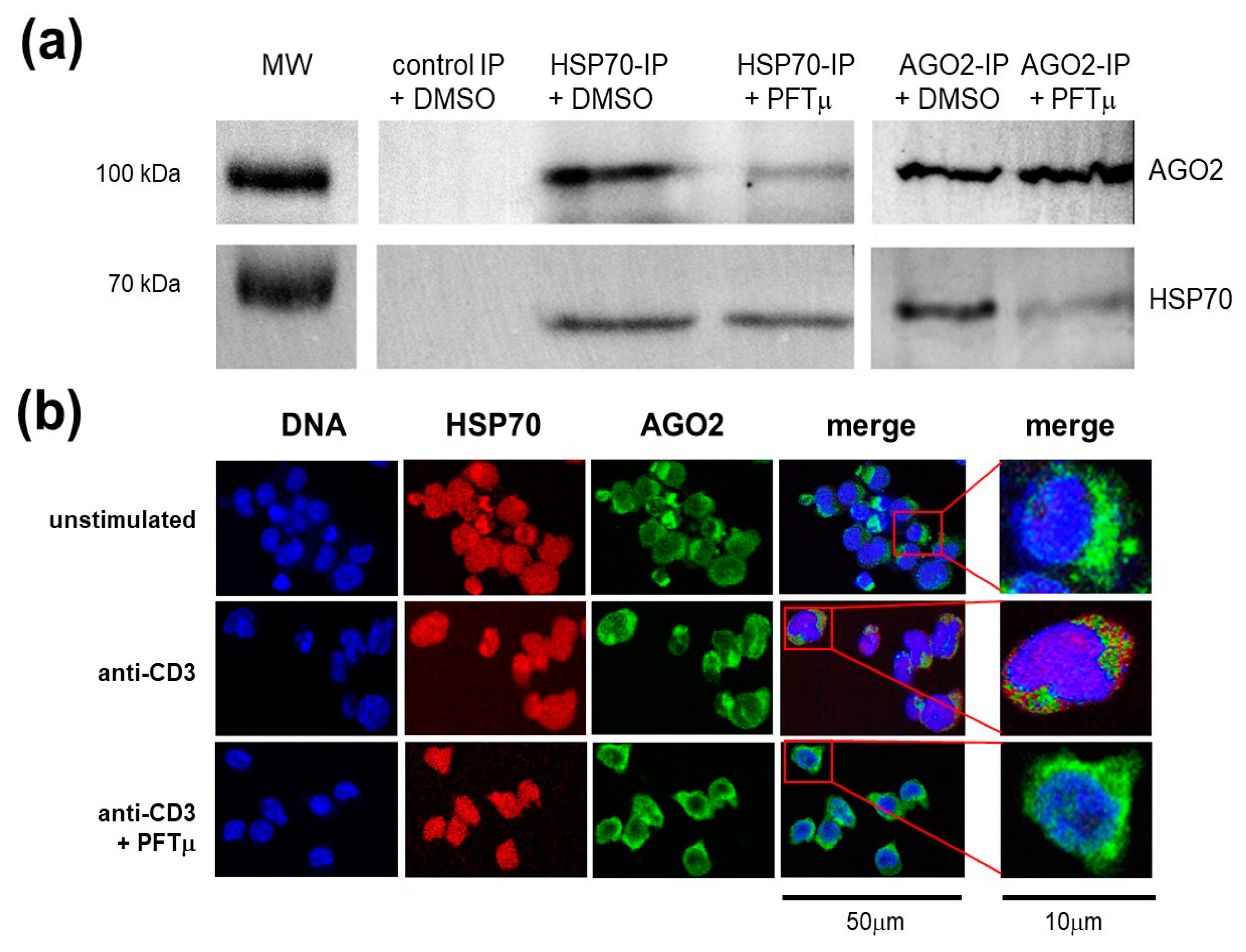
2.4. HSP70 Interacts with a RNA-induced Silencing Complex Protein AGO2
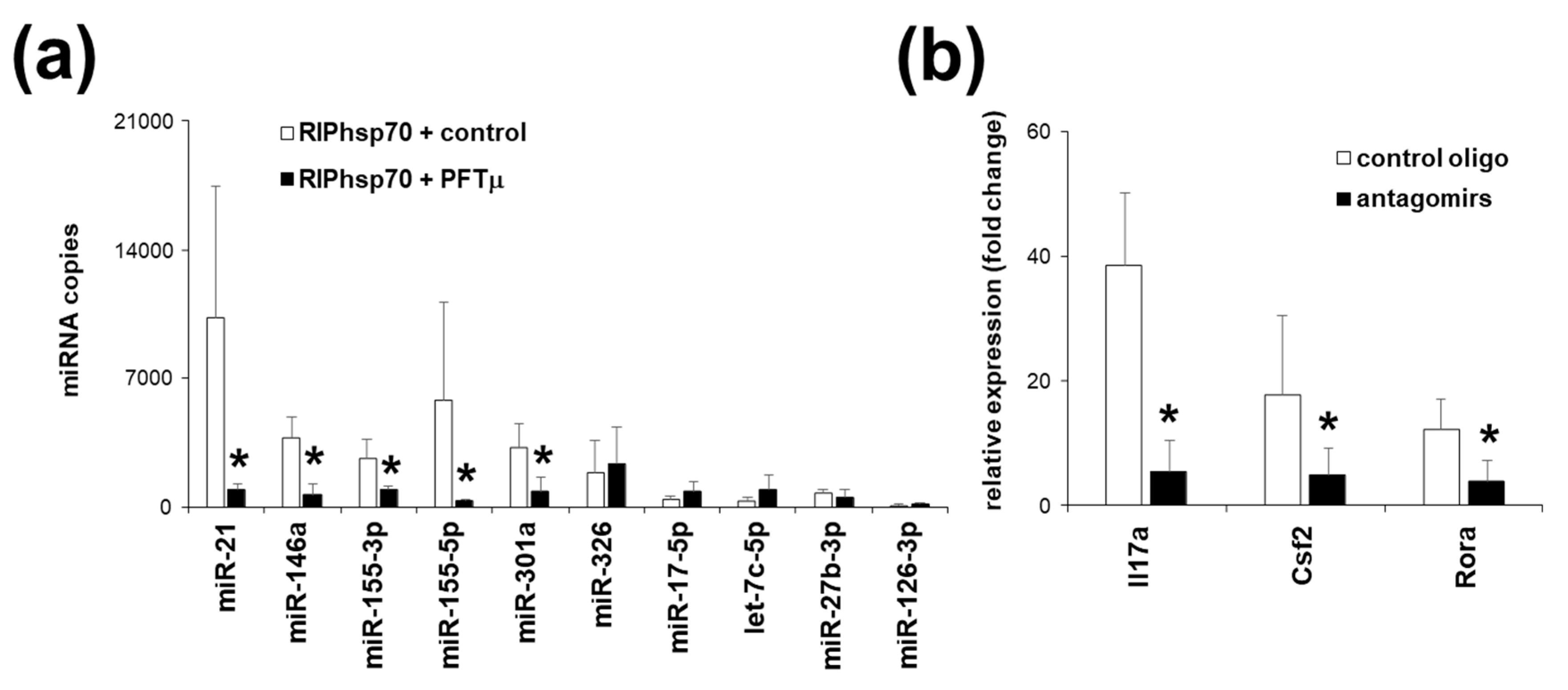
2.5. Th17 Specific miRNA Co-Precipitate with HSP70
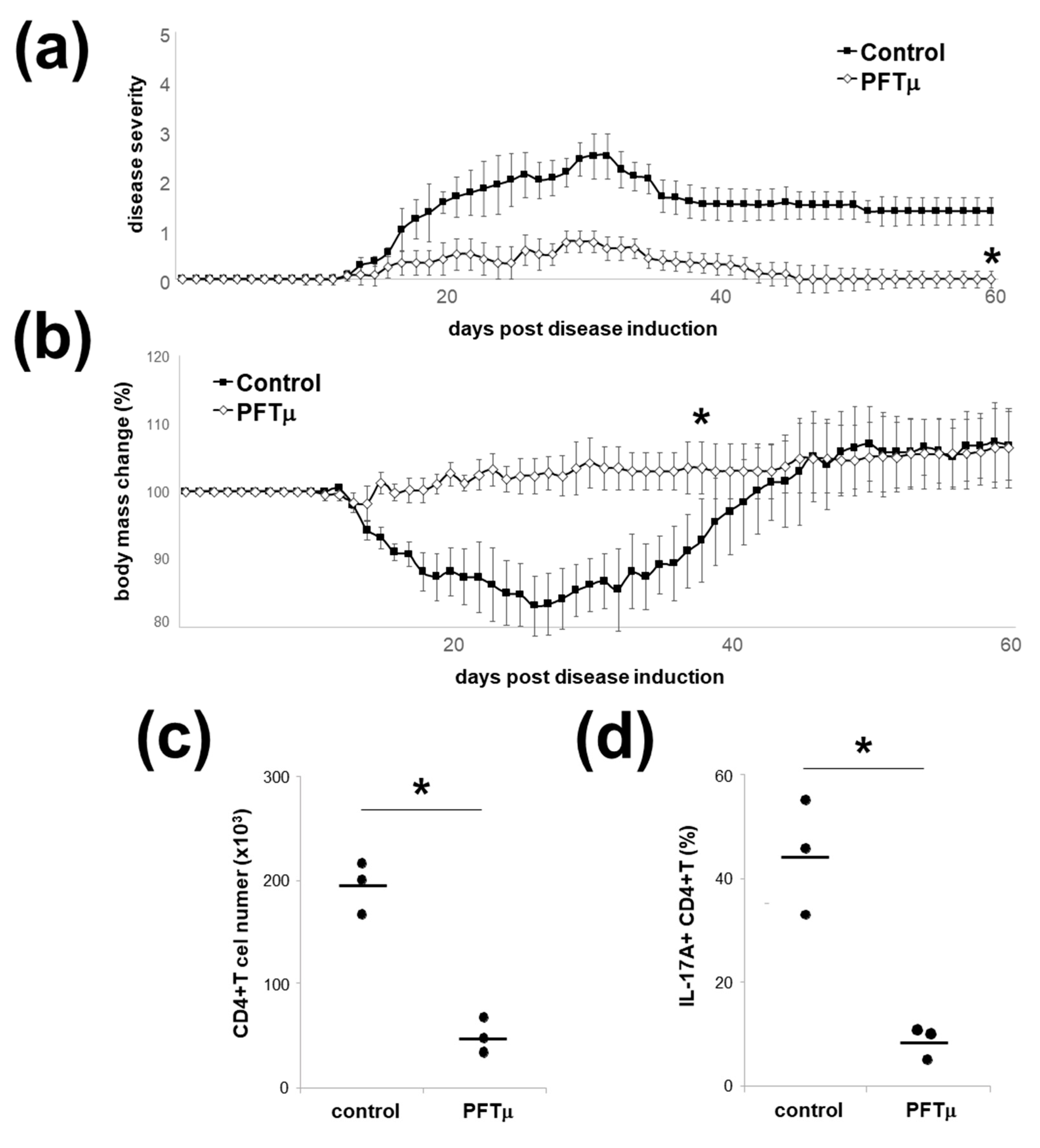
2.6. HSP70 Inhibition In Vivo Downregulates Experimental Autoimmune Encephalomyelitis
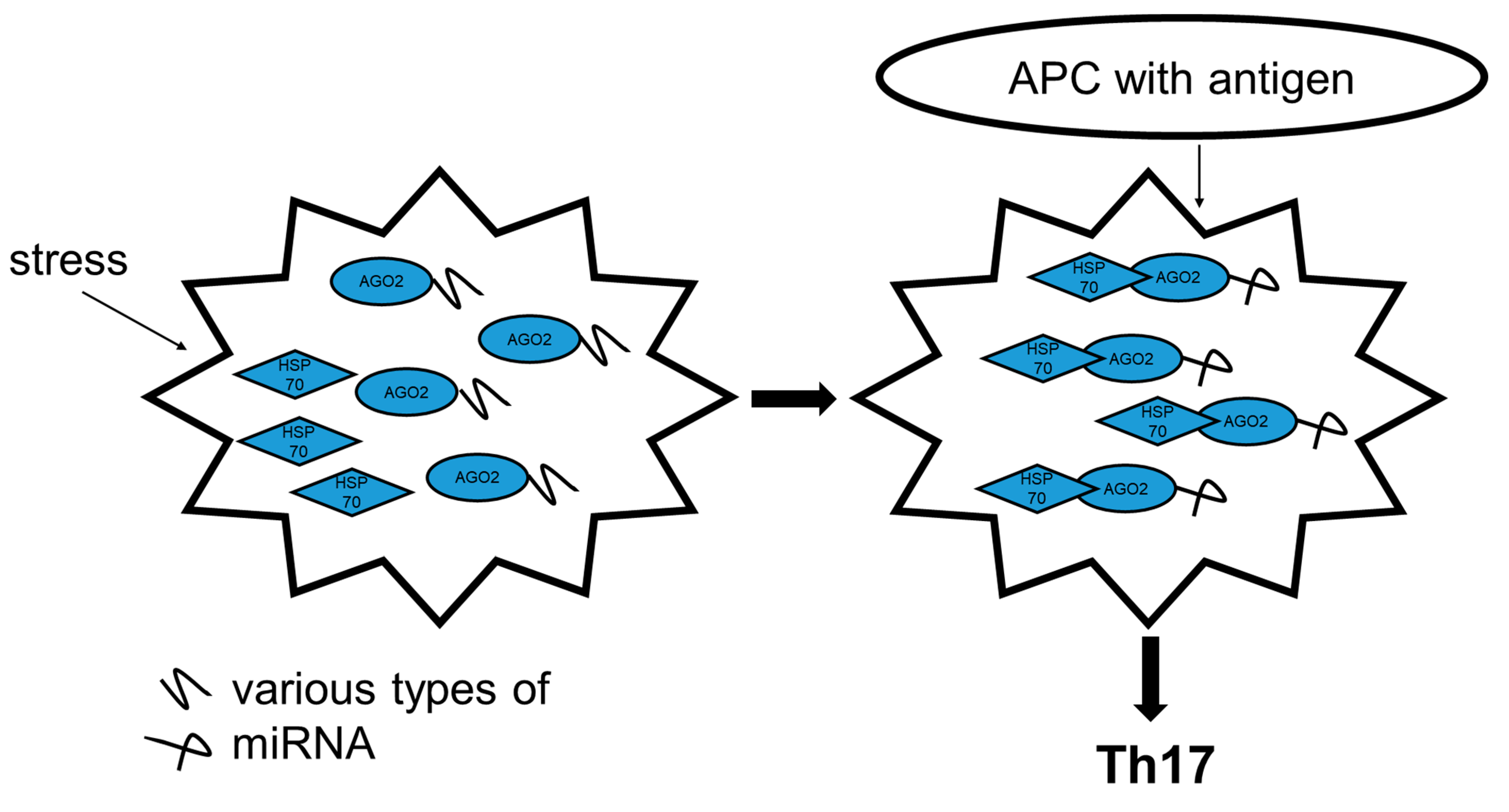
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Cell Sorting and Reagents
4.2. Mice
4.3. Western Blot Analysis
4.4. Co-immunoprecipitation and RNA-binding Protein Immunoprecipitation (RIP)
4.5. Th17 In Vitro Differentiation
4.6. Transfections of Antagomirs against miR-21, miR-146a, miR-155-5p, miR-155-3p and miR-301a and HSP70 Expression Silencing
4.7. Extraction of RNA and mRNA Expression Analysis
4.8. miRNA Quantification
4.9. Immunocytochemistry
4.10. Induction of EAE and PFTμ Treatment
4.11. Isolation of CNS-infiltrating Cells and Analysis of Infiltrating CD4+ T Cells
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Rajewsky, K. MicroRNA control in the immune system: Basic principles. Cell 2009, 136, 26–36. [Google Scholar] [CrossRef]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Zurawska, A.; Mycko, M.P.; Selmaj, K.W. Circular RNAs as a novel layer of regulatory mechanism in multiple sclerosis. J. Neuroimmunol. 2019, 334, 576971. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 2009, 10, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Mycko, M.P.; Cichalewska, M.; Machlanska, A.; Cwiklinska, H.; Mariasiewicz, M.; Selmaj, K.W. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2012, 109, E1248–E1257. [Google Scholar] [CrossRef] [PubMed]
- Mycko, M.P.; Cichalewska, M.; Cwiklinska, H.; Selmaj, K.W. miR-155-3p Drives the Development of Autoimmune Demyelination by Regulation of Heat Shock Protein 40. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 16504–16515. [Google Scholar] [CrossRef] [PubMed]
- Murugaiyan, G.; Beynon, V.; Mittal, A.; Joller, N.; Weiner, H.L. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 2213–2221. [Google Scholar] [CrossRef]
- Li, B.; Wang, X.; Choi, I.Y.; Wang, Y.C.; Liu, S.; Pham, A.T.; Moon, H.; Smith, D.J.; Rao, D.S.; Boldin, M.P.; et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J. Clin. Investig. 2017, 127, 3702–3716. [Google Scholar] [CrossRef]
- Young, J.C.; Agashe, V.R.; Siegers, K.; Hartl, F.U. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 2004, 5, 781–791. [Google Scholar] [CrossRef]
- Demand, J.; Luders, J.; Hohfeld, J. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol. Cell. Biol. 1998, 18, 2023–2028. [Google Scholar] [CrossRef]
- Hohfeld, J.; Hartl, F.U. Post-translational protein import and folding. Curr. Opin. Cell Biol. 1994, 6, 499–509. [Google Scholar] [CrossRef]
- Lanneau, D.; Wettstein, G.; Bonniaud, P.; Garrido, C. Heat shock proteins: Cell protection through protein triage. Sci. World J. 2010, 10, 1543–1552. [Google Scholar] [CrossRef]
- Mycko, M.P.; Cwiklinska, H.; Walczak, A.; Libert, C.; Raine, C.S.; Selmaj, K.W. A heat shock protein gene (Hsp70.1) is critically involved in the generation of the immune response to myelin antigen. Eur. J. Immunol. 2008, 38, 1999–2013. [Google Scholar] [CrossRef]
- Cwiklinska, H.; Mycko, M.P.; Luvsannorov, O.; Walkowiak, B.; Brosnan, C.F.; Raine, C.S.; Selmaj, K.W. Heat shock protein 70 associations with myelin basic protein and proteolipid protein in multiple sclerosis brains. Int. Immunol. 2003, 15, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Cwiklinska, H.; Mycko, M.P.; Szymanska, B.; Matysiak, M.; Selmaj, K.W. Aberrant stress-induced Hsp70 expression in immune cells in multiple sclerosis. J. Neurosci. Res. 2010, 88, 3102–3110. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Muromoto, R.; Takahashi, M.; Takeuchi, S.; Takeda, Y.; Jetten, A.M.; Matsuda, T. Inhibitory effects of azole-type fungicides on interleukin-17 gene expression via retinoic acid receptor-related orphan receptors alpha and gamma. Toxicol. Appl. Pharmacol. 2012, 259, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, K.; Hashimoto, M.; Sekiya, T.; Nakagawa, R.; Wakabayashi, Y.; Sugiyama, Y.; Komai, K.; Saba, I.; Moroy, T.; Yoshimura, A. Gfi1 negatively regulates T(h)17 differentiation by inhibiting RORgammat activity. Int. Immunol. 2009, 21, 881–889. [Google Scholar] [CrossRef]
- Chang, M.R.; Lyda, B.; Kamenecka, T.M.; Griffin, P.R. Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor gamma is therapeutic in the collagen-induced arthritis experimental model. Arthritis Rheumatol. 2014, 66, 579–588. [Google Scholar] [CrossRef]
- Baumjohann, D.; Ansel, K.M. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat. Rev. Immunol. 2013, 13, 666–678. [Google Scholar] [CrossRef]
- Matysiak, M.; Fortak-Michalska, M.; Szymanska, B.; Orlowski, W.; Jurewicz, A.; Selmaj, K. MicroRNA-146a negatively regulates the immunoregulatory activity of bone marrow stem cells by targeting prostaglandin E2 synthase-2. J. Immunol. 2013, 190, 5102–5109. [Google Scholar] [CrossRef]
- Lee, Y.; Mitsdoerffer, M.; Xiao, S.; Gu, G.; Sobel, R.A.; Kuchroo, V.K. IL-21R signaling is critical for induction of spontaneous experimental autoimmune encephalomyelitis. J. Clin. Investig. 2015, 125, 4011–4020. [Google Scholar] [CrossRef]
- Basu, R.; Whitley, S.K.; Bhaumik, S.; Zindl, C.L.; Schoeb, T.R.; Benveniste, E.N.; Pear, W.S.; Hatton, R.D.; Weaver, C.T. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat. Immunol. 2015, 16, 286–295. [Google Scholar] [CrossRef]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.M.; Jack, R.S.; Wunderlich, F.T.; Bruning, J.C.; Muller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef]
- Bailis, W.; Yashiro-Ohtani, Y.; Fang, T.C.; Hatton, R.D.; Weaver, C.T.; Artis, D.; Pear, W.S. Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity 2013, 39, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Meyer Zu Horste, G.; Wu, C.; Wang, C.; Cong, L.; Pawlak, M.; Lee, Y.; Elyaman, W.; Xiao, S.; Regev, A.; Kuchroo, V.K. RBPJ Controls Development of Pathogenic Th17 Cells by Regulating IL-23 Receptor Expression. Cell Rep. 2016, 16, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Hasan, Z.; Koizumi, S.I.; Sasaki, D.; Yamada, H.; Arakaki, N.; Fujihara, Y.; Okitsu, S.; Shirahata, H.; Ishikawa, H. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat. Commun. 2017, 8, 15628. [Google Scholar] [CrossRef] [PubMed]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef]
- Brucklacher-Waldert, V.; Ferreira, C.; Stebegg, M.; Fesneau, O.; Innocentin, S.; Marie, J.C.; Veldhoen, M. Cellular Stress in the Context of an Inflammatory Environment Supports TGF-beta-Independent T Helper-17 Differentiation. Cell Rep. 2017, 19, 2357–2370. [Google Scholar] [CrossRef]
- Segal, L.N.; Clemente, J.C.; Tsay, J.C.; Koralov, S.B.; Keller, B.C.; Wu, B.G.; Li, Y.; Shen, N.; Ghedin, E.; Morris, A.; et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 2016, 1, 16031. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef]
- Dutzan, N.; Abusleme, L.; Bridgeman, H.; Greenwell-Wild, T.; Zangerle-Murray, T.; Fife, M.E.; Bouladoux, N.; Linley, H.; Brenchley, L.; Wemyss, K.; et al. On-going Mechanical Damage from Mastication Drives Homeostatic Th17 Cell Responses at the Oral Barrier. Immunity 2017, 46, 133–147. [Google Scholar] [CrossRef]
- Lewkowicz, P.; Cwiklinska, H.; Mycko, M.P.; Cichalewska, M.; Domowicz, M.; Lewkowicz, N.; Jurewicz, A.; Selmaj, K.W. Dysregulated RNA-Induced Silencing Complex (RISC) Assembly within CNS Corresponds with Abnormal miRNA Expression during Autoimmune Demyelination. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 7521–7537. [Google Scholar] [CrossRef]
- Pare, J.M.; Tahbaz, N.; Lopez-Orozco, J.; LaPointe, P.; Lasko, P.; Hobman, T.C. Hsp90 regulates the function of argonaute 2 and its recruitment to stress granules and P-bodies. Mol. Biol. Cell 2009, 20, 3273–3284. [Google Scholar] [CrossRef]
- Jang, J.H.; Jung, J.S.; Choi, J.I.; Kang, S.K. Nuclear Ago2/HSP60 contributes to broad spectrum of hATSCs function via Oct4 regulation. Antioxid. Redox Signal. 2012, 16, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R. mRNP granules. Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Naruse, K.; Matsuura-Suzuki, E.; Watanabe, M.; Iwasaki, S.; Tomari, Y. In vitro reconstitution of chaperone-mediated human RISC assembly. RNA 2018, 24, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Tsuboyama, K.; Tadakuma, H.; Tomari, Y. Conformational Activation of Argonaute by Distinct yet Coordinated Actions of the Hsp70 and Hsp90 Chaperone Systems. Mol. Cell 2018, 70, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Noubade, R.; Eidenschenk, C.; Ota, N.; Zeng, W.; Zheng, Y.; Hackney, J.; Ding, J.; Singh, H.; Ouyang, W. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat. Immunol. 2011, 12, 1238–1245. [Google Scholar] [CrossRef]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Raveney, B.J.; Oki, S.; Hohjoh, H.; Nakamura, M.; Sato, W.; Murata, M.; Yamamura, T. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nat. Commun. 2015, 6, 8437. [Google Scholar] [CrossRef]
- Jain, R.; Chen, Y.; Kanno, Y.; Joyce-Shaikh, B.; Vahedi, G.; Hirahara, K.; Blumenschein, W.M.; Sukumar, S.; Haines, C.J.; Sadekova, S.; et al. Interleukin-23-Induced Transcription Factor Blimp-1 Promotes Pathogenicity of T Helper 17 Cells. Immunity 2016, 44, 131–142. [Google Scholar] [CrossRef]
- Juryńczyk, M.; Lewkowicz, P.; Domowicz, M.; Mycko, M.P.; Selmaj, K.W. Heat shock protein 70 (Hsp70) interacts with the Notch1 intracellular domain and contributes to the activity of Notch signaling in myelin-reactive CD4 T cells. J Neuroimmunol. 2015, 287, 19–26. [Google Scholar]
- Mycko, M.P.; Cwiklinska, H.; Szymanski, J.; Szymanska, B.; Kudla, G.; Kilianek, L.; Odyniec, A.; Brosnan, C.F.; Selmaj, K.W. Inducible heat shock protein 70 promotes myelin autoantigen presentation by the HLA class II. J. Immunol. 2004, 172, 202–213. [Google Scholar] [CrossRef]
- Page, N.; Gros, F.; Schall, N.; Decossas, M.; Bagnard, D.; Briand, J.P.; Muller, S. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, N.; Akbari, O.; Garcia, S.; Brazil, M.; Stockinger, B. The HSC73 molecular chaperone: Involvement in MHC class II antigen presentation. J. Immunol. 1999, 163, 1936–1942. [Google Scholar] [PubMed]
- Galazka, G.; Jurewicz, A.; Domowicz, M.; Cannella, B.; Raine, C.S.; Selmaj, K. HINT1 peptide/Hsp70 complex induces NK-cell-dependent immunoregulation in a model of autoimmune demyelination. Eur. J. Immunol. 2014, 44, 3026–3044. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Li, Y.; Cui, Y.; Guo, Y.; Dong, N.; Li, G.; Dai, Y.; Ji, L. Down-regulated miR-374c and Hsp70 promote Th17 cell differentiation by inducing Fas expression in experimental autoimmune encephalomyelitis. Int. J. Biol. Macromol. 2019. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, X.; Cai, X.; Tian, Y.; Wang, D.; Qi, J.; Teng, Z.; Lu, G.; Ni, Q.; Wang, S.; et al. Pifithrin-mu incorporated in gold nanoparticle amplifies pro-apoptotic unfolded protein response cascades to potentiate synergistic glioblastoma therapy. Biomaterials 2020, 232, 119677. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cwiklinska, H.; Cichalewska-Studzinska, M.; Selmaj, K.W.; Mycko, M.P. The Heat Shock Protein HSP70 Promotes Th17 Genes’ Expression via Specific Regulation of microRNA. Int. J. Mol. Sci. 2020, 21, 2823. https://doi.org/10.3390/ijms21082823
Cwiklinska H, Cichalewska-Studzinska M, Selmaj KW, Mycko MP. The Heat Shock Protein HSP70 Promotes Th17 Genes’ Expression via Specific Regulation of microRNA. International Journal of Molecular Sciences. 2020; 21(8):2823. https://doi.org/10.3390/ijms21082823
Chicago/Turabian StyleCwiklinska, Hanna, Maria Cichalewska-Studzinska, Krzysztof W. Selmaj, and Marcin P. Mycko. 2020. "The Heat Shock Protein HSP70 Promotes Th17 Genes’ Expression via Specific Regulation of microRNA" International Journal of Molecular Sciences 21, no. 8: 2823. https://doi.org/10.3390/ijms21082823
APA StyleCwiklinska, H., Cichalewska-Studzinska, M., Selmaj, K. W., & Mycko, M. P. (2020). The Heat Shock Protein HSP70 Promotes Th17 Genes’ Expression via Specific Regulation of microRNA. International Journal of Molecular Sciences, 21(8), 2823. https://doi.org/10.3390/ijms21082823