The Contribution of Astrocyte Autophagy to Systemic Metabolism



Abstract
1. Central Regulation of Metabolism
2. Astrocyte Functions and Their Importance in Systemic Metabolism
3. Autophagy Importance in Metabolism
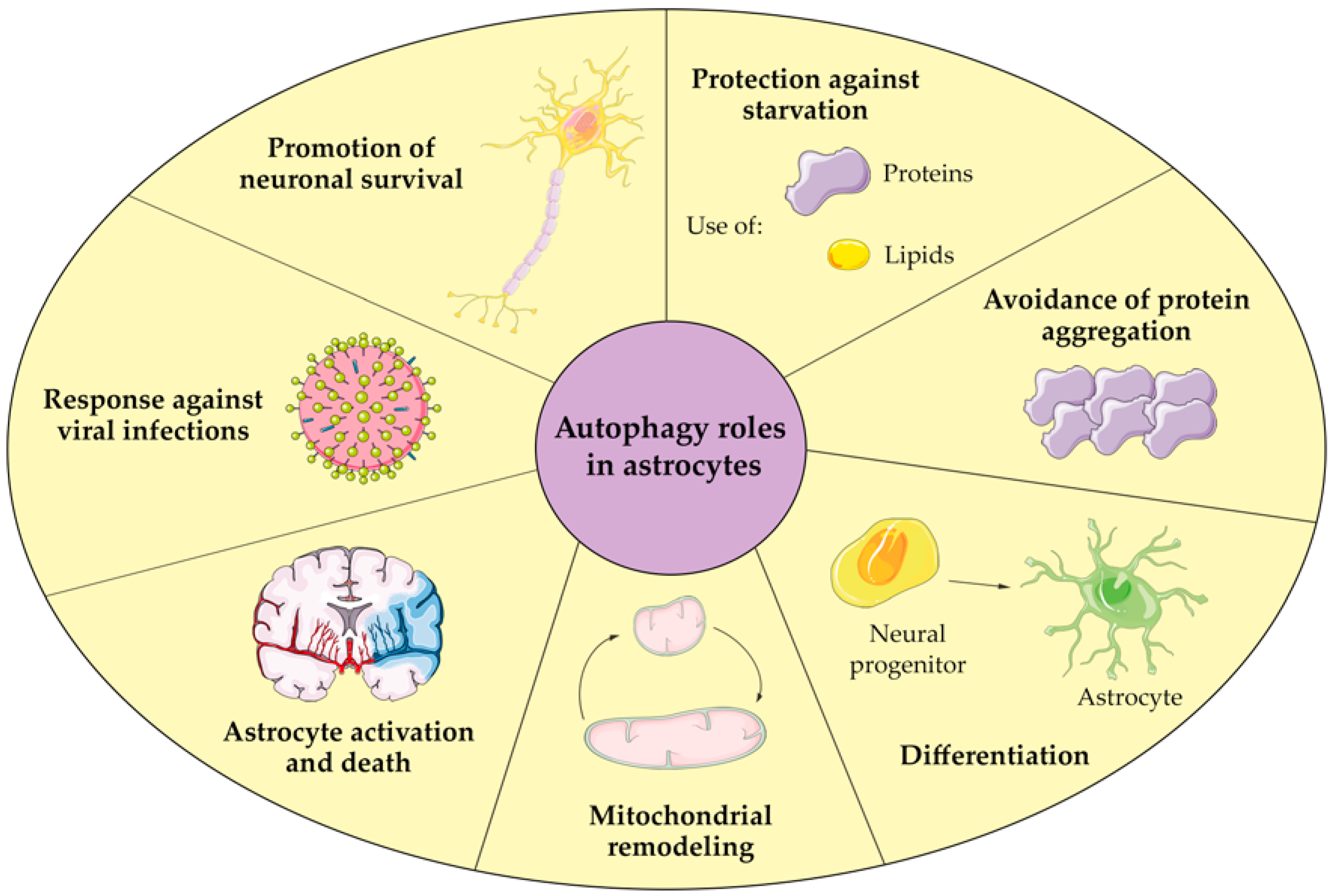
4. Autophagy in Astrocytes
5. Involvement of Astrocyte Autophagy in Systemic Metabolism
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| α-MSH | α melanocyte-stimulating hormone |
| AgRP | Agouti-related protein |
| APOE | Apolipoprotein E |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| GFAP | Glial fibrillary acidic protein |
| HFD | High fat diet |
| HIV | Human immunodeficiency virus |
| LAMP-2A | Lysosome-associated membrane protein 2A |
| LC3 | Microtubule-associated protein 1A/1B light chain 3B |
| LRRK2 | Leucine-rich repeat kinase 2 |
| NPY | Neuropeptide Y |
| PA | Palmitic acid |
| POMC | Proopiomelanocortin |
| ROS | Reactive oxygen species |
| TFEB | Transcription factor EB |
| ULK2 | Unc-51 like autophagy activating kinase 2 |
References
- Seeley, R.J.; Woods, S.C. Monitoring of stored and available fuel by the CNS: Implications for obesity. Nat. Rev. Neurosci. 2003, 4, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Porte, D. Diabetes, obesity, and the brain. Science 2005, 307, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- García-Cáceres, C.; Balland, E.; Prevot, V.; Luquet, S.; Woods, S.C.; Koch, M.; Horvath, T.L.; Yi, C.X.; Chowen, J.A.; Verkhratsky, A.; et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat. Neurosci. 2019, 22, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Waterson, M.J.; Horvath, T.L. Neuronal regulation of energy homeostasis: Beyond the hypothalamus and feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Marina, N.; Turovsky, E.; Christie, I.N.; Hosford, P.S.; Hadjihambi, A.; Korsak, A.; Ang, R.; Mastitskaya, S.; Sheikhbahaei, S.; Theparambil, S.M.; et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia 2018, 66, 1185–1199. [Google Scholar] [CrossRef]
- Petzold, G.C.; Murthy, V.N. Role of astrocytes in neurovascular coupling. Neuron 2011, 71, 782–797. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Broadwell, R.D. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions: I. Neurons and glia. J. Electron. Microsc. Tech. 1986, 3, 413–437. [Google Scholar] [CrossRef]
- Brown, A.M.; Ransom, B.R. Astrocyte glycogen and brain energy metabolism. Glia 2007, 55, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Mederos, S.; González-Arias, C.; Perea, G. Astrocyte–neuron networks: A multilane highway of signaling for homeostatic brain function. Front. Synaptic Neurosci. 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, S.; Almeida, A.; Bolaños, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994, 62, 45–53. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- Chari, M.; Yang, C.S.; Lam, C.K.; Lee, K.; Mighiu, P.; Kokorovic, A.; Cheung, G.W.; Lai, T.Y.; Wang, P.Y.; Lam, T.K. Glucose transporter-1 in the hypothalamic glial cells mediates glucose sensing to regulate glucose production in vivo. Diabetes 2011, 60, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Marty, N.; Dallaporta, M.; Foretz, M.; Emery, M.; Tarussio, D.; Bady, I.; Binnert, C.; Beermann, F.; Thorens, B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J. Clin. Investig. 2005, 115, 3545–3553. [Google Scholar] [CrossRef]
- Young, J.K.; McKenzie, J.C. GLUT2 immunoreactivity in Gomori-positive astrocytes of the hypothalamus. J. Histochem. Cytochem. 2004, 52, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Allard, C.; Carneiro, L.; Grall, S.; Cline, B.H.; Fioramonti, X.; Chrétien, C.; Baba-Aissa, F.; Giaume, C.; Pénicaud, L.; Leloup, C. Hypothalamic astroglial connexins are required for brain glucose sensing-induced insulin secretion. J. Cereb. Blood Flow Metab. 2014, 34, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Blázquez, C.; Galve-Roperh, I.; Guzmán, M. De novo-synthesized ceramide signals apoptosis in astrocytes via extracellular signal-regulated kinase. FASEB J. 2000, 14, 2315–2322. [Google Scholar] [CrossRef]
- Le Foll, C.; Dunn-Meynell, A.A.; Miziorko, H.M.; Levin, B.E. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes 2014, 63, 1259–1269. [Google Scholar] [CrossRef]
- Diano, S.; Kalra, S.P.; Horvath, T.L. Leptin receptor immunoreactivity is associated with the Golgi apparatus of hypothalamic neurons and glial cells. J. Neuroendocrinol. 1998, 10, 647–650. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Fuente-Martín, E.; García-Cáceres, C.; Granado, M.; de Ceballos, M.L.; Sánchez-Garrido, M.Á.; Sarman, B.; Liu, Z.W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizón, P.; et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef]
- Kim, J.G.; Suyama, S.; Koch, M.; Jin, S.; Argente-Arizon, P.; Argente, J.; Liu, Z.W.; Zimmer, M.R.; Jeong, J.K.; Szigeti-Buck, K.; et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci. 2014, 17, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hipothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- Horvath, T.L.; Sarman, B.; García-Cáceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D. Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure, and body weight. Cell Metab. 2017, 25, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Buckman, L.B.; Thompson, M.M.; Lippert, R.N.; Blackwell, T.S.; Yull, F.E.; Ellacott, K.L. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol. Metab. 2015, 4, 58–63. [Google Scholar] [CrossRef]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKB/NF-kB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; De Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef]
- Hubbard, V.M.; Valdor, R.; Patel, B.; Singh, R.; Cuervo, A.M.; Macian, F. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol. 2010, 185, 7349–7357. [Google Scholar] [CrossRef]
- Meng, Q.; Cai, D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IκB kinase β(IKKβ)/NF-κB pathway. J. Biol. Chem. 2011, 286, 32324–32332. [Google Scholar] [CrossRef]
- Coupé, B.; Ishii, Y.; Dietrich, M.O.; Komatsu, M.; Horvath, T.L.; Bouret, S.G. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012, 15, 247–255. [Google Scholar] [CrossRef]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef]
- Kaushik, S.; Arias, E.; Kwon, H.; Lopez, N.M.; Athonvarangkul, D.; Sahu, S.; Schwartz, G.J.; Pessin, J.E.; Singh, R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012, 13, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Kim, H.K.; Moon, E.Y.; Kim, S.S.; Choi, C.S.; Komatsu, M.; Jeong, Y.T.; Lee, M.K.; Kim, K.W.; Kim, M.S.; et al. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 2012, 153, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Garcia-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016, 23, 113–127. [Google Scholar] [CrossRef]
- Ignacio-Souza, L.M.; Bombassaro, B.; Pascoal, L.B.; Portovedo, M.A.; Razolli, D.S.; Coope, A.; Victorio, S.C.; De Moura, R.F.; Nascimento, L.F.; Arruda, A.P.; et al. Defective regulation of the ubiquitin/proteasome system in the hypothalamus of obese male mice. Endocrinology 2014, 155, 2831–2844. [Google Scholar] [CrossRef]
- Portovedo, M.; Ignacio-Souza, L.M.; Bombassaro, B.; Coope, A.; Reginato, A.; Razolli, D.S.; Torsoni, M.A.; Torsoni, A.S.; Leal, R.F.; Velloso, L.A.; et al. Saturated fatty acids modulate autophagy’s proteins in the hypothalamus. PLoS ONE 2015, 10, e0119850. [Google Scholar] [CrossRef]
- Morselli, E.; Criollo, A.; Rodriguez-Navas, C.; Clegg, D.J. Chronic high fat diet consumption impairs metabolic health of male mice. Inflamm. Cell Signal. 2015, 1, e561. [Google Scholar] [CrossRef]
- Reginato, A.; de Fante, T.; Portovedo, M.; da Costa, N.F.; Payolla, T.B.; Miyamotto, J.É.; Simino, L.A.; Ignácio-Souza, L.M.; Torsoni, M.A.; Torsoni, A.S.; et al. Autophagy proteins are modulated in the liver and hypothalamus of the offspring of mice with diet-induced obesity. J. Nutr. Biochem. 2016, 34, 30–41. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired autophagy in APOE4 astrocytes. J. Alzheimer’s Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef]
- Pamenter, M.E.; Perkins, G.A.; McGinness, A.K.; Gu, X.Q.; Ellisman, M.H.; Haddad, G.G. Autophagy and apoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS ONE 2012, 7, e51469. [Google Scholar] [CrossRef]
- Korenić, A.; Andjus, P.; Radenović, L.; Spasojević, I. The role of autophagy and lipolysis in survival of astrocytes under nutrient deprivation. Neurosci. Lett. 2015, 595, 128–133. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Lee, V.M.; Trojanowski, J.Q. Protein astrogliopathies (PAG) in human neurodegenerative diseases and aging. Brain Pathol. 2017, 27, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Jänen, S.B.; Chaachouay, H.; Richter-Landsberg, C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 2010, 58, 1766–1774. [Google Scholar] [CrossRef]
- Tang, G.; Yue, Z.; Talloczy, Z.; Hagemann, T.; Cho, W.; Messing, A.; Sulzer, D.I.; Goldman, J.E. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum. Mol. Genet. 2008, 17, 1540–1555. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Lu, S.Z.; Guo, Y.S.; Liang, P.Z.; Zhang, S.Z.; Yin, S.; Yin, Y.Q.; Wang, X.M.; Ding, F.; Gu, X.S.; Zhou, J.W. Suppression of astrocytic autophagy by αB-crystallin contributes to α-synuclein inclusion formation. Transl. Neurodegener. 2019, 8, 3. [Google Scholar] [CrossRef]
- Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 155–162. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Wang, S.; Li, B.; Qiao, H.; Lv, X.; Liang, Q.; Shi, Z.; Xia, W.; Ji, F.; Jiao, J. Autophagy-related gene Atg5 is essential for astrocyte differentiation in the developing mouse cortex. EMBO Rep. 2014, 15, 1053–1061. [Google Scholar] [CrossRef]
- Ha, S.; Jeong, S.H.; Yi, K.; Chu, J.J.M.; Kim, S.; Kim, E.K.; Yu, S.W. Autophagy mediates astrogenesis in adult hippocampal neural stem cells. Exp. Neurobiol. 2019, 28, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Patric, I.R.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation silencing of ULK2, an autophagy gene, is essential for astrocyte transformation and tumor growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ikeshima-Kataoka, H.; Kreft, M.; Vardjan, N.; Zorec, R.; Noda, M. Metabolic plasticity of astrocytes and aging of the brain. Int. J. Mol. Sci. 2019, 20, 941. [Google Scholar] [CrossRef]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.K.; Götz, M.; et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef]
- Qin, A.P.; Liu, C.F.; Qin, Y.Y.; Hong, L.Z.; Xu, M.; Yang, L.; Liu, J.; Qin, Z.H.; Zhang, H.L. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 2010, 6, 738–753. [Google Scholar] [CrossRef]
- Han, B.; Zhang, Y.; Zhang, Y.; Bai, Y.; Chen, X.; Huang, R.; Wu, F.; Leng, S.; Chao, J.; Zhang, J.H.; et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: Implications for cerebral ischemic stroke. Autophagy 2018, 14, 1164–1184. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, T.H.; Yang, L.Y.; Shih, C.M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef]
- Gray, L.R.; Roche, M.; Flynn, J.K.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. Is the central nervous system a reservoir of HIV-1? Curr. Opin. HIV AIDS 2014, 9, 552–558. [Google Scholar] [CrossRef]
- Li, G.H.; Henderson, L.; Nath, A. Astrocytes as an HIV reservoir: Mechanism of HIV infection. Curr. HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef]
- Ojeda, D.S.; Grasso, D.; Urquiza, J.; Till, A.; Vaccaro, M.I.; Quarleri, J. Cell death is counteracted by mitophagy in HIV-productively infected astrocytes but is promoted by inflammasome activation among non-productively infected cells. Front. Immunol. 2018, 9, 2633. [Google Scholar] [CrossRef]
- Mehla, R.; Chauhan, A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J. Neuroimmunol. 2015, 285, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Liu, Y.; Zhang, G.; Wu, H.; Hou, Y. Progesterone suppresses Aβ42-induced neuroinflammation by enhancing autophagy in astrocytes. Int. Immunopharmacol. 2018, 54, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc. Natl. Acad. Sci. USA 2012, 109, E2334–E2342. [Google Scholar] [CrossRef]
- Liu, X.; Tian, F.; Wang, S.; Wang, F.; Xiong, L. Astrocyte autophagy flux protects neurons against oxygen-glucose deprivation and ischemic/reperfusion injury. Rejuvenation Res. 2018, 21, 405–415. [Google Scholar] [CrossRef]
- Kulkarni, A.; Dong, A.; Kulkarni, V.V.; Chen, J.; Laxton, O.; Anand, A.; Maday, S. Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy 2019, 1–17. [Google Scholar] [CrossRef]
- Pla, A.; Pascual, M.; Guerri, C. Autophagy constitutes a protective mechanism against ethanol toxicity in mouse astrocytes and neurons. PLoS ONE 2016, 11, e0153097. [Google Scholar] [CrossRef]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Subramani, S.; Malhotra, V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013, 14, 143–151. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Chen, Z.; Nie, S.D.; Qu, M.L.; Zhou, D.; Wu, L.Y.; Shi, X.J.; Ma, L.R.; Li, X.; Zhou, S.L.; Wang, S.; et al. The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Fuente-Martin, E.; Finan, B.; Kim, M.; Frank, A.; Garcia-Caceres, C.; Navas, C.R.; Gordillo, R.; Neinast, M.; Kalainayakan, S.P.; et al. Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 2014, 9, 633–645. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef]
- Karmi, A.; Iozzo, P.; Viljanen, A.; Hirvonen, J.; Fielding, B.A.; Virtanen, K.; Oikonen, V.; Kemppainen, J.; Viljanen, T.; Guiducci, L.; et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes 2010, 59, 2171–2177. [Google Scholar] [CrossRef]
- Patil, S.; Melrose, J.; Chan, C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef]
- Wong, K.L.; Wu, Y.R.; Cheng, K.S.; Chan, P.; Cheung, C.W.; Lu, D.Y.; Su, T.H.; Liu, Z.M.; Leung, Y.M. Palmitic acid-induced lipotoxicity and protection by (+)-catechin in rat cortical astrocytes. Pharmacol. Rep. 2014, 66, 1106–1113. [Google Scholar] [CrossRef]
- González-Giraldo, Y.; Garcia-Segura, L.M.; Echeverria, V.; Barreto, G.E. Tibolone preserves mitochondrial functionality and cell morphology in astrocytic cells treated with palmitic acid. Mol. Neurobiol. 2018, 55, 4453–4462. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Wang, J.; Liu, S.; Gao, M.; Ling, E.A.; Hao, A. Cytoprotective effects of melatonin on astroglial cells subjected to palmitic acid treatment in vitro. J. Pineal Res. 2012, 52, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Rodriguez, A.; Acaz-Fonseca, E.; Boya, P.; Arevalo, M.A.; Garcia-Segura, L.M. Lipotoxic effects of palmitic acid on astrocytes are associated with autophagy impairment. Mol. Neurobiol. 2019, 56, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cáceres, M.P.; Toledo-Valenzuela, L.; Díaz-Castro, F.; Ávalos, Y.; Burgos, P.; Narro, C.; Peña-Oyarzun, D.; Espinoza-Caicedo, J.; Cifuentes-Araneda, F.; Navarro-Aguad, F.; et al. Palmitic acid reduces the autophagic flux and insulin sensitivity through the activation of the Free Fatty Acid Receptor 1 (FFAR1) in the hypothalamic neuronal cell line N43/5. Front. Endocrinol. (Lausanne) 2019, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.A.B.; Melo, N.F.M.; Vieira, É.D.S.; Nogueira, P.A.S.; Coope, A.; Velloso, L.A.; Dezonne, R.S.; Ueira-Vieira, C.; Botelho, F.V.; Gomes, J.A.S.; et al. Palmitate treated-astrocyte conditioned medium contains increased glutathione and interferes in hypothalamic synaptic network in vitro. Neurochem. Int. 2018, 120, 140–148. [Google Scholar] [CrossRef]


{kind=link}
| Blood–brain barrier (BBB) formation and maintenance | [6] |
| Nutrient transport to neurons | [7,8] |
| Regulation of cerebral blood flow depending on neuronal activity | [9] |
| Glycogen storage | [10,11] |
| Control of ion and water homeostasis | [12] |
| Modulation of synaptic activity | [13,14,15] |
| Antioxidant defense | [16,17,18] |
| Response against CNS injuries | [19,20] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Rodriguez, A.; Arevalo, M.-A. The Contribution of Astrocyte Autophagy to Systemic Metabolism. Int. J. Mol. Sci. 2020, 21, 2479. https://doi.org/10.3390/ijms21072479
Ortiz-Rodriguez A, Arevalo M-A. The Contribution of Astrocyte Autophagy to Systemic Metabolism. International Journal of Molecular Sciences. 2020; 21(7):2479. https://doi.org/10.3390/ijms21072479
Chicago/Turabian StyleOrtiz-Rodriguez, Ana, and Maria-Angeles Arevalo. 2020. "The Contribution of Astrocyte Autophagy to Systemic Metabolism" International Journal of Molecular Sciences 21, no. 7: 2479. https://doi.org/10.3390/ijms21072479
APA StyleOrtiz-Rodriguez, A., & Arevalo, M.-A. (2020). The Contribution of Astrocyte Autophagy to Systemic Metabolism. International Journal of Molecular Sciences, 21(7), 2479. https://doi.org/10.3390/ijms21072479