Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
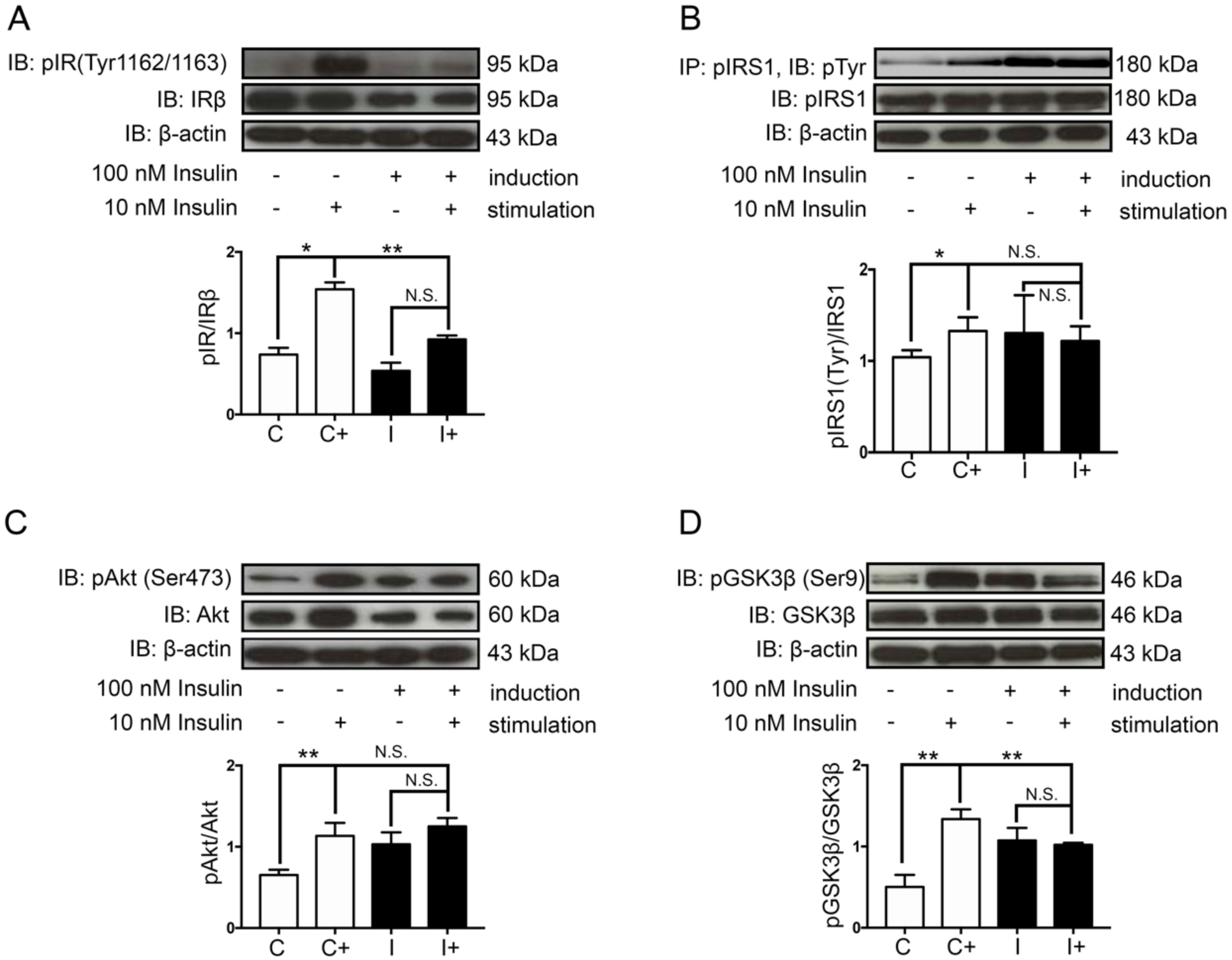
2.1. Hyperinsulinemic Condition Induced Neuronal Insulin Resistance in a Human Neuroblastoma Cell Line, SH-SY5Y
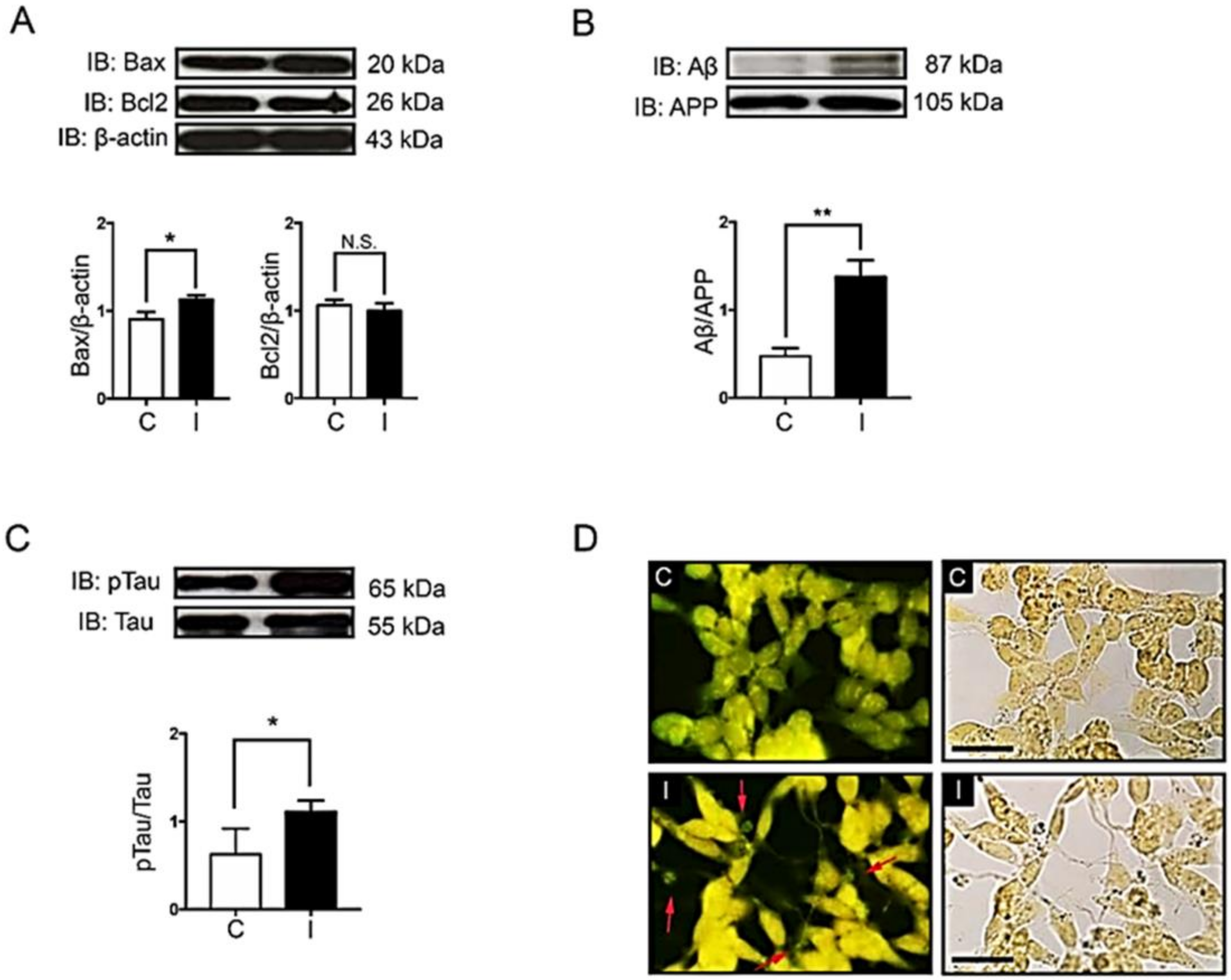
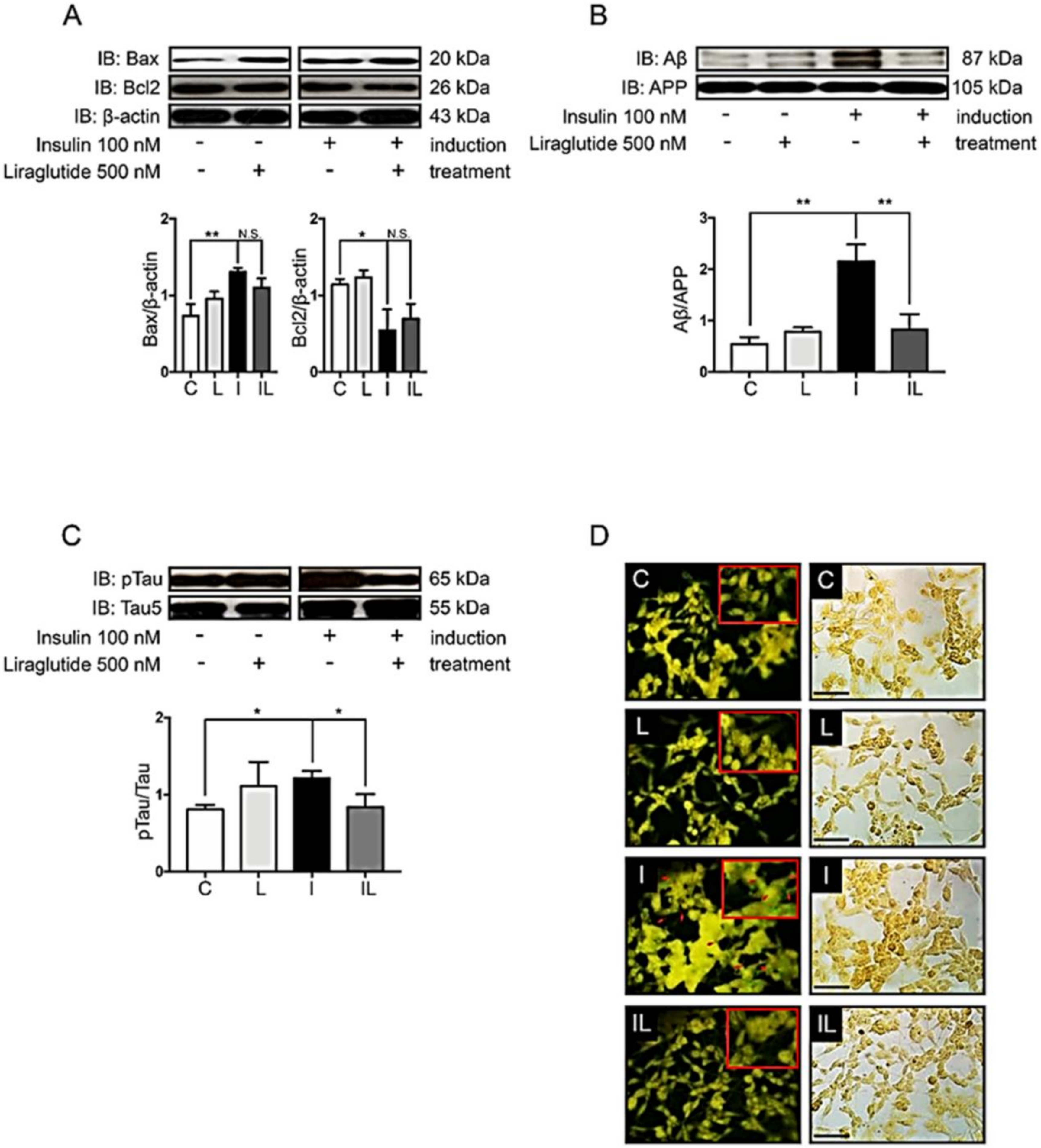
2.2. Hyperinsulinemic Condition Increased the Expression of Apoptotic Protein Markers and the Formation of Alzheimer’s Markers in Neuronal Cells
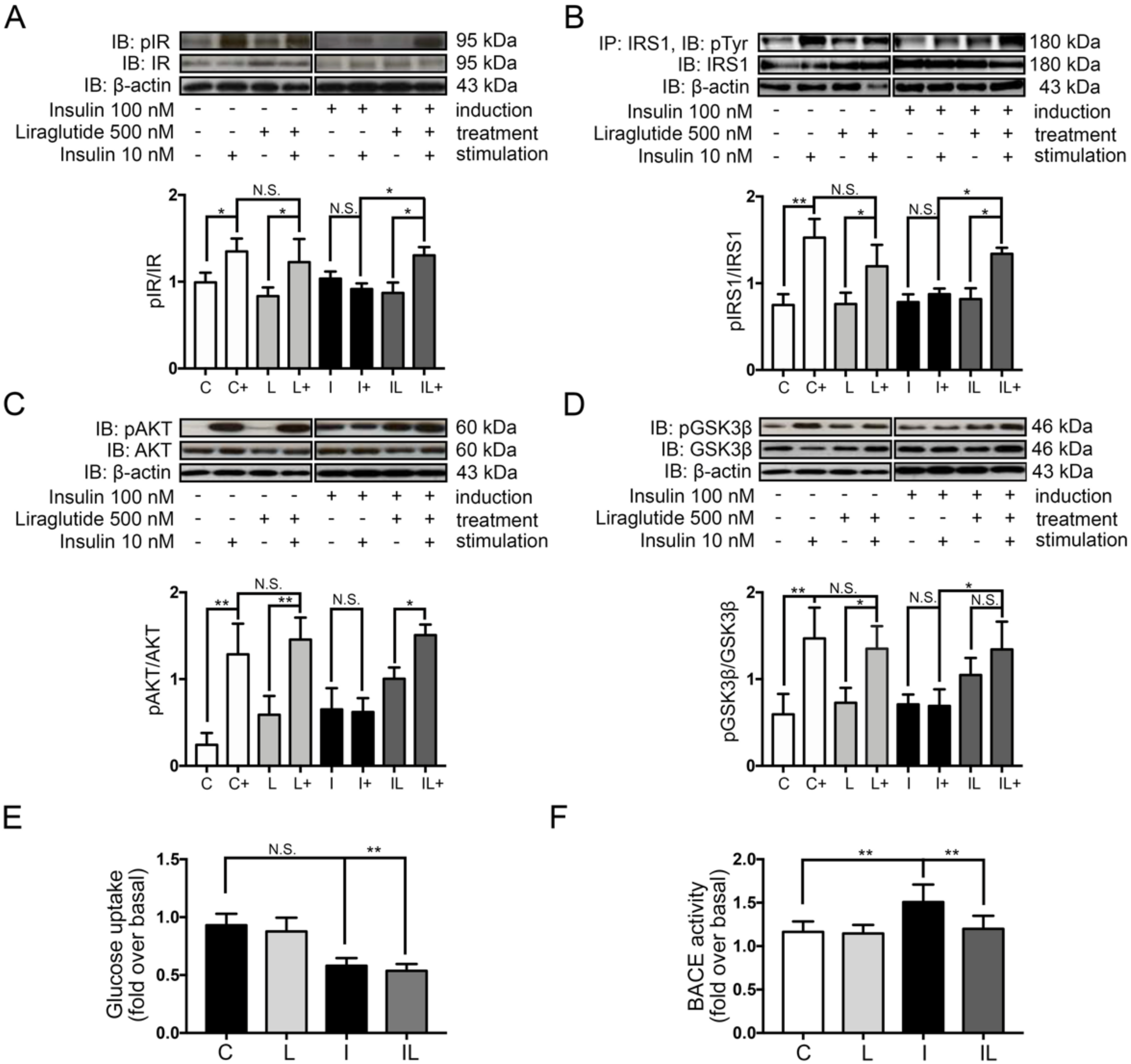
2.3. Liraglutide Restored Neuronal Insulin Sensitivity in Hyperinsulinemic Conditions
2.4. Liraglutide Decreased the Formation of Alzheimer’s Markers and Plaque
2.5. Liraglutide Inhibited Beta Secretase 1 (BACE-1) Activity in Insulin-Induced Insulin Resistance in Neuronal Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
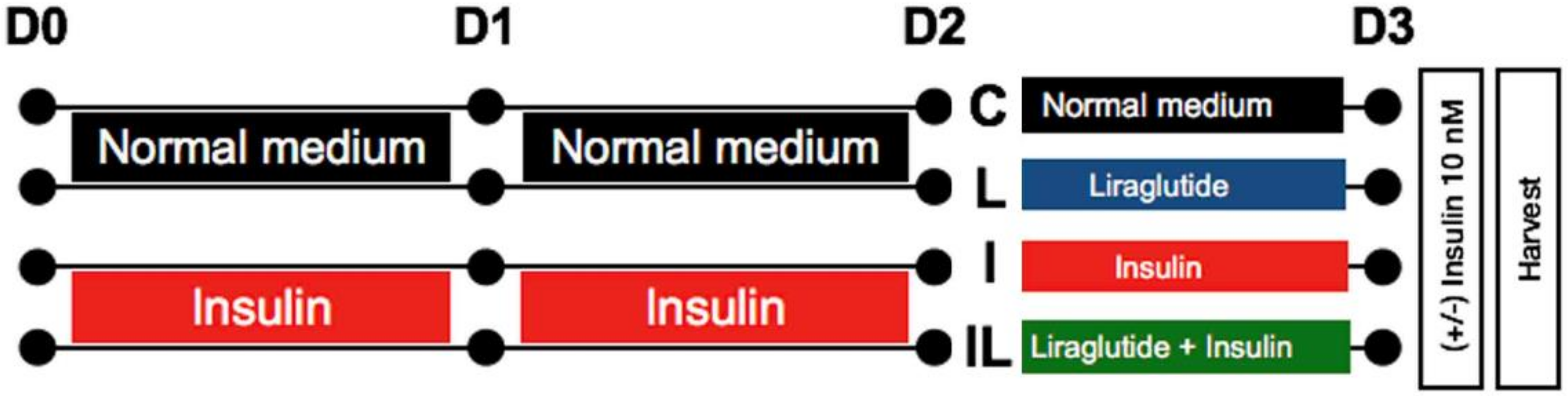
4.2. Neuronal Insulin-Resistant Induction and Liraglutide Treatment
4.3. Cell Viability Assay
4.4. Glucose Uptake Assay
4.5. Determination of Beta-Secretase (BACE-1) Activity
4.6. Immunoprecipitation
4.7. Western Blot Analysis
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| GLP-1 | Glucagon-like peptide-1 |
| Aβ | Amyloid-beta |
| T2 DM | Diabetes mellitus type 2 |
| IRs | Insulin receptors |
| IRS-1 | Insulin receptor substrate 1 |
| GSK-3β | Glycogen synthase kinase-3 beta |
| BACE-1 | Beta secretase 1 or β-site amyloid precursor protein cleaving enzyme 1 |
| PKB | Protein kinase B |
| APP | Amyloid Precursor Protein |
| NFTs | Neurofibrillary tangles |
References
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef]
- Dou, J.-T.; Chen, M.; Dufour, F.; Alkon, D.L.; Zhao, W.-Q. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 2005, 12, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.-L.; Chen, C.-M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers. Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. Insulin resistance and Alzheimer’s disease pathogenesis: Potential mechanisms and implications for treatment. Curr. Alzheimer Res. 2007, 4, 147–152. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Hölscher, C.; Li, L. New roles for insulin-like hormones in neuronal signalling and protection: New hopes for novel treatments of Alzheimer’s disease? Neurobiol. Aging 2010, 31, 1495–1502. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.-X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers. Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. The Link Between Tau and Insulin Signaling: Implications for Alzheimer’s Disease and Other Tauopathies. Front. Cell. Neurosci. 2019, 13, 17. [Google Scholar] [CrossRef]
- Starks, E.J.; Patrick O’Grady, J.; Hoscheidt, S.M.; Racine, A.M.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Okonkwo, O.C.; Puglielli, L.; Asthana, S.; et al. Insulin Resistance is Associated with Higher Cerebrospinal Fluid Tau Levels in Asymptomatic APOE ɛ4 Carriers. J. Alzheimer’s Dis. 2015, 46, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Alonso, A.D.C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Z.; Xia, Y.-Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers. Dis. 2013, 33 (Suppl. 1), S123–S139. [Google Scholar] [CrossRef]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef]
- Zhao, L. Insulin-Degrading Enzyme as a Downstream Target of Insulin Receptor Signaling Cascade: Implications for Alzheimer’s Disease Intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Welsh, G.I.; Proud, C.G. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2 B. Biochem. J. 1993, 294, 625–629. [Google Scholar] [CrossRef]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- Aravind, S.R. Adding a DPP-4 inhibitor to metformin therapy may be safer than you think. Curr. Med. Res. Opin. 2014, 30, 791–794. [Google Scholar] [CrossRef]
- Das, S.; Roy, P.; Pal, R.; Auddy, R.G.; Chakraborti, A.S.; Mukherjee, A. Engineered silybin nanoparticles educe efficient control in experimental diabetes. PLoS ONE 2014, 9, e101818. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, N. Review of current and emerging therapies in type 2 diabetes mellitus. Am. J. Manag. Care 2012, 18, S17–S26. [Google Scholar] [PubMed]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Jellinger, P.S. Focus on incretin-based therapies: Targeting the core defects of type 2 diabetes. Postgrad. Med. 2011, 123, 53–65. [Google Scholar] [CrossRef]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef]
- Han, W.-N.; Hölscher, C.; Yuan, L.; Yang, W.; Wang, X.-H.; Wu, M.-N.; Qi, J.-S. Liraglutide protects against amyloid-β protein-induced impairment of spatial learning and memory in rats. Neurobiol. Aging 2012, 34, 576–588. [Google Scholar] [CrossRef]
- Porter, D.W.; Kerr, B.D.; Flatt, P.R.; Holscher, C.; Gault, V.A. Four weeks administration of Liraglutide improves memory and learning as well as glycaemic control in mice with high fat dietary-induced obesity and insulin resistance. Diabetes. Obes. Metab. 2010, 12, 891–899. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Z.-F.; Holscher, C.; Gao, C.; Jiang, Y.-H.; Liu, Y.-Z. (Val8) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur. J. Pharmacol. 2012, 674, 280–286. [Google Scholar] [CrossRef]
- Smith, D.L.; Stinefelt, B.M.; Blemings, K.P.; Wilson, M.E. Diet-induced alterations in progesterone clearance appear to be mediated by insulin signaling in hepatocytes. J. Anim. Sci. 2006, 84, 1102–1109. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2013; pp. 295–300. [Google Scholar]
- Craft, S. Insulin resistance syndrome and Alzheimer disease: Pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis. Assoc. Disord. 2006, 20, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Craft, S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: Implications for treatment. CNS Drugs 2003, 17, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin resistance in Alzheimer’s disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. BACE1: The β-Secretase Enzyme in Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Trudeau, F.; Gagnon, S.; Massicotte, G. Hippocampal synaptic plasticity and glutamate receptor regulation: Influences of diabetes mellitus. Eur. J. Pharmacol. 2004, 490, 177–186. [Google Scholar] [CrossRef]
- Plyte, S.E.; Hughes, K.; Nikolakaki, E.; Pulverer, B.J.; Woodgett, J.R. Glycogen synthase kinase-3: Functions in oncogenesis and development. Biochim. Biophys. Acta 1992, 1114, 147–162. [Google Scholar] [CrossRef]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Zhang, X.; Song, W. The role of APP and BACE1 trafficking in APP processing and amyloid-β generation. Alzheimers. Res. Ther. 2013, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Sharma, M.K.; Jalewa, J.; Hölscher, C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5 Y cells exposed to methylglyoxal stress. J. Neurochem. 2014, 128, 459–471. [Google Scholar] [CrossRef]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Vilsbøll, T.; Deacon, C.F. The incretin system and its role in type 2 diabetes mellitus. Mol. Cell. Endocrinol. 2009, 297, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Patterson, S.; Porter, D.; Gault, V.A.; Holscher, C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J. Neurosci. Res. 2011, 89, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Greig, N.H. The glucagon-like peptides: A new genre in therapeutic targets for intervention in Alzheimer’s disease. J. Alzheimers. Dis. 2002, 4, 487–496. [Google Scholar] [CrossRef]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimers. Dis. 2010, 19, 1205–1219. [Google Scholar] [CrossRef]
- Perry, T.; Lahiri, D.K.; Chen, D.; Zhou, J.I.E.; Shaw, K.T.Y.; Egan, J.M.; Greig, N.H. A Novel Neurotrophic Property of Glucagon-Like Peptide 1: A Promoter of Nerve Growth Factor-Mediated Differentiation in PC12 Cells. J. Pharmacol. Exp. Ther. 2002, 300, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, J.; Ma, D.; Zhang, M.; Hu, S.; Shao, S.; Gong, C.-X. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J. Alzheimers. Dis. 2013, 37, 637–648. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Hölscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76, 57–67. [Google Scholar] [CrossRef]
- Potikanond, S.; Sookkhee, S.; Na Takuathung, M.; Mungkornasawakul, P.; Wikan, N.; Smith, D.R.; Nimlamool, W. Kaempferia parviflora Extract Exhibits Anti-cancer Activity against HeLa Cervical Cancer Cells. Front. Pharmacol. 2017, 8, 630. [Google Scholar] [CrossRef]
- Pitman, R.T.; Fong, J.T.; Billman, P.; Puri, N. Knockdown of the fat mass and obesity gene disrupts cellular energy balance in a cell-type specific manner. PLoS ONE 2012, 7, e38444. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jantrapirom, S.; Nimlamool, W.; Chattipakorn, N.; Chattipakorn, S.; Temviriyanukul, P.; Inthachat, W.; Govitrapong, P.; Potikanond, S. Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity. Int. J. Mol. Sci. 2020, 21, 1725. https://doi.org/10.3390/ijms21051725
Jantrapirom S, Nimlamool W, Chattipakorn N, Chattipakorn S, Temviriyanukul P, Inthachat W, Govitrapong P, Potikanond S. Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity. International Journal of Molecular Sciences. 2020; 21(5):1725. https://doi.org/10.3390/ijms21051725
Chicago/Turabian StyleJantrapirom, Salinee, Wutigri Nimlamool, Nipon Chattipakorn, Siriporn Chattipakorn, Piya Temviriyanukul, Woorawee Inthachat, Piyarat Govitrapong, and Saranyapin Potikanond. 2020. "Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity" International Journal of Molecular Sciences 21, no. 5: 1725. https://doi.org/10.3390/ijms21051725
APA StyleJantrapirom, S., Nimlamool, W., Chattipakorn, N., Chattipakorn, S., Temviriyanukul, P., Inthachat, W., Govitrapong, P., & Potikanond, S. (2020). Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity. International Journal of Molecular Sciences, 21(5), 1725. https://doi.org/10.3390/ijms21051725