Computational Models Using Multiple Machine Learning Algorithms for Predicting Drug Hepatotoxicity with the DILIrank Dataset



 ,
, 
Abstract
1. Introduction
2. Results
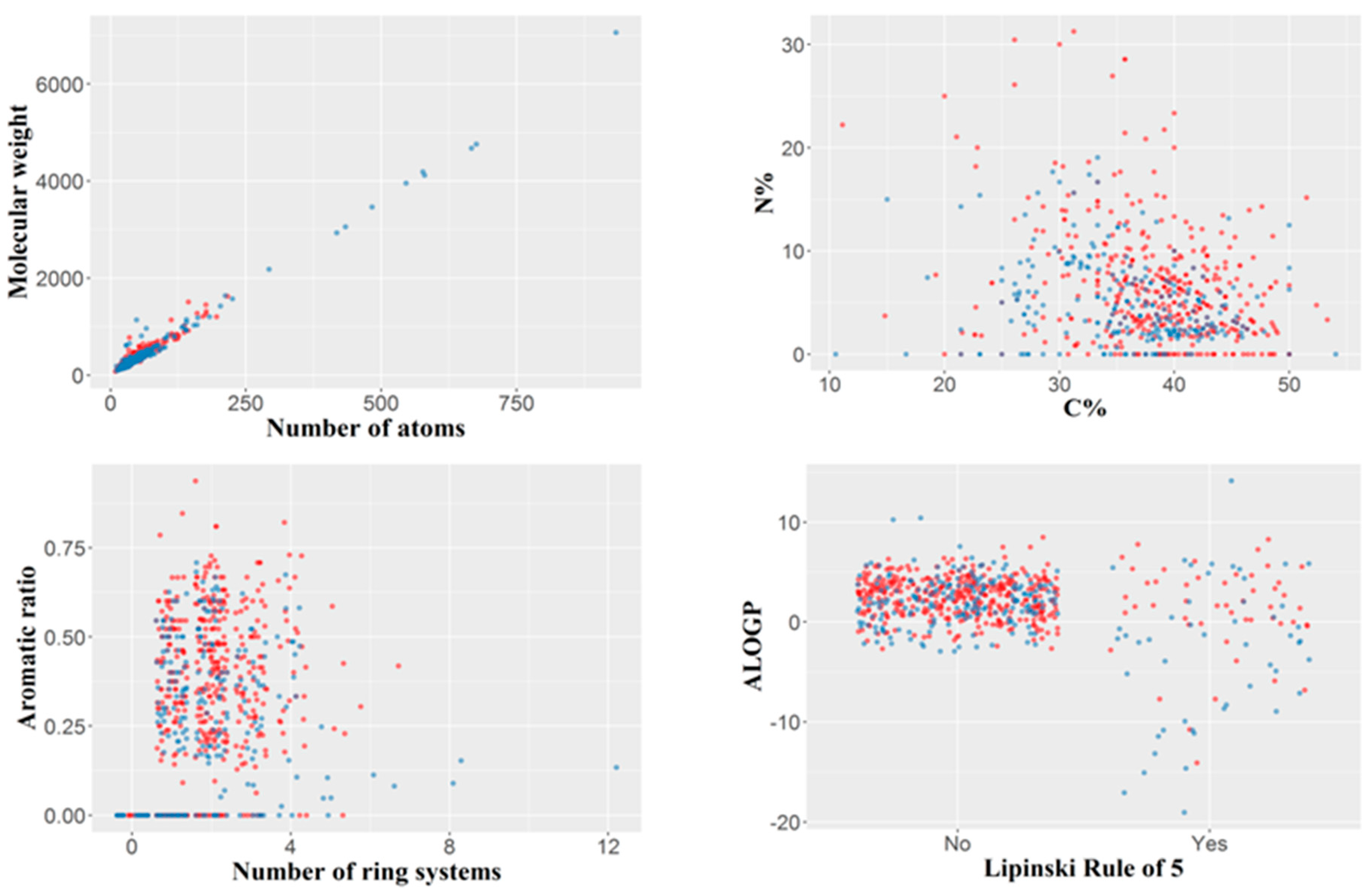
2.1. Dataset Analysis
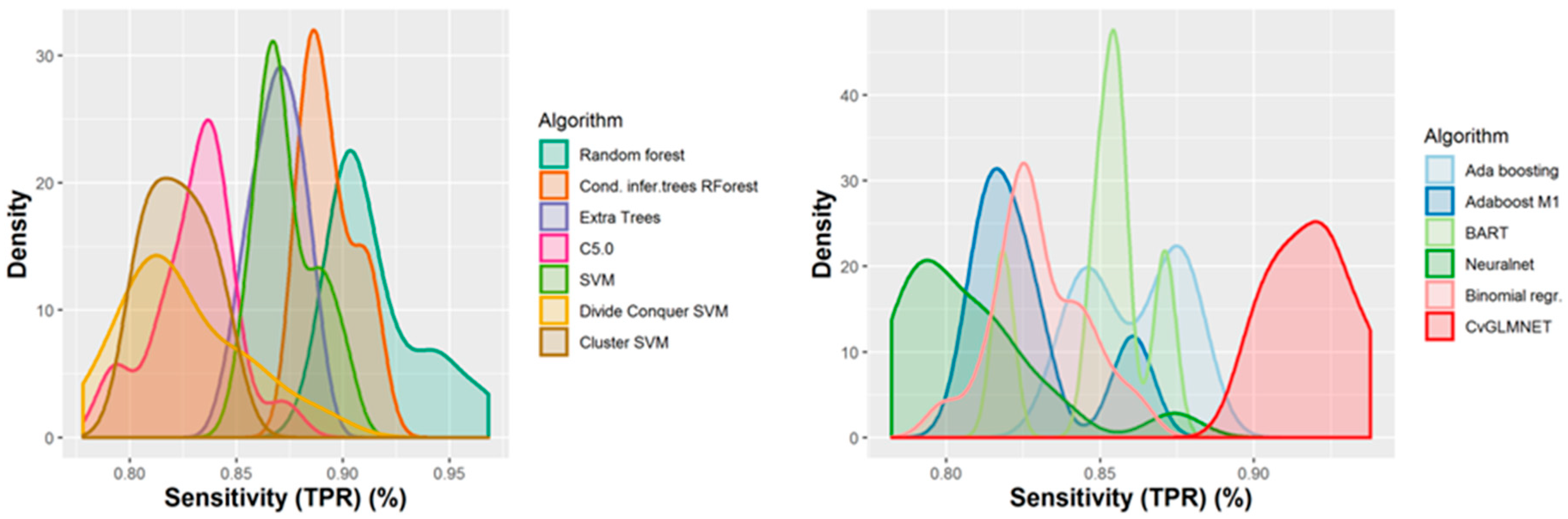
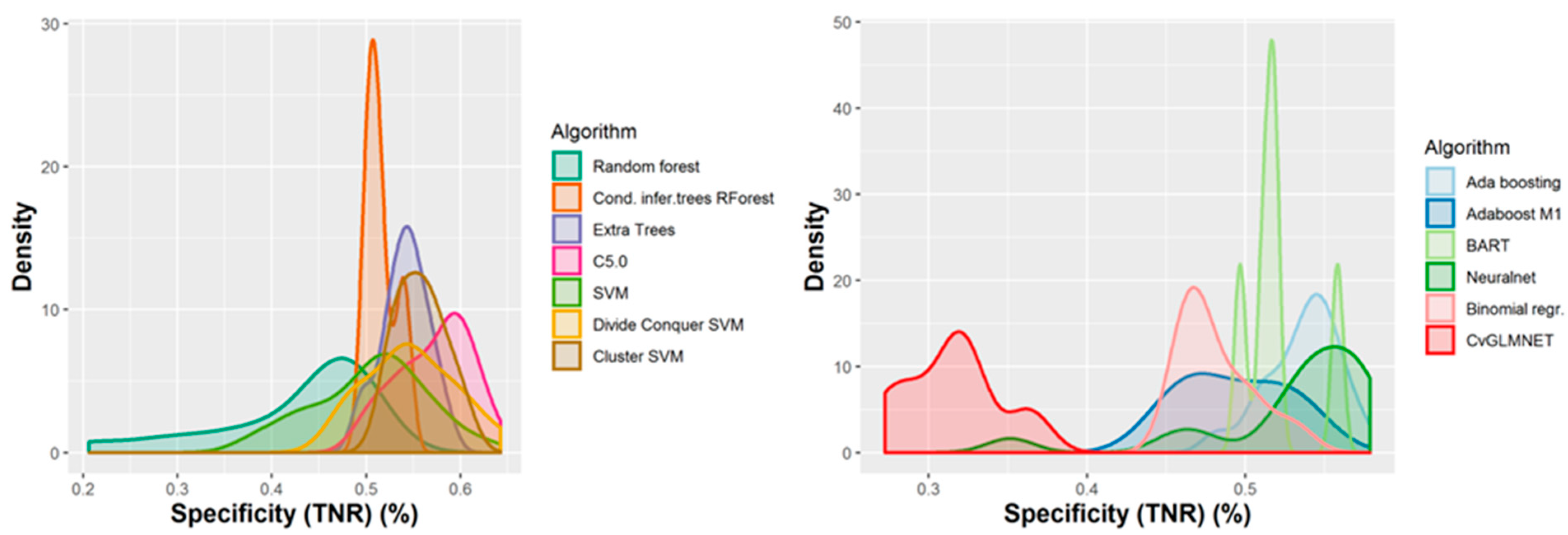
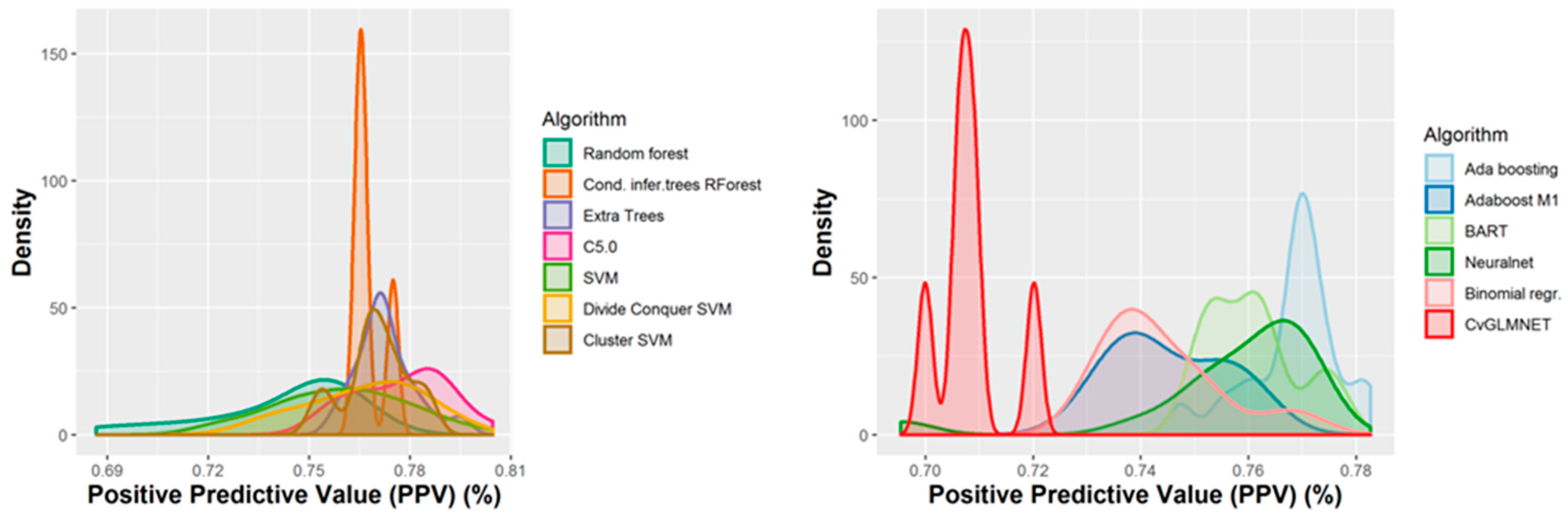
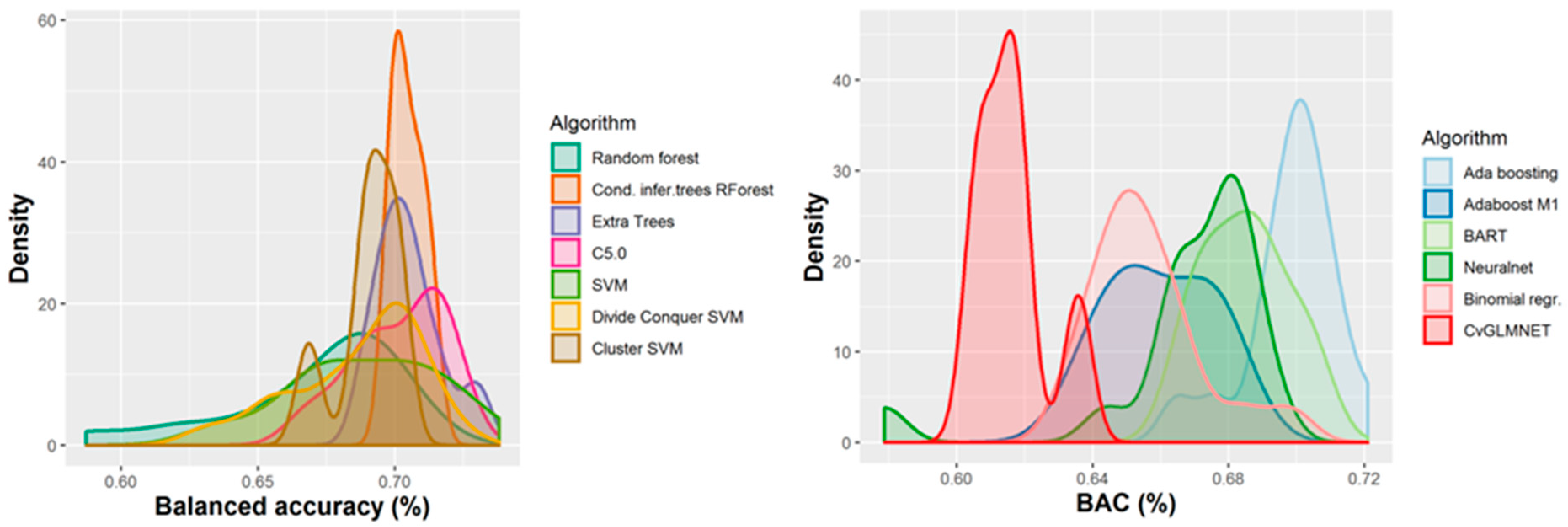
2.2. Performances of Models
2.3. y-Randomization Test
2.4. Descriptors Associated with Hepatotoxicity
2.5. Virtual Screening
2.6. Outliers, Applicability Domain and Wrongly Classified Drugs
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Descriptors
4.3. Feature Selection
4.4. Classification Algorithms
4.5. Performance Evaluation
4.6. Virtual Screening
- (a)
- By a majority vote applied to the classification performed by each model;
- (b)
- By computing the average of the probabilities outputted by each model and then applying the 50% threshold to classify the compound as being of concern or of no concern (only 72 models outputted probabilities, 6 only made binary predictions);
- (c)
- By developing meta-models using the predictions of the best 50 models (selected with the help of the same selection algorithms as for the building of the individual models) as independent variables for the final classification. We evaluated models based exclusively on the 50 best-performing individual models. We also built models that additionally included the dose and duration of treatment as supplementary features for the improvement of the performance.
4.7. Outliers and Applicability Domain
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Applicability Domain |
| ANNs | Artificial Neural Networks |
| AUC | Area Under the Receiver Operating Characteristics Curve |
| BA | Balanced Accuracy |
| BART | Bayesian Additive Regression Trees |
| CV | Cross-Validation |
| DC-SVM | Divide-and-Conquer SVM |
| DILI | Drug induced liver injury |
| ECHA | European Chemicals Agency |
| ERT | Extremely Randomized Trees |
| FPR | False Positive Rate |
| IBk | an RWeka implementation of the knn algorithm |
| IFOREST | Isolation Forest |
| knn | k-nearest neighbours |
| LDA | Linear Discriminant Analysis |
| MMCE | Mean MisClassification Error |
| NIDDK | National Institute of Diabetes and Digestive and Kidney Diseases |
| OECD | The Organisation for Economic Co-operation and Development |
| PPV | Positive Predictive Value |
| QSAR | Quantitative Structure–Activity Relationship |
| RDA | Regularized Discriminant Analysis |
| RF | Random Forests |
| SOD | Subspace Outlier Detection |
| SVM | Support Vector Machines |
| TNR | True Negative Rate |
| TPR | True Positive Rate |
References
- Temple, R.J.; Himmel, M.H. Safety of newly approved drugs: Implications for prescribing. JAMA 2002, 287, 2273–2275. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, W.; Kappenberg, F.; Brecklinghaus, T.; Stoeber, R.; Marchan, R.; Zhang, M.; Ebbert, K.; Kirschner, H.; Grinberg, M.; Leist, M.; et al. Prediction of human drug-induced liver injury (DILI) in relation to oral doses and blood concentrations. Arch. Toxicol. 2019, 93, 1609–1637. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Nelson, L.J.; Gómez Del Moral, M.; Martínez-Naves, E.; Cubero, F.J. Dissecting the molecular pathophysiology of drug-induced liver injury. World J. Gastroenterol. 2018, 24, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Dara, L. Cell death in drug-induced liver injury. In Advances in Pharmacology; Elsevier: Cambridge, MA, USA, 2019; Volume 85, pp. 31–74. ISBN 9780128167595. [Google Scholar]
- Boelsterli, U.A.; Zimmerman, H.J.; Kretz-Rommel, A. Idiosyncratic liver toxicity of nonsteroidal antiinflammatory drugs: Molecular mechanisms and pathology. Crit. Rev. Toxicol. 1995, 25, 207–235. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.H. Drug-Induced Liver Injury Throughout the Drug Development Life Cycle: Where We Have Been, Where We are Now, and Where We are Headed. Perspectives of a Clinical Hepatologist. Pharm. Med. 2013, 27, 165–191. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Ballet, F. Preventing Drug-Induced Liver Injury: How Useful Are Animal Models? Dig. Dis. 2015, 33, 477–485. [Google Scholar] [CrossRef]
- Negres, S.; Dinu, M.; Ancuceanu, R.; Olaru, T.O.; Ghica, M.V.; Seremet, O.C.; Zbarcea, C.E.; Velescu, B.S.; Stefanescu, E.; Chirita, C. Correlations in silico/in vitro/in vivo regarding determinating acute toxicity in non-clinical experimental trial, according to bioethic regulations inforced by the European Union. Farmacia 2015, 63, 877–885. [Google Scholar]
- Toropova, A.P.; Toropov, A.A. CORAL: Binary classifications (active/inactive) for drug-induced liver injury. Toxicol. Lett. 2017, 268, 51–57. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Suzuki, A.; Thakkar, S.; Yu, K.; Hu, C.; Tong, W. DILIrank: The largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today 2016, 21, 648–653. [Google Scholar] [CrossRef]
- Hong, H.; Thakkar, S.; Chen, M.; Tong, W. Development of Decision Forest Models for Prediction of Drug-Induced Liver Injury in Humans Using A Large Set of FDA-approved Drugs. Sci. Rep. 2017, 7, 17311. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fu, L.; Zhang, J.-W.; Wei, H.; Ye, W.-L.; Deng, Z.-K.; Zhang, L.; Cheng, Y.; Ouyang, D.; Cao, Q.; et al. Three-Level Hepatotoxicity Prediction System Based on Adverse Hepatic Effects. Mol. Pharm. 2019, 16, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, Q.; Chen, P.; Wang, B. In Silico Prediction of Drug-Induced Liver Injury Based on Ensemble Classifier Method. Int. J. Mol. Sci. 2019, 20, 4106. [Google Scholar] [CrossRef]
- He, S.; Ye, T.; Wang, R.; Zhang, C.; Zhang, X.; Sun, G.; Sun, X. An In Silico Model for Predicting Drug-Induced Hepatotoxicity. Int. J. Mol. Sci. 2019, 20, 1897. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P. The “double cross-validation” software tool for MLR QSAR model development. Chemom. Intell. Lab. Syst. 2016, 159, 108–126. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Gower, J.C. A general coefficient of similarity and some of its properties. Biometrics 1971, 857–871. [Google Scholar] [CrossRef]
- Korotcov, A.; Tkachenko, V.; Russo, D.P.; Ekins, S. Comparison of Deep Learning With Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol. Pharm. 2017, 14, 4462–4475. [Google Scholar] [CrossRef]
- Palomba, D.; Martínez, M.J.; Ponzoni, I.; Díaz, M.F.; Vazquez, G.E.; Soto, A.J. QSPR models for predicting log P(liver) values for volatile organic compounds combining statistical methods and domain knowledge. Molecules 2012, 17, 14937–14953. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Ishwaran, H.; Kogalur, U.B. Fast Unified Random Forests for Survival, Regression, and Classification (RF-SRC); Manual; 2019; Available online: https://cran.r-project.org/web/packages/randomForestSRC/randomForestSRC.pdf (accessed on 19 March 2019).
- Ben-Gal, I. Outlier Detection. In Data Mining and Knowledge Discovery Handbook; Maimon, O., Rokach, L., Eds.; Springer-Verlag: New York, NY, USA, 2005; pp. 131–146. ISBN 9780387244358. [Google Scholar]
- Domingues, R.; Filippone, M.; Michiardi, P.; Zouaoui, J. A comparative evaluation of outlier detection algorithms: Experiments and analyses. Pattern Recognit. 2018, 74, 406–421. [Google Scholar] [CrossRef]
- Campos, G.O.; Zimek, A.; Sander, J.; Campello, R.J.G.B.; Micenková, B.; Schubert, E.; Assent, I.; Houle, M.E. On the evaluation of unsupervised outlier detection: Measures, datasets, and an empirical study. Data Min. Knowl. Discov. 2016, 30, 891–927. [Google Scholar] [CrossRef]
- Berenger, F.; Yamanishi, Y. A Distance-Based Boolean Applicability Domain for Classification of High Throughput Screening Data. J. Chem. Inf. Model. 2019, 59, 463–476. [Google Scholar] [CrossRef]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of Different Approaches to Define the Applicability Domain of QSAR Models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef]
- Jin, W.; Tung, A.K.H.; Han, J.; Wang, W. Ranking Outliers Using Symmetric Neighborhood Relationship. In Advances in Knowledge Discovery and Data Mining; Ng, W.-K., Kitsuregawa, M., Li, J., Chang, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 3918, pp. 577–593. ISBN 9783540332060. [Google Scholar]
- Tang, J.; Chen, Z.; Fu, A.W.; Cheung, D.W. Enhancing Effectiveness of Outlier Detections for Low Density Patterns. In Advances in Knowledge Discovery and Data Mining; Chen, M.-S., Yu, P.S., Liu, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 2336, pp. 535–548. ISBN 9783540437048. [Google Scholar]
- Ramappa, V.; Aithal, G.P. Hepatotoxicity Related to Anti-tuberculosis Drugs: Mechanisms and Management. J. Clin. Exp. Hepatol. 2013, 3, 37–49. [Google Scholar] [CrossRef]
- Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Ethambutol. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548745/ (accessed on 19 March 2019).
- Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Dactinomycin. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548778/ (accessed on 19 March 2019).
- Kibleur, Y.; Brochart, H.; Schaaf, H.S.; Diacon, A.H.; Donald, P.R. Dose regimen of para-aminosalicylic acid gastro-resistant formulation (PAS-GR) in multidrug-resistant tuberculosis. Clin. Drug Investig. 2014, 34, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Acetaminophen. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548162/ (accessed on 19 March 2019).
- Pessayre, D.; Larrey, D. Acute and chronic drug-induced hepatitis. Baillieres Clin. Gastroenterol. 1988, 2, 385–422. [Google Scholar] [CrossRef]
- Gunawan, B.K.; Kaplowitz, N. Mechanisms of drug-induced liver disease. Clin. Liver Dis. 2007, 11, 459–475. [Google Scholar] [CrossRef]
- Katarey, D.; Verma, S. Drug-induced liver injury. Clin. Med. 2016, 16, s104–s109. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kaplowitz, N. Diagnosis, management and prevention of drug-induced liver injury. Gut 2009, 58, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, S.; Li, T.; Liu, Z.; Wu, L.; Roberts, R.; Tong, W. Drug-induced liver injury severity and toxicity (DILIst): Binary classification of 1279 drugs by human hepatotoxicity. Drug Discov. Today 2019. [Google Scholar] [CrossRef]
- Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Leuprolide. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548676/ (accessed on 19 March 2012).
- He, S.; Zhang, C.; Zhou, P.; Zhang, X.; Ye, T.; Wang, R.; Sun, G.; Sun, X. Herb-Induced Liver Injury: Phylogenetic Relationship, Structure-Toxicity Relationship, and Herb-Ingredient Network Analysis. Int. J. Mol. Sci. 2019, 20, 3633. [Google Scholar] [CrossRef] [PubMed]
- Prins, B.; Dartee, W.P.; Verboom, W.; Reinhoudt, D.N.; Koster, A.S. Quantitative structure activity relationship for the acute cytotoxicity of 13 (bis) aziridinyl-benzoquinones: Relation to cellular ATP depletion. Arch. Toxicol. 1994, 68, 255–260. [Google Scholar] [CrossRef][Green Version]
- Siraki, A.G.; Chevaldina, T.; O’Brien, P.J. Application of quantitative structure-toxicity relationships for acute NSAID cytotoxicity in rat hepatocytes. Chem. Biol. Interact. 2005, 151, 177–191. [Google Scholar] [CrossRef]
- Rodgers, A.D.; Zhu, H.; Fourches, D.; Rusyn, I.; Tropsha, A. Modeling liver-related adverse effects of drugs using knearest neighbor quantitative structure-activity relationship method. Chem. Res. Toxicol. 2010, 23, 724–732. [Google Scholar] [CrossRef]
- Low, Y.; Uehara, T.; Minowa, Y.; Yamada, H.; Ohno, Y.; Urushidani, T.; Sedykh, A.; Muratov, E.; Kuz’min, V.; Fourches, D.; et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 2011, 24, 1251–1262. [Google Scholar] [CrossRef]
- Huang, S.-H.; Tung, C.-W.; Fülöp, F.; Li, J.-H. Developing a QSAR model for hepatotoxicity screening of the active compounds in traditional Chinese medicines. Food Chem. Toxicol. 2015, 78, 71–77. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P.; Rasulev, B.F.; Benfenati, E.; Gini, G.; Leszczynska, D.; Leszczynski, J. CORAL: Binary classifications (active/inactive) for Liver-Related Adverse Effects of Drugs. Curr. Drug Saf. 2012, 7, 257–261. [Google Scholar] [CrossRef]
- Matthews, E.J.; Ursem, C.J.; Kruhlak, N.L.; Benz, R.D.; Sabaté, D.A.; Yang, C.; Klopman, G.; Contrera, J.F. Identification of structure-activity relationships for adverse effects of pharmaceuticals in humans: Part B. Use of (Q)SAR systems for early detection of drug-induced hepatobiliary and urinary tract toxicities. Regul. Toxicol. Pharmacol. 2009, 54, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cheng, F.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico Prediction of Drug Induced Liver Toxicity Using Substructure Pattern Recognition Method. Mol. Inf. 2016, 35, 136–144. [Google Scholar] [CrossRef]
- Ai, H.; Chen, W.; Zhang, L.; Huang, L.; Yin, Z.; Hu, H.; Zhao, Q.; Zhao, J.; Liu, H. Predicting Drug-Induced Liver Injury Using Ensemble Learning Methods and Molecular Fingerprints. Toxicol. Sci. 2018, 165, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Bisgin, H.; Tong, L.; Hong, H.; Fang, H.; Borlak, J.; Tong, W. Toward predictive models for drug-induced liver injury in humans: Are we there yet? Biomark. Med. 2014, 8, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Dehnbostel, F.O.; Preissner, R. Prediction Is a Balancing Act: Importance of Sampling Methods to Balance Sensitivity and Specificity of Predictive Models Based on Imbalanced Chemical Data Sets. Front. Chem. 2018, 6, 362. [Google Scholar] [CrossRef]
- Satalkar, V.; Kulkarni, S.; Joshi, D. QSAR based analysis of fatal drug induced renal toxicity. J. Comput. Methods Mol. Des. 2015, 5, 24–32. [Google Scholar]
- Kotsampasakou, E.; Ecker, G.F. Predicting Drug-Induced Cholestasis with the Help of Hepatic Transporters-An in Silico Modeling Approach. J. Chem. Inf. Model. 2017, 57, 608–615. [Google Scholar] [CrossRef]
- Jiang, L.; He, Y.; Zhang, Y. Prediction of hepatotoxicity of traditional Chinese medicine compounds by support vector machine approach. In Proceedings of the 8th International Conference on Systems Biology (ISB), Qingdao, China, 24–27 October 2014; pp. 27–30. [Google Scholar]
- Zheng, M.; McErlane, K.M.; Ong, M.C. Hydromorphone metabolites: Isolation and identification from pooled urine samples of a cancer patient. Xenobiotica 2002, 32, 427–439. [Google Scholar] [CrossRef]
- Leeson, P.D. Impact of Physicochemical Properties on Dose and Hepatotoxicity of Oral Drugs. Chem. Res. Toxicol. 2018, 31, 494–505. [Google Scholar] [CrossRef]
- Sieber, S.M.; Correa, P.; Dalgard, D.W.; McIntire, K.R.; Adamson, R.H. Carcinogenicity and hepatotoxicity of cycasin and its aglycone methylazoxymethanol acetate in nonhuman primates. J. Natl. Cancer Inst. 1980, 65, 177–189. [Google Scholar]
- Cai, J.; Luo, J.; Wang, S.; Yang, S. Feature selection in machine learning: A new perspective. Neurocomputing 2018, 300, 70–79. [Google Scholar] [CrossRef]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature Selection: A Data Perspective. ACM Comput. Surv. 2017, 50, 1–45. [Google Scholar] [CrossRef]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [PubMed]
- Bischl, B.; Lang, M.; Kotthoff, L.; Schiffner, J.; Richter, J.; Studerus, E.; Casalicchio, G.; Jones, Z.M. mlr: Machine Learning in R. J. Mach. Learn. Res. 2016, 17, 1–5. [Google Scholar]
- Romanski, P.; Kotthoff, L. FSelector: Selecting Attributes. 2018. Available online: https://cran.r-project.org/web/packages/FSelector/index.html (accessed on 19 March 2019).
- Strobl, C.; Boulesteix, A.-L.; Kneib, T.; Augustin, T.; Zeileis, A. Conditional Variable Importance for Random Forests. BMC Bioinform. 2008, 9, 307. [Google Scholar] [CrossRef]
- Wright, M.N.; Ziegler, A. Ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77, 1–17. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Veselinović, J.B.; Đorđević, V.; Bogdanović, M.; Morić, I.; Veselinović, A.M. QSAR modeling of dihydrofolate reductase inhibitors as a therapeutic target for multiresistant bacteria. Struct. Chem. 2018, 29, 541–551. [Google Scholar] [CrossRef]
- R Core Team. R Foundation for Statistical Computing. In R: A Language and Environment for Statistical Computing; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Bischl, B.; Lang, M. ParallelMap: Unified Interface to Parallelization Back-Ends; The R Foundation: Vienna, Austria, 2015. [Google Scholar]
- Wing, M.K.C.J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; Team, R.C.; Benesty, M.; et al. Caret: Classification and Regression Training; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Dieguez-Santana, K.; Pham-The, H.; Rivera-Borroto, O.M.; Puris, A.; Le-Thi-Thu, H.; Casanola-Martin, G.M. A Two QSAR Way for Antidiabetic Agents Targeting Using α-Amylase and α-Glucosidase Inhibitors: Model Parameters Settings in Artificial Intelligence Techniques. Lett. Drug Des. Discov. 2017, 14, 862–868. [Google Scholar] [CrossRef]
- Raevsky, O.A.; Grigorev, V.Y.; Yarkov, A.V.; Polianczyk, D.E.; Tarasov, V.V.; Bovina, E.V.; Bryzhakina, E.N.; Dearden, J.C.; Avila-Rodriguez, M.; Aliev, G. Classification (Agonist/Antagonist) and Regression “Structure-Activity” Models of Drug Interaction with 5-HT6. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 213–221. [Google Scholar] [CrossRef]
- Barrett, T.S.; Lockhart, G. Efficient Exploration of Many Variables and Interactions Using Regularized Regression. Prev. Sci. 2019, 20, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Ahmadi, E.; Weckman, G.R.; Masel, D.T. Decision making model to predict presence of coronary artery disease using neural network and C5.0 decision tree. J. Ambient Intell. Humaniz. Comput. 2018, 9, 999–1011. [Google Scholar] [CrossRef]
- Schöning, V.; Hammann, F. How far have decision tree models come for data mining in drug discovery? Expert Opin. Drug Discov. 2018, 13, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Quinlan, R. C50: C5.0 Decision Trees and Rule-Based Models; The R Foundation: Vienna, Austria, 2018. [Google Scholar]
- Hdoufane, I.; Bjij, I.; Soliman, M.; Tadjer, A.; Villemin, D.; Bogdanov, J.; Cherqaoui, D. In Silico SAR Studies of HIV-1 Inhibitors. Pharmaceuticals 2018, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Ancuceanu, R.; Dinu, M.; Neaga, I.; Laszlo, F.; Boda, D. Development of QSAR machine learning-based models to forecast the effect of substances on malignant melanoma cells. Oncol. Lett. 2019, 17, 4188–4196. [Google Scholar] [CrossRef]
- Alfaro, E.; Gámez, M.; García, N. Ensemble Classifiers Methods. In Ensemble Classification Methods with Applicationsin R; Alfaro, E., Gámez, M., García, N., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2018; pp. 31–50. ISBN 9781119421566. [Google Scholar]
- Deng, H.; Runger, G. Feature selection via regularized trees. In Proceedings of the 2012 International Joint Conference on Neural Networks (IJCNN), Brisbane, Australia, 10–15 June 2012; pp. 1–8. [Google Scholar]
- Deng, H. Guided Random Forest in the RRF Package. arXiv 2013, arXiv:1306.0237. [Google Scholar]
- Hothorn, T.; Hornik, K.; Zeileis, A. Unbiased Recursive Partitioning: A Conditional Inference Framework. J. Comput. Graph. Stat. 2006, 15, 651–674. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation Forest: A New Classifier Ensemble Method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef]
- Ballings, M.; Poel, D.V. RotationForest: Fit and Deploy Rotation Forest Models; The R Foundation: Vienna, Austria, 2017. [Google Scholar]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. AtbPpred: A Robust Sequence-Based Prediction of Anti-Tubercular Peptides Using Extremely Randomized Trees. Comput. Struct. Biotechnol. J. 2019, 17, 972–981. [Google Scholar] [CrossRef]
- Simm, J.; Abril, I.M.; Sugiyama, M. Tree-Based Ensemble Multi-Task Learning Method for Classification and Regression; The Institute of Electronics, Information and Communication Engineers: Tokyo, Japan, 2014; Volume 97. [Google Scholar]
- Chipman, H.A.; George, E.I.; McCulloch, R.E. BART: Bayesian additive regression trees. Ann. Appl. Stat. 2010, 4, 266–298. [Google Scholar] [CrossRef]
- Feng, D.; Svetnik, V.; Liaw, A.; Pratola, M.; Sheridan, R.P. Building Quantitative Structure-Activity Relationship Models Using Bayesian Additive Regression Trees. J. Chem. Inf. Model. 2019, 59, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Hernández, B.; Raftery, A.E.; Pennington, S.R.; Parnell, A.C. Bayesian Additive Regression Trees using Bayesian model averaging. Stat. Comput. 2018, 28, 869–890. [Google Scholar] [CrossRef] [PubMed]
- Kapelner, A.; Bleich, J. bartMachine: Machine Learning with Bayesian Additive Regression Trees. J. Stat. Softw. 2016, 70, 1–40. [Google Scholar] [CrossRef]
- Saritas, M.M.; Yasar, A. Performance Analysis of ANN and Naive Bayes Classification Algorithm for Data Classification. Int. J. Intell. Syst. Appl. Eng. 2019, 7, 88–91. [Google Scholar] [CrossRef]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. e1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Idakwo, G.; Luttrell, J.; Chen, M.; Hong, H.; Zhou, Z.; Gong, P.; Zhang, C. A review on machine learning methods for in silico toxicity prediction. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2018, 36, 169–191. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, S.; Li, Y.; Wang, Q. Development of quantitative structure-activity relationship models to predict potential nephrotoxic ingredients in traditional Chinese medicines. Food Chem. Toxicol. 2019, 128, 163–170. [Google Scholar] [CrossRef]
- Chen, H.; Chen, L. Support Vector Machine Classification of Drunk Driving Behaviour. Int. J. Environ. Res. Public Health 2017, 14, 108. [Google Scholar] [CrossRef]
- Gu, Q.; Han, J. Clustered Support Vector Machines. In Proceedings of the 16th International Conference on Artificial Intelligence and Statistics (AISTATS), Scottsdale, AZ, USA, 29 April–1 May 2013; Volume 31, pp. 307–315. [Google Scholar]
- Hsieh, C.-J.; Si, S.; Dhillon, I. A Divide-and-Conquer Solver for Kernel Support Vector Machines. In Proceedings of the 31st International Conference on Machine Learning, PMLR, Beijing, China, 22–24 June 2014; Volume 32, pp. 566–574. [Google Scholar]
- He, T.; Demircioglu, A. SwarmSVM: Ensemble Learning Algorithms Based on Support Vector Machines; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Freund, Y.; Schapire, R.E. A Short Introduction to Boosting. J. Jpn. Soc. Artif. Intell. 1999, 14, 771–780. [Google Scholar]
- Culp, M.; Johnson, K.; Michailidis, G. ada: The R Package Ada for Stochastic Boosting; The R Foundation: Vienna, Austria, 2016. [Google Scholar]
- Hornik, K.; Buchta, C.; Zeileis, A. Open-Source Machine Learning: R Meets Weka. Comput. Stat. 2009, 24, 225–232. [Google Scholar] [CrossRef]
- Bruce, P.C.; Bruce, A. Practical Statistics for Data Scientists: 50 Essential Concepts, 1st ed.; O’Reilly: Sebastopol, CA, USA, 2017; ISBN 9781491952962. [Google Scholar]
- Zheng, S.; Ding, C.; Nie, F.; Huang, H. Harmonic Mean Linear Discriminant Analysis. IEEE Trans. Knowl. Data Eng. 2019, 31, 1520–1531. [Google Scholar] [CrossRef]
- Yang, X.; Elkhalil, K.; Kammoun, A.; Al-Naffouri, T.Y.; Alouini, M.-S. Regularized Discriminant Analysis: A Large Dimensional Study. In Proceedings of the 2018 IEEE International Symposium on Information Theory (ISIT), Vail, CO, USA, 17–22 June 2018; pp. 536–540. [Google Scholar]
- Weihs, C.; Ligges, U.; Luebke, K.; Raabe, N. klaR Analyzing German Business Cycles. In Proceedings of the Data Analysis and Decision Support; Baier, D., Decker, R., Schmidt-Thieme, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 335–343. [Google Scholar]
- Du, K.-L.; Swamy, M.N.S. Fundamentals of Machine Learning. In Neural Networks and Statistical Learning; Springer: London, UK, 2014; pp. 15–65. ISBN 9781447155706. [Google Scholar]
- Rashka, S. Python Machine Learning; Packt Publishing: Birmingham-Mumbay, UK, 2015. [Google Scholar]
- Boudergua, S.; Alloui, M.; Belaidi, S.; Al Mogren, M.M.; Ellatif Ibrahim, U.A.A.; Hochlaf, M. QSAR Modeling and Drug-Likeness Screening for Antioxidant Activity of Benzofuran Derivatives. J. Mol. Struct. 2019, 1189, 307–314. [Google Scholar] [CrossRef]
- Fritsch, S.; Guenther, F.; Wright, M.N. Neuralnet: Training of Neural Networks; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Rong, X. Deepnet: Deep Learning Toolkit in R; The R Foundation: Vienna, Austria, 2014. [Google Scholar]
- Deng, Z.; Zhu, X.; Cheng, D.; Zong, M.; Zhang, S. Efficient k NN classification algorithm for big data. Neurocomputing 2016, 195, 143–148. [Google Scholar] [CrossRef]
- Shameera Ahamed, T.K.; Rajan, V.K.; Sabira, K.; Muraleedharan, K. QSAR classification-based virtual screening followed by molecular docking studies for identification of potential inhibitors of 5-lipoxygenase. Comput. Biol. Chem. 2018, 77, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Chitre, T.S.; Asgaonkar, K.D.; Patil, S.M.; Kumar, S.; Khedkar, V.M.; Garud, D.R. QSAR, docking studies of 1,3-thiazinan-3-yl isonicotinamide derivatives for antitubercular activity. Comput. Biol. Chem. 2017, 68, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.; Hechenbichler, K. kknn: Weighted k-Nearest Neighbors; The R Foundation: Vienna, Austria, 2016. [Google Scholar]
- Baumann, D.; Baumann, K. Reliable estimation of prediction errors for QSAR models under model uncertainty using double cross-validation. J. Cheminform. 2014, 6, 47. [Google Scholar] [CrossRef]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Romanova, M.A.; Zadorozhny, A.D.; Kurilenko, N.S.; Shilov, B.V.; Pogodin, P.V.; Ivanov, S.M.; Filimonov, D.A.; Poroikov, V.V. Comparison of Quantitative and Qualitative (Q)SAR Models Created for the Prediction of Ki and IC50 Values of Antitarget Inhibitors. Front. Pharmacol. 2018, 9, 1136. [Google Scholar] [CrossRef]
- Capuzzi, S.J.; Sun, W.; Muratov, E.N.; Martínez-Romero, C.; He, S.; Zhu, W.; Li, H.; Tawa, G.; Fisher, E.G.; Xu, M.; et al. Computer-Aided Discovery and Characterization of Novel Ebola Virus Inhibitors. J. Med. Chem. 2018, 61, 3582–3594. [Google Scholar] [CrossRef]
- Yang, H.; Du, Z.; Lv, W.-J.; Zhang, X.-Y.; Zhai, H.-L. In silico toxicity evaluation of dioxins using structure–activity relationship (SAR) and two-dimensional quantitative structure–activity relationship (2D-QSAR). Arch. Toxicol. 2019, 93, 3207–3218. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, K.S. Solitude: An Implementation of Isolation Forest; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Fan, C. HighDimOut: Outlier Detection Algorithms for High-Dimensional Data; The R Foundation: Vienna, Austria, 2015. [Google Scholar]
- Ancuceanu, R.; Tamba, B.; Stoicescu, C.S.; Dinu, M. Use of QSAR Global Models and Molecular Docking for Developing New Inhibitors of c-src Tyrosine Kinase. Int. J. Mol. Sci. 2019, 21, 19. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Interpretation | Descriptor Block (group) | Frequency Occurring Among the First 5 Most Important Features | Sense of the Contribution * |
|---|---|---|---|---|
| Mp | mean atomic polarizability (scaled on Carbon atom) | Constitutional indices | 12 (70.59%) | + |
| H% | percentage of H atoms | Constitutional indices | 12 (70.59%) | − |
| GATS1m | Geary autocorrelation of lag 1 weighted by mass | 2D autocorrelations | 12 (70.59%) | − |
| SpPosA_B(m) | normalized spectral positive sum from Burden matrix weighted by mass | 2D matrix-based descriptors | 10 (58.82%) | + |
| MLOGP | Moriguchi octanol-water partition coeff. (logP) | Molecular properties | 4 (23.53%) | + |
| PCR | ratio of multiple path count over path count | Walk and path counts | 3 (17.65%) | + |
| totalcharge | total charge | Constitutional indices | 2 (11.76%) | − |
| SM1_Dz.m. | spectral moment of order 1 from Barysz matrix weighted by mass | 2D matrix-based descriptors | 2 (11.76%) | + |
| SIC1 | Structural Information Content index (neighborhood symmetry of 1-order) | Information indices | 2 (11.76%) | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ancuceanu, R.; Hovanet, M.V.; Anghel, A.I.; Furtunescu, F.; Neagu, M.; Constantin, C.; Dinu, M. Computational Models Using Multiple Machine Learning Algorithms for Predicting Drug Hepatotoxicity with the DILIrank Dataset. Int. J. Mol. Sci. 2020, 21, 2114. https://doi.org/10.3390/ijms21062114
Ancuceanu R, Hovanet MV, Anghel AI, Furtunescu F, Neagu M, Constantin C, Dinu M. Computational Models Using Multiple Machine Learning Algorithms for Predicting Drug Hepatotoxicity with the DILIrank Dataset. International Journal of Molecular Sciences. 2020; 21(6):2114. https://doi.org/10.3390/ijms21062114
Chicago/Turabian StyleAncuceanu, Robert, Marilena Viorica Hovanet, Adriana Iuliana Anghel, Florentina Furtunescu, Monica Neagu, Carolina Constantin, and Mihaela Dinu. 2020. "Computational Models Using Multiple Machine Learning Algorithms for Predicting Drug Hepatotoxicity with the DILIrank Dataset" International Journal of Molecular Sciences 21, no. 6: 2114. https://doi.org/10.3390/ijms21062114
APA StyleAncuceanu, R., Hovanet, M. V., Anghel, A. I., Furtunescu, F., Neagu, M., Constantin, C., & Dinu, M. (2020). Computational Models Using Multiple Machine Learning Algorithms for Predicting Drug Hepatotoxicity with the DILIrank Dataset. International Journal of Molecular Sciences, 21(6), 2114. https://doi.org/10.3390/ijms21062114