1. Introduction
Monoclonal antibodies display exquisite targeting properties and have dramatically changed the way we diagnose and treat cancer. Labelling of antibodies with radiometals for imaging and radionuclide therapy traditionally requires antibody conjugation to a bifunctional chelator (BFC) able to bind covalently to an amino acid residue or glycan side-chain at one end and to efficiently coordinate the radiometal at the other. Examples of FDA-approved radiolabelled antibodies include the
90Y-labelled Ibritumomab tiuxetan for the radioimmunotherapy of B-cell non-Hodgkin lymphoma [
1] and the
111In-labelled capromab pendetide for prostate cancer radioimmunoscintigraphy [
2]. The concept has been extended in recent years to include radioimmunoPET by virtue of long-lived positron-emitting radionuclides such as zirconium-89 [
3].
One major limitation of radiolabelled antibodies is slow clearance arising from their large size (approximately 150 KDa, compared to ≤1 KDa for the radiolabelled BFC) and lack of domains that facilitate clearance by molecular recognition pathways. Together these result in longer blood half-life and slower extravasation and tissue penetration compared to the radiolabelled chelator on its own. Hence, the antibody circulates in the blood for days (48 h or even more) before reaching an acceptable tumour-to-blood ratio, leading to delayed imaging and prolonged radiation exposure of healthy tissues. In addition, this limits the choice of radiometals that can be used for imaging with antibodies to those possessing a long half-life comparable with the blood half-life of the immunoconjugate so that the activity has not decayed by the time sufficient contrast is achieved.
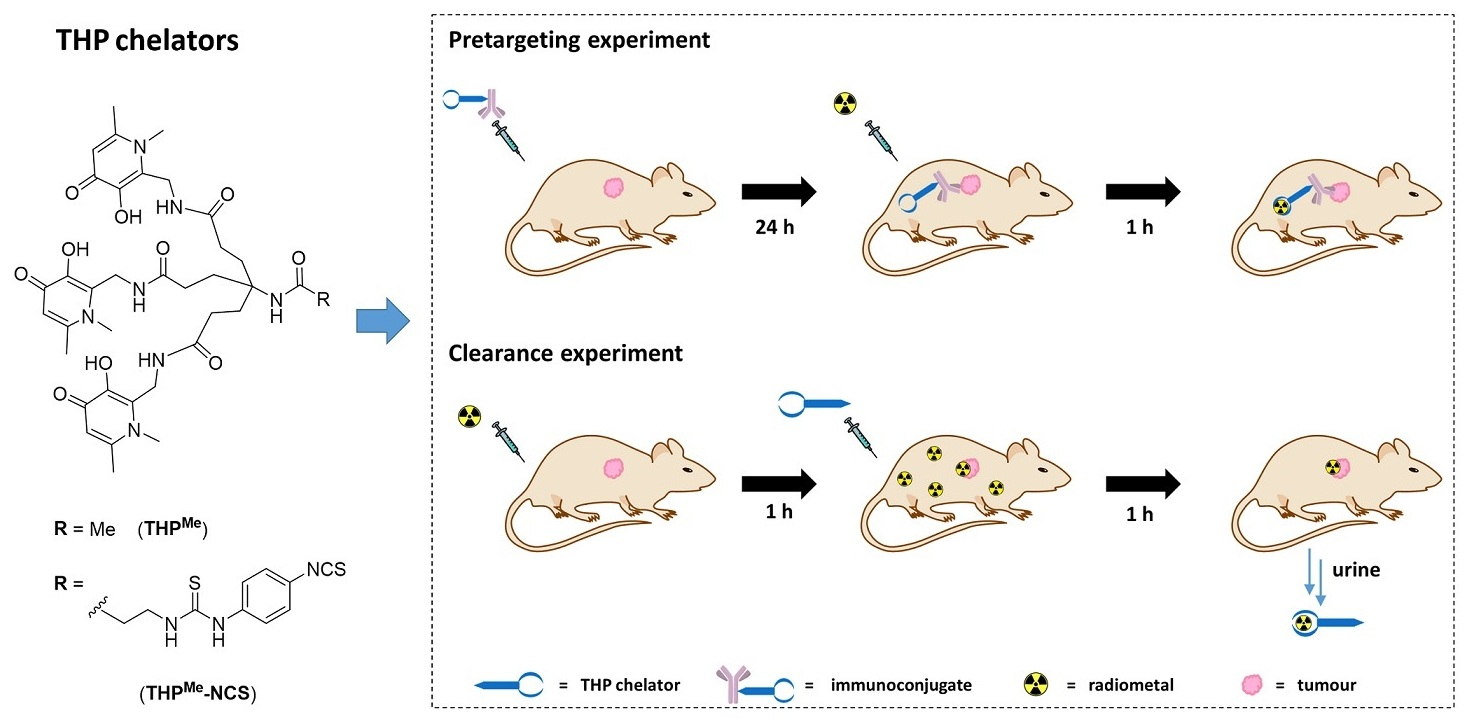
Pretargeting provides a unique approach to tackle this issue, by separating the inevitably slow targeting/clearance of the antibody from the delivery of the radioactive probe, which, owing to its smaller size, can be considerably quicker (minutes to hours). For a pretargeting approach to be successful, these two components should have great affinity for one another and very fast (preferably diffusion-limited) chemical association so that they are able to combine in vivo, at the tumour site, to give the fully functional radiolabelled antibody [
4].
Some pretargeting strategies that have been successfully investigated and translated into clinical trials have used bispecific antibodies/haptens or biotin/avidin as the high-affinity chemical pair [
5]. Other promising approaches are based on complementary oligonucleotide pairs, and bio-orthogonal click chemistry [
6]. Notably, the common feature of all these strategies is that part of the connecting moiety is incorporated into the antibody and the other (chelator) part is radiolabelled separately. This radiolabelled probe is administered at a later stage when the excess circulating antibody has cleared from the blood. The optimal time between the two administrations can be estimated by imaging with a directly radiolabelled antibody.
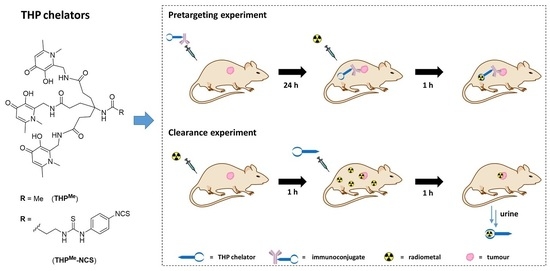
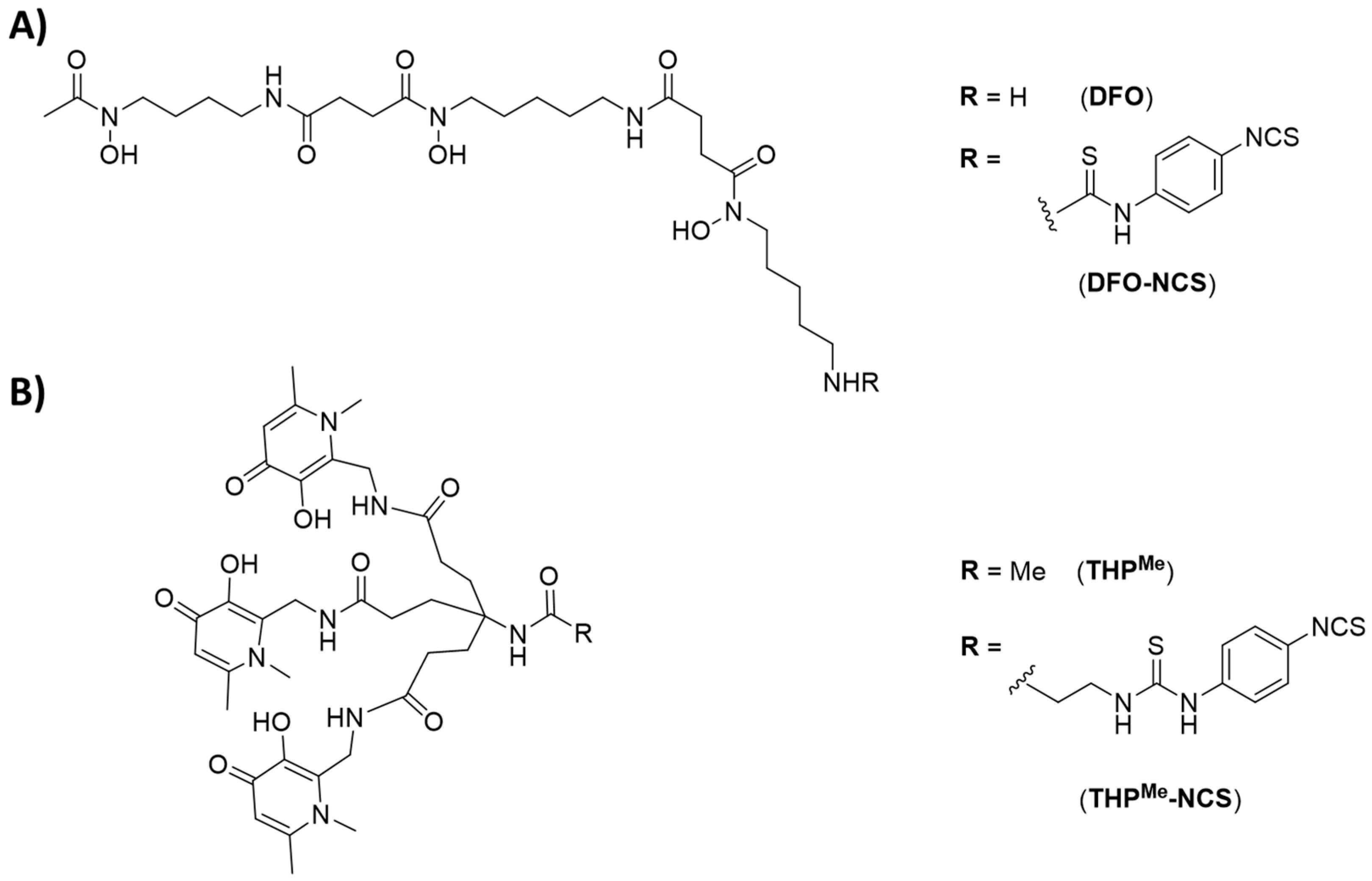
We previously demonstrated that the tris(hydroxypyridinone) chelator THP
Me (
Figure 1A) and its second-generation analogue THP
H possess high thermodynamic affinity for the short-lived radiometal
68Ga (pGa = 30.0 for THP
Me at physiological pH) [
7,
8] and were able to complex
68Ga very rapidly at neutral pH even at very low chelator concentration. Notably, THP
Me was able to bind
68Ga in serum, and the radiolabelled complex [
68Ga(THP
Me)] is stable in the presence of apotransferrin [
7,
9]. These outstanding radiolabelling properties, already exploited as a basis for rapid, one-step kit-based
68Ga radiolabelling of both small-molecule and protein-based PET radiopharmaceuticals [
10,
11,
12], allow them to bind
68Ga
3+ in vivo, when administered i.v. to mice previously injected with
68Ga (as confirmed by radioHPLC analysis of the urine) despite the in vivo conditions of extreme dilution and competing affinities [
8]. Thus,
68Ga
3+ and THP
Me (or THP
H) represent a potential chemical pretargeting pair. Notably, several BFC have been developed based on THP
Me, including isothiocyanate (THP
Me-NCS,
Figure 1A) and maleimide derivatives that can effectively bind to lysine or cysteine side chains, respectively, in mild conditions (aqueous solution, mildly basic pH, room temperature), allowing facile conjugation to antibodies [
10,
13,
14].
We therefore hypothesised that a different pretargeting strategy, based on metal chelation, could be feasible using a radiometal (
68Ga
3+) and a BFC (THP
Me-NCS,
Figure 1A) as a high-affinity chemical pair. In this pretargeting approach, the whole BFC would be attached to the antibody and then radiolabelled in vivo at the tumour site as schematically represented in
Figure 2A. This strategy would minimise handling of the radioactivity, compared to traditional pretargeting approaches, since no radiosynthetic work is required other than buffering the radiometal. This would in turn reduce operators’ exposure and greatly simplify the clinical protocol so that it could be performed in hospitals lacking extensive radiochemistry facilities and expertise.
Herein, we tested the pretargeting by metal chelation hypothesis in a murine xenograft model of human colorectal cancer (SW1222) utilising THP
Me-NCS-conjugated huA33 antibody as a pretargeting vector and acetate-buffered
68Ga as the radioactive species. The transmembrane glycoprotein A33 targeted by huA33 is expressed in 95% of colorectal carcinomas [
15] (including the colorectal cancer cell line SW1222 used in this work). It remains on the cell surface upon binding to the antibody and is not internalised or shed. Therefore, this antibody/antigen couple represents an ideal model for pretargeting, as already demonstrated in preclinical studies of the bio-orthogonal click chemistry pretargeting approach [
16,
17,
18]. We also evaluated the potential of THP
Me as a means of quickly removing background radioactivity after administration of
68Ga
3+ salts, which similarly relies on efficient in vivo chelation of
68Ga
3+ by THP
Me (
Figure 2B).
2. Results
DFO is an FDA-approved treatment for iron overload [
19] and had also been investigated in the past as a blood-clearance agent for
67Ga imaging, owing to the similarity between high-spin Fe(III) and Ga(III) [
20,
21]. In competition studies in vitro, THP
Me was shown to out-compete DFO for binding to
68Ga
3+ [
22]. Nevertheless, it was important to investigate, prior to undertaking pretargeting studies, whether DFO would be a better candidate than THP
Me to test the pretargeting via metal chelation hypothesis in vivo. The in vivo Ga(III) binding abilities of THP
Me and DFO were compared by injecting mice with each of these chelators in an equimolar dose (24 nmol), or saline as a negative control, 1 h after
68Ga
3+ administration.
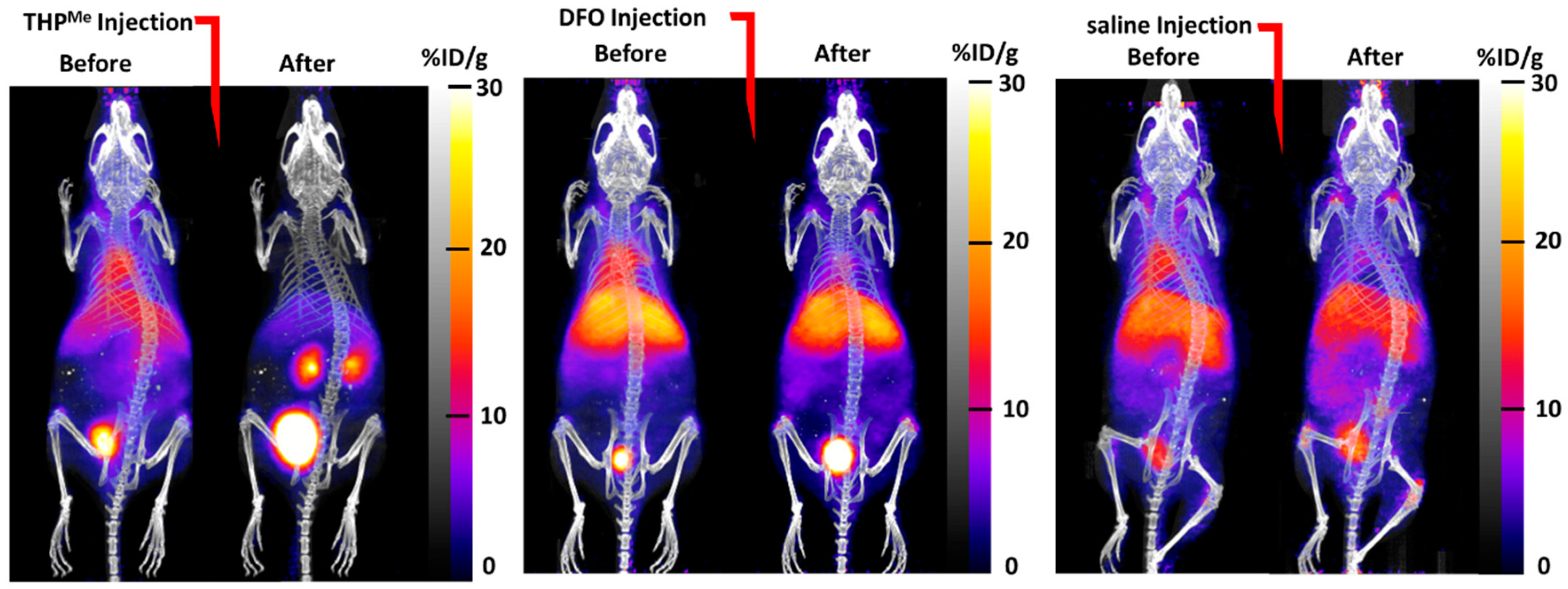
The maximum intensity projection (MIP) images (
Figure 3) show how biodistribution of the activity was markedly affected by injection of THP
Me compared to saline: there was a sudden and dramatic increase in renal clearance, while the effect of DFO was less visible. When comparing ex vivo biodistribution, THP
Me showed a statistically significant increase in
68Ga accumulation in kidneys compared to saline (%ID/g = 112 ± 43 for THP
Me treated mice vs. 6.2 ± 2.4 for saline group), while no such effect was visible for DFO (%ID/g in kidneys = 6.7 ± 2.9). On the other hand, DFO caused a reduced and less variable gallium accumulation in femur and liver (%ID/g in femur = 6.8 ± 1.2, liver = 17.5 ± 7.6) compared to the control group (%ID/g in femur = 15 ± 11, liver = 22 ± 19), although to a lesser extent than for THP
Me (%ID/g in femur = 2.8 ± 0.5, liver = 12.5 ± 3.0) (
Figure S1). Even though no statistically significant difference was detected between the chelators and control groups for these organs, likely due to the large variability observed for the saline group, these data suggest that DFO is also, to some extent, able to bind circulating
68Ga under these conditions.
Overall, these results support our hypothesis that THPMe is equal or superior to DFO in binding 68Ga3+ effectively in vivo and clearing it renally, and suggest that THPMe would be a promising candidate for testing our pretargeting via metal-chelation approach.
A dose-finding study was also conducted to verify that THP
Me maintained its ability to bind
68Ga in vivo even when its concentration was significantly reduced. Mice were injected with
68Ga and then with different concentrations of the chelator. Notably, a significantly different
68Ga biodistribution compared to the saline-only control was observed even for the lowest dose considered of THP
Me (1 μg, 1.2 nmol), and almost no difference was found between the 5 μg (6 nmol) and 20 μg (24 nmol) groups for most organs (
Figure S2). This indicated that THP
Me radiolabelling in vivo was effective even at low micromolar concentration of chelator, suggesting that radiolabelling of a pretargeted tumour would be feasible.
2.1. Immunoconjugate Characterisation and Radiolabelling
Conjugation of THP
Me-NCS to the huA33 was initially performed using 5–10 molar equivalents of bifunctional chelator, following modified published procedures [
23]. Conjugation yield was calculated based on measurements of the antibody concentration after immunoconjugate purification via size exclusion chromatography (
Table S1). Matrix-assisted laser desorption-ionisation (MALDI) analysis of the immunoconjugates compared to the native antibodies (
Figure S3) allowed us to determine average chelator-to-antibody ratios (degree of labelling, DOL) of 0.6 and 0.4 achieved for the 10 and 5 molar equivalents batches, respectively (
Table S1).
Radiolabelling of the THP
Me-NCS-huA33 conjugate was initially performed at a slightly acidic pH (pH 5.5–6, as measured using pH paper) obtained by neutralisation of the generator eluate
68Ga with ammonium acetate buffer, as previously reported for this chelator [
7,
13]. The radiochemical yield of the crude product was measured by ITLC using 50 mM EDTA as a mobile phase (
Figure S4). In addition, the radiolabelled immunoconjugate was run through a PD10 size exclusion column to rule out the presence of scarcely soluble Ga(OH)
3 species, which would not have been revealed under the ITLC conditions utilised, but would be trapped on the size exclusion column. Quantitative radiolabelling was obtained for both 5 molar equivalents and 10 molar equivalents batches at antibody concentrations as low as 1 μM, confirming successful conjugation. Under the same conditions the native antibody did not show radiolabelling, confirming that the THP
Me- NCS chelator is necessary for efficient
68Ga binding to the antibody.
Radiolabelling of the immunoconjugate was also investigated in conditions that more closely resemble those of preclinical experiments. Neutral pH is desirable for intravenous injection in conscious mice and the pH experienced by the radiometal in vivo is also close to neutral. Therefore, the effect of eluate neutralisation to pH 7–7.2 on radiolabelling was evaluated. In addition, to take into account the time interval between eluate neutralisation and injection of the mice, the effect of a 45 min delay between eluate neutralisation and radiolabelling of the 10 equivalent batch was investigated (
Figure S5). Remarkably, even in this case radiolabelling was quantitative with radiochemical yield >95%, consistent with previous reports of highly efficient neutral-pH labelling of proteins conjugated with THP
Me [
10].
Finally, to verify that conjugation to the chelator did not impair the antibody’s ability to bind to its target receptor, the immunoreactive fraction was determined for both the 5 and 10 molar equivalents batches via an immunoreactivity assay (
Table S1) [
24]. In this experiment, a large excess of antigen is employed, able to bind the totality of the added radioimmunoconjugate (≈ 100% uptake) if its immunoreactivity is undiminished compared to the unmodified antibody. Accordingly, the observed uptake percentage reflects the immunoreactive fraction of the radioimmunoconjugate batch. For both the batches used in pretargeting experiments, only marginal detriment of the immunoreactivity was found, with immunoreactivity fractions equal or superior to 90%. Based on these results, the 10 molar equivalents batch was selected to perform preclinical experiments.
2.2. In Vivo Pretargeting Experiments
Preclinical experiments were performed in nude mice bearing human SW1222 colon cancer xenografts. Different groups, of five mice each, were considered (
Table 1), as discussed below.
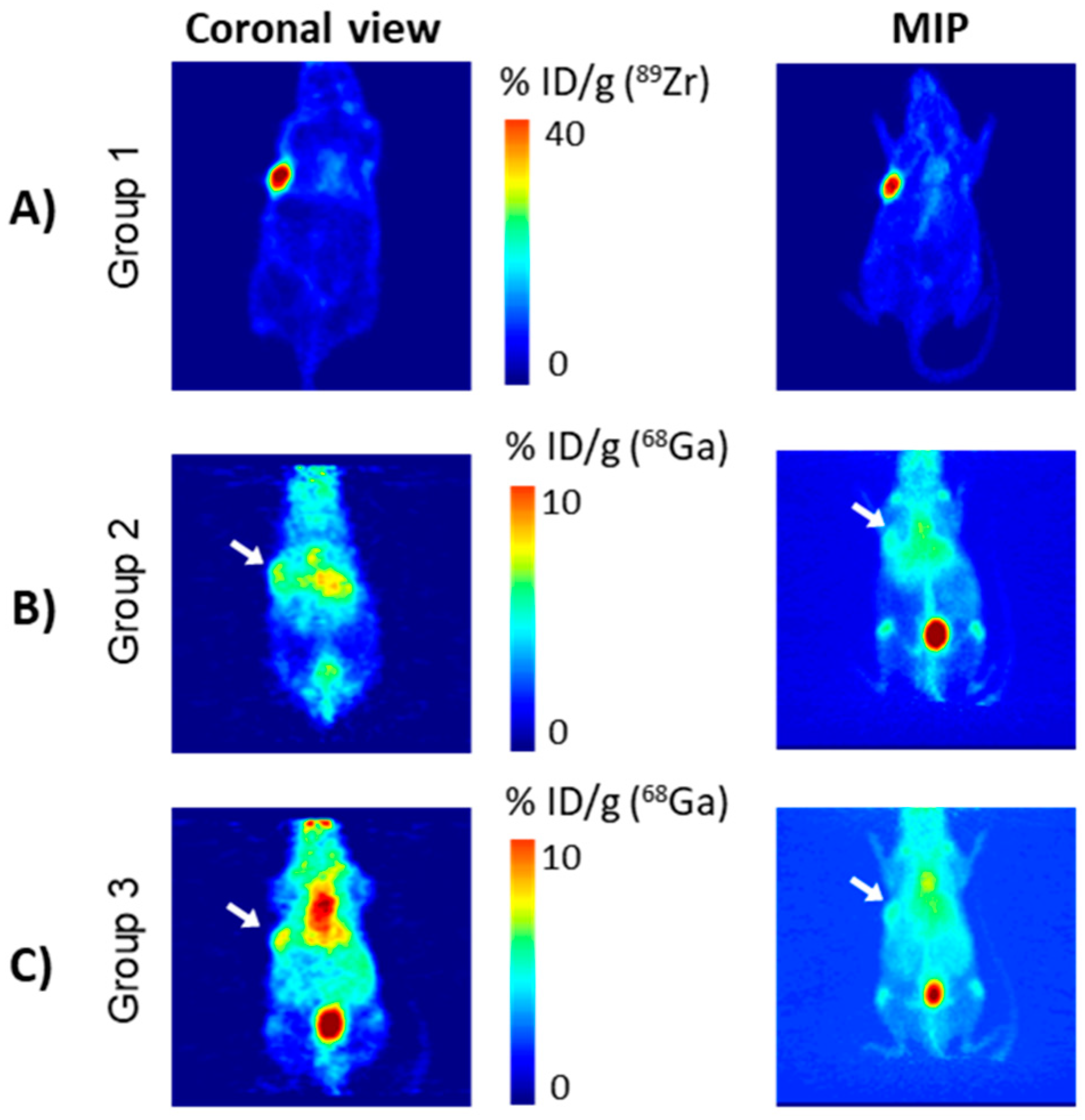
Notably, the use of directly radiolabelled 68Ga-THPMe-NCS-huA33 as a positive control was precluded by the short half-life of the radiometal. Therefore, prior to conducting pretargeting experiments using 68Ga and THPMe-NCS-huA33, PET imaging of the same tumour model was undertaken with directly labelled 89Zr-DFO-NCS-huA33 immunoconjugate, to verify that the xenografts expressed the A33 antigen targeted by the huA33 antibody and that 24 h was sufficient time for the radioimmunoconjugate to localise at the tumour site and clear from circulation.
Radiolabelling of the DFO-NCS-huA33 immunoconjugate with
89Zr was achieved in nearly quantitative yield and purity, as confirmed by ITLC analysis of the fractions eluted from post-radiolabelling size-exclusion chromatography columns (
Figure S6). Imaging of mice injected with
89Zr-labelled DFO-NCS-huA33 immunoconjugate (group 1), performed at 25 h following radioimmunoconjugate administration, showed selective tumour localisation of the radioimmunoconjugate (
Figure 4A).
Mice in group 2 (the pretargeting group) were pretreated with 100 μg of the THP
Me-NCS-huA33 conjugate (10 eq. batch) followed, after 24 h, by acetate-buffered
68Ga (pH ≈ 7) generator eluate. To ensure uniformity in the Ga
3+ speciation at the time of injection, all mice were injected with
68Ga at the same time (10 min range). Therefore, imaging of the mice had to be staggered with respect to the injection times, with the first mouse scanned at 50 min after the injection of
68Ga and the last scanned at 90 min after injection. The scanning was then repeated after one further hour to investigate the effect of a longer delay between
68Ga injection and PET imaging on image quality and contrast. A representative PET scan at 130 min after
68Ga administration is shown in
Figure 4B; the 70 min time point for the same mouse and a representative PET scan for a second mouse are also reported in the
supplementary information (Figure S7). Ex vivo biodistribution was then determined at 3 h after
68Ga injection and is reported in
Figure 5 (red histograms). Both the PET scans and the ex vivo biodistribution data showed some accumulation of
68Ga in the tumour (≈11 %ID/g). However, the tissue with the highest radioactivity concentration was blood even at 3 h after the
68Ga injection. Uptake in non-target organs, such as liver and bones (typical of the in vivo behaviour of unchelated gallium [
25]), was also visible.
As a negative control, five mice (group 3) were injected with acetate buffered
68Ga, without prior injection of antibody-chelator conjugate. Imaging and ex vivo biodistribution were performed as in the pretargeting group (group 2) and, notably, showed very similar
68Ga biodistribution to those in group 2, as evident in the PET images (
Figure 4C and
Figure S8).
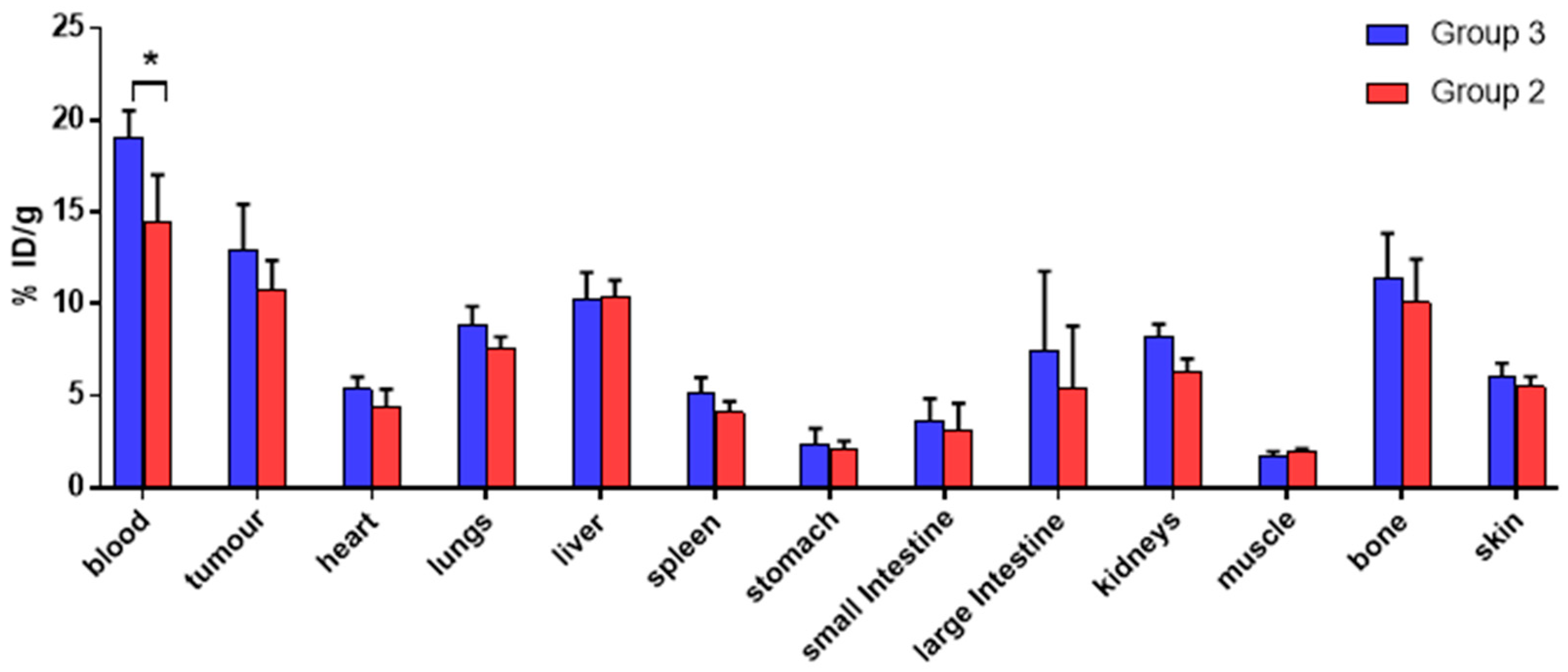
A comparison of the ex vivo biodistribution of
68Ga in groups 2 and 3 (
Figure 5,
t-test performed for each organ) also revealed no significant difference in tumour
68Ga uptake for the two groups (10.8 ± 1.5 %ID/g in group 2 vs. 12.9 ± 2.5 %ID/g in group 3). The only significant difference among the organs examined was found for blood, whose uptake was higher for group 3 (19.1 ± 1.4 %ID/g) compared to group 2 (14.4 ± 2.6 %ID/g). Overall, these results suggest that the observed activity accumulation in the tumour xenografts in group 2 is due to direct uptake of unchelated “free”
68Ga and is not mediated by antibody pretargeting and subsequent in vivo radiolabelling of the antibody-chelator conjugate.
As in vivo radiolabelling efficacy is expected to be highly dependent on local accessible chelator concentration, we hypothesised that failure to observe a pretargeting effect could be explained by insufficient local concentration of the chelator on the tumour surface. Therefore, in a second attempt at pretargeting we repeated the immunoconjugation with higher molar equivalents of chelator, aiming to increase the average chelator/antibody ratio of the conjugate and, hence, the concentration of chelator available at the tumour cell surface for in vivo radiolabelling. A first attempt at antibody-chelator conjugation using 50 molar equivalents of chelators resulted in precipitation of the immunoconjugate and was therefore abandoned; while an immunoconjugation performed using 30 equivalents of THP
Me-NCS was successful. MALDI revealed a modest increase in the average chelator-to-antibody ratio of the resulting immunoconjugate from 0.6 (for the 10 molar equivalents batch) to 1.0, the highest ratio achievable without precipitation (
Table S1). As a result, this new batch was utilised in a second pretargeting experiment. However, no significant difference was found in the tumour uptake comparing the 30 molar equivalents pretargeting group (group 4) and the group 3 controls (
Figures S9 and S10). Instead, ANOVA analysis of the data revealed lower bone uptake for the pretargeting 30 eq. group (6.0 ± 1.1 %ID/g) compared to both the control group 3 (11.4 ± 2.4 %ID/g) and to group 2 (10.1 ± 2.4 %ID/g).
2.3. Exploiting the In Vivo Chelation Strategy to Increase Contrast
Although our initial pretargeting by metal chelation strategy proved unpromising in this experimental setup, it was interesting to notice the relatively high tumour accumulation of free gallium in this xenograft model (>10 %ID/g), comparable to values observed when targeting tumours with
68Ga-radiolabelled peptide (see for example [
12]), although the tumour/blood ratio was nevertheless <1. Noting that our previous studies showed that the in vivo radiolabelling of THP
Me had a profound impact on the biodistribution of
68Ga
3+ [
8], significantly accelerating its renal clearance in the urine, we decided to evaluate the use of THP
Me as a contrast-enhancing agent in the same model. In this additional experiment, five mice (group 5) were administered acetate-buffered
68Ga followed, at 1 h post injection, by THP
Me (20 μg, 24 nmol), aiming to clear the radiometal selectively from blood, thus increasing the tumour-to-blood activity ratio.
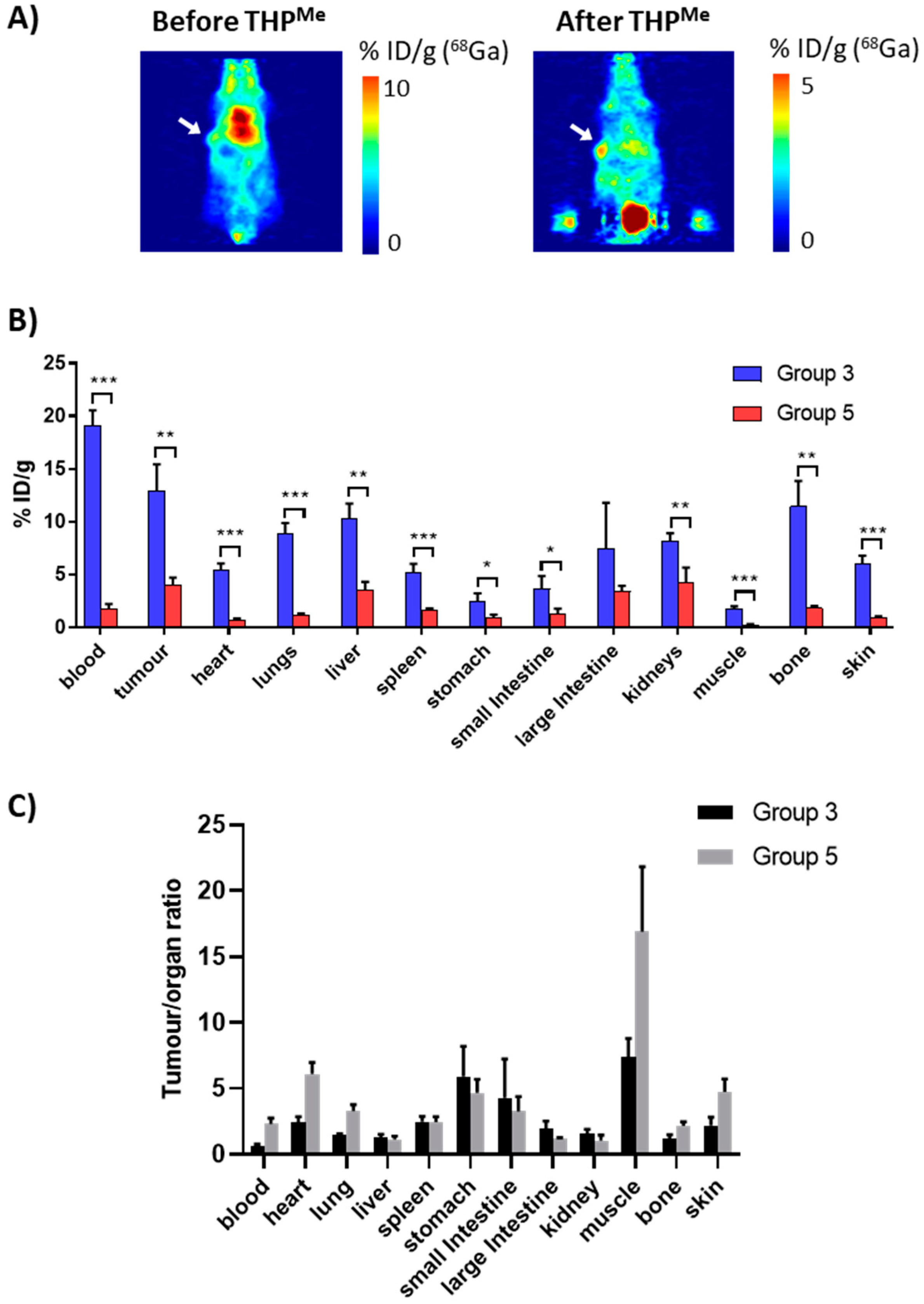
Mice were imaged from 20 min after THP
Me injection, with two of them also imaged between the
68Ga and the THP
Me injection, in order to compare the PET images before and after THP
Me-mediated blood clearance in the same animal. An exemplar PET image is presented in
Figure 6, together with the ex vivo biodistribution (3 h after
68Ga injection) compared to the
68Ga control group (group 3). Calculated
p-values and tumour/organ ratios for each organ are reported in
Table S2.
Notably, both PET images and ex vivo biodistribution data revealed the dramatic effect of THP
Me injection, which significantly decreased the uptake of
68Ga in most organs. A
t-test revealed significantly reduced uptake in all organs except the large intestine, probably as a result of the larger variability in the %ID/g for this organ. Interestingly, the clearance effect is far more noticeable for the blood than for the tumour and many other tissues (
Figure 6B); the tumour-to-blood ratio increased from 0.7 in the
68Ga-only control to 2.3 in the blood clearance experiment (> threefold increase) and similarly the tumour-to-muscle ratio from 7.4 to 16.9 (
Figure 6C).
3. Discussion
In this work we performed an in vivo investigation of a new pretargeting hypothesis based on metal chelation, where the high-affinity chemical pair is represented by a radiometal and a chelator anchored to the antibody. The in vivo biodistribution of radiolabelled antibody
89Zr-DFO-NCS-huA33, whose ability to target A33 has been extensively validated [
17], was used here as a positive control. This experiment confirmed that the xenografts expressed the desired glycoprotein and that the batch of huA33 antibody used in this work was able to selectively accumulate in the tumour tissue, and to do so in the 25 h between injection of the antibody and imaging, in agreement with previous findings [
17]. The radiometal-chelator chemical pair utilised for the pretargeting studies consisted of
68Ga
3+, administered in an acetate buffered solution, and the THP
Me-based chelator THP
Me-NCS coupled to the antibody [
7,
13]. The ability of THP
Me to capture
68Ga
3+ in vivo has been previously reported and was corroborated here with a more thorough examination [
8], including a comparison with the clinically established iron chelator DFO. This highlighted the superior Ga
3+ binding ability of THP
Me, while a dose-finding study confirmed successful in vivo chelation of
68Ga(III) even at a very small (1.2 nmol) dose of chelator.
Amongst the several available bifunctional derivatives of THP
Me [
7,
13], for our pretargeting studies we decided to use the isothiocyanate derivative THP
Me-NCS [
13], which can non-specifically bind to lysine residues in the antibody. This approach was preferred to site-specific conjugation because it allows, in principle, the attachment of a higher number of chelators to the antibody. In addition, previous pretargeting studies found no improvement in image contrast and quality when site-specific rather than non-site-specific conjugation was used in the same animal model [
16].
Conjugation of the selected antibody was initially performed using 5 and 10 molar equivalents of THP
Me-NCS and its success was demonstrated by efficient
68Ga radiolabelling of the purified immunoconjugates, but not of the native antibody, in the mild conditions typically used for
68Ga labelling of THP
Me bioconjugates [
7,
13]. The modified antibody was radiolabelled quantitatively, without need of purification, at neutral pH and immunoconjugate concentration as low as 1 μM. Moreover, the radiolabelling efficiency was maintained after pre-neutralisation of the eluate 45 min before radiolabelling—conditions that mimic the conditions of the in vivo pretargeting experiment and that often impair radiolabelling when other chelators are used. Testing under these conditions was important to rule out any effect of different gallium speciation (e.g., colloid formation) over the pH range or time when considering the results of preclinical experiments. In fact, in our experience (unpublished results), unchelated gallium often displays a variable in vivo behaviour after the pH has been raised above 3, presumably owing to the multitude of species, including potentially colloidal gallium hydroxides, that may be present in these conditions. An immunoreactivity assay also gave encouraging results, showing only a marginal impairment of the immunoreactivity towards SW1222 cells for both the radiolabelled immunoconjugate batches. MALDI analysis of the immunoconjugates showed that, despite the excess of chelator used in the conjugation process, a modest chelator/antibody ratio of 0.6 was achieved when 10 molar equivalents of the THP
Me-NCS were used. This is an average value, and likely includes immunoconjugate molecules with fewer and more chelators attached.
In view of its higher chelator/antibody ratio compared to the 5 molar equivalents batch, and its similar immunoreactivity, the 10 molar equivalents batch was used for a first pretargeting experiment. Based on our prior experience with the click chemistry-based pretargeting strategy [
17], it was decided to use a 100 μg dose of the immunoconjugate. Unfortunately, neither in vivo imaging nor ex vivo biodistribution highlighted any significant difference between the pretargeted and the negative control (
68Ga-only) group, suggesting that the tumour uptake first noticed in the pretargeting group was solely attributable to uptake of unchelated
68Ga in the tumour. Accumulation of gallium in some tumours is a known phenomenon, probably in part due to its similarity to Fe(III), which allows gallium to bind to transferrin and hence delivery to tissues overexpressing transferrin receptors [
26]. This effect has been exploited in the past to image some types of cancer, as well as inflammation/infections, with
67Ga scintigraphy or SPECT [
27]. The long-lived
67Ga isotope (in the form of
67Ga citrate) is preferred for these scans since a substantial target accumulation and contrast is only achieved after several hours or days. The use of
68Ga-citrate in PET imaging of lung and prostate cancer, and of bone infections, has been recently evaluated in clinical trials [
28,
29,
30].
In an attempt to increase chelator concentration at the tumour site, a second pretargeting experiment was performed using a new immunoconjugate batch (30 molar equivalents) with an average chelator/antibody ratio of 1.0, the maximum achievable without partial precipitation of the immunoconjugate. This did not lead to any increase in the tumour uptake of 68Ga compared to the 68Ga-only control. On the other hand, a decreased uptake in bones was evident for this fourth group of animals and uptake in liver and large intestines was also reduced although to a lesser extent. Since all these organs are targets for accumulation of free gallium (especially bone), the decreased uptake suggests that part of the gallium may actually bind the antibody in vivo, but in circulation rather than at the tumour site. This effect could be exacerbated by the non-homogeneous chelator-to-antibody ratio of the immunoconjugate: although MALDI measurements gave a single average value, this is likely to represent a mixture of molecules with higher and lower ratios. Antibodies with more chelators attached are likely to have a lower immunoreactivity compared to those with a lower number of chelators and so are less likely to bind to their target antigen, but more likely to bind 68Ga. These immunoconjugate molecules could be radiolabelled while in circulation, diminishing 68Ga accumulation in bones/liver/intestine without significantly altering the tumour uptake, which is largely attributable to unchelated 68Ga.
Overall, no pretargeting-related tumour uptake was observed in these in vivo experiments and notably, no improvement in tumour uptake was visible in group 4 (DOL = 1) compared to group 2 (DOL = 0.6). If failure to see any pretargeted 68Ga uptake were a consequence of impaired affinity of the immunoconjugate, that would rule out the THPMe-NCS chelator for pretargeting purposes. If instead, this was due to an insufficient number of chelator molecules per antibody, no immediate solution presents itself, as attempted conjugation of more than one THPMe-NCS moiety per antibody resulted in precipitation of the immunoconjugate, likely due to aggregation. Therefore, we can conclude that the pretargeting via 68Ga chelation is not a viable option when using THPMe-NCS.
As a possible strategy to achieve higher DOL in future investigations, the use of the dendritic THP
Me system could be explored, to conjugate multiple chelators at a single point of attachment, thus increasing the concentration of chelator available at the tumour site [
31].
Notably, although unsuccessful from a pretargeting point of view, these studies highlighted a significant amount of uptake of unchelated gallium in SW1222 tumours as early as 1–2 h after injection (≈13 %ID/g). This led us to evaluate the use of THP
Me as a blood clearance agent to increase the tumour-to-blood activity (and hence tumour-to-background) ratio. Notably, in this experiment THP
Me was able to clear
68Ga from the blood, leading to renal excretion, much more efficiently than from the tumours, resulting in a threefold enhancement of the tumour-to-blood activity ratio (shifting its value from <1 to >2) as calculated from the ex vivo biodistribution data at 3 h; ratios of tumour to other tissues were also enhanced. In agreement with these data, PET images of the mice before and after THP
Me treatment showed increased image contrast and better tumour visualisation. These results together suggest that the SW1222 colon carcinoma could be a good model to further investigate the use of unchelated
68Ga to image tumours with THP
Me as a blood-clearing agent. While THP
Me itself has not been evaluated in humans yet, one of its derivatives, THP
Me-PSMA labelled with
68Ga, is in phase III clinical trials for imaging prostate cancer suggesting low toxicity of the THP
Me family and potential for clinical translation. The results also suggest that the hydroxypyridinone prototype deferiprone, an iron-chelating drug used safely in patients to treat iron overload, might likewise warrant evaluation as Ga
3+ blood clearance agent as previously investigated [
32].
Overall, the use of a Ga3+ blood-clearing agent could potentially be very useful in a clinical setting, especially for those countries relying on 67Ga-citrate imaging, allowing better contrast to be achieved, reducing time between radiotracer administration and imaging and perhaps allowing the tumour-targeting properties of gallium citrate to be exploited for PET imaging with 68Ga.
4. Materials and Methods
4.1. General Equipment and Consumables
THP
Me-NH
2 was obtained from Chematech (Dijon, France). THP
Me was synthesised according to published procedures [
7]. The fully humanised IgG1 huA33 was obtained from the Ludwig Institute and the Olivia Newton-John Cancer Research Institute [
33]. DFO-NCS was obtained from Macrocyclics Inc. (Plano, TX, USA) and the DFO-NCS-huA33 conjugate used for
89Zr radiolabelling was produced according to published procedures [
17]. Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich and used without further purification. All the buffers used for the huA33 experiments were prepared using Chelex-treated deionised water.
NMR spectra were acquired on a Bruker Advance 400 spectrometer (Bruker, Billerica, MA, USA) with a 5 mm QNP probe at 400.13 MHz, with chemical shifts referenced to the solvent peak. High resolution ESI-MS data were acquired on an Exactive Orbitrap Mass Spectrometer (Thermo Scientific, Waltham, MA, USA). Data were acquired and reference mass-corrected via a dual-spray electrospray ionisation source, using the factory-defined calibration procedure. Preparative HPLC was carried out using an Agilent Zorbax XDB-C18 column (21.2 × 150 mm, 5 µm) with a 5 mL/min flow rate and UV detection at 214 nm on an Agilent 1200 LC system (Agilent, Santa Clara, CA, USA). Mobile phase A was water with 0.1% trifluoroacetic acid (TFA), and mobile phase B was acetonitrile with 0.1% TFA. A gradient from 0% to 100% B (1%/min) was employed.
68Ga was obtained from Eckert and Ziegler generator, by elution with 5 mL of 0.1 M ultrapure HCl (Fluka-Honeywell, Charlotte, NC, USA). The eluate was collected in ≈1 mL fractions and the activity of all fractions was measured with a dose calibrator (Capintec, Florham Park, NJ, USA). The highest activity fraction was used for radiolabelling.
89Zr was produced at Memorial Sloan Kettering Cancer Center (MSKCC, New York, NY USA), through proton-beam bombardment of yttrium foil, and isolated as
89Zr-oxalate [
34]. Uptake of radioactivity in SW1222 cells and organs was measured using a LKB Wallac 1282 Compugamma Gamma-counter (LKB, Vienna, Austria) at King’s College London (KCL), an Automatic Wizard gamma-counter (Perkin-Elmer, Waltham, MA, USA) at MSKCC.
4.2. Synthesis of THPMe-NCS
Bn-THP
Me-NCS (now commercially available from Chematech, Dijon, France) was synthesised according to published procedures [
13]. Briefly,
p-phenylene-NCS (135 mg, 0.77 mmol) and DIPEA (75 μL, 0.44 mmol) were added to a solution of THP
Me-NH
2 (75 mg, 0.077 mmol) in DMF (2 mL) under stirring. After 15 min the DMF was removed under high-vacuum and the crude residue purified by preparative HPLC, yielding 43.7 mg of Bn-THP
Me-NCS (0.035 mmol, 45% yield). Benzyl deprotection was then performed by addition of 5 mL of BCl
3 solution (1 M in dichloromethane) and stirring under nitrogen followed by quenching with methanol on ice, solvent removal by rotary evaporation and purification via preparative HPLC to give 27.2 mg of THP
Me-NCS (0.028 mmol, 80% yield). Product purity was verified by
1H NMR and high-resolution mass spectrometry.
1H NMR (400 Mhz, CD3OD) δ: 1.89 (t, 6H, CH2-CH2CONH-pyridinone), 2.14 (t, 6H, CH2-CH2-CONH-pyridinone), 2.40 (t, 2H, SCNH-CH2-CH2-CONH), 2.48 (s, 9H, 6-CH3), 3.83 (s broad: 9H, N-CH3, and 2H, SCNH-CH2-CH2-CONH), 4.59 (s, 6H, CONH-CH2-2), 6.86 (s, 3H, 5-H (pyridinone)), 7.12 (d, 2H, SCN-mPh), 7.33 (d: 2 H, SCN-oPh).
High. Res. ESI-MS: m/z for C45H56N10O10S2 [M+H]+ calc: 961.3695, found: 961.3684, [M+2H]+ calc: 481.1884, found: 481.1877. No other peaks were observed.
4.3. HuA33 Conjugation to THPMe-NCS
A solution of huA33 in PBS (7 μM, 0.8 mL, pH 7.4) was adjusted to pH 8.8–9.0 with a 0.1 M solution of Na
2CO
3, and 5, 10, 30 or 50 equivalents of THP
Me-NCS (10 mM in DMSO) were added in small aliquots to prevent precipitation. The reaction mixture was shaken for 1 h at room temperature, then purified by size-exclusion chromatography on a PD-10 column (Sephadex G-25M PD-10 column, GE Healthcare, Chicago, IL, USA) eluting with ammonium acetate 0.2 M (for preliminary radiolabelling) or PBS (for preclinical experiments) and concentrated using centrifugal filtration units with a 50,000 molecular weight cut off (Amicon
TM ultra 2 mL, Millipore Corp, Burlington, MA, USA). The final concentration of the immunoconjugate was measured spectrophotometrically using a Biospec nano (Shimadzu, Kyoto, Japan), considering ε
280 = 2.1 × 10
5 L/mol·cm, and was used to determine the conjugation yield (
Table S1).
4.4. MALDI-ToF Mass Spectrometry
THPMe-NCS-huA33 immunoconjugates were analysed by MALDI-ToF MS/MS using a Bruker ultraflex MALDI-ToF/ToF (Bruker Daltonic GmbH, Fällanden, Switzerland) according to the following procedure: 1 μL of each sample (1 mg/mL) was mixed with 1 μL of sinapic acid (10 mg/mL in 50% acetonitrile: water and 0.1% TFA). Then, 1 μL of the sample/matrix solution was spotted onto a stainless-steel target plate and allowed to dry. Ions were analysed in positive mode and external calibration was performed by use of a standard protein mixture (Bovine Serum Albumin). The measure was repeated three times for both the native antibody and each conjugation batch. The difference between the average mass of the immunoconjugate, as determined by a profile measurement of the sample and that of the native antibody (mode of distribution taken as an estimate of the mean) was divided by the exact mass of the chelator to determine the average number of chelators per antibody (DOL).
4.5. THPMe-NCS-HuA33 Radiolabelling
Radiolabelling of the immunoconjugate at a final concentration of 1 μM was performed in two different sets of conditions as described below. The radiolabelling mixture was incubated for 5 min at 37 °C. Success of the radiolabelling was verified by ITLC-SG (Pall Corp, New York, NY, USA) using 50 mM EDTA in water as a mobile phase and analysed on an AR-2000 ITLC plate reader (Bioscan Inc, Washington, DC, USA). The radiolabelling mixture was also applied to a PD10 column preconditioned with ammonium acetate 0.2 M (or PBS, depending on the radiolabelling mixture) to trap residual radiolabelled THPMe-NCS and scarcely soluble 68Ga species. The eluted fractions were analysed by ITLC and the residual activity trapped on the PD10 column was also measured.
Radiolabelling conditions tested:
pH 5.5–6.0 (verification of conjugation): 400 μL of the highest activity fraction (≈88 MBq) were mixed with 100 μL of ammonium acetate 4 M, and 42 μL of the neutralised solution (≈4 MBq) added to an immunconjugate solution (3 μL, 15 μM in ammonium acetate 0.2 M). Radiochemical yield (from ITLC): 95%; activity retained on PD10 column: 3%.
pH 7 (evaluation of the radiolabelling in the conditions required for the in vivo experiment): 410 μL of the highest activity fraction was adjusted to pH 7 using ammonium acetate (0.2 M, 400 μL) and sodium carbonate (0.1 M, 400 μL). Then, 40 μL of this solution (≈5 MBq) was added to 40 μL of immunoconjugate (2 μM in ammonium acetate 0.2 M or PBS). In a variation of the experiment, the effect of a 45 min delay between gallium eluate neutralisation and radiolabelling was also investigated. Radiochemical yield (from ITLC): 98 %; activity retained on PD10 column: 2.6% (3.0% when 45 min delay was applied).
A similar radiolabelling procedure was also applied to the native antibody (≈1 MBq, final antibody concentration = 7.5 μM) to verify that presence of chelator was necessary for successful radiolabelling. Radiochemical yield (from ITLC): 14%; activity retained on PD10 column: 97%.
4.6. DFO-NCS-huA33 Radiolabelling with89Zr
89Zr-oxalate (50 μL, 57 MBq), was diluted with 1 M oxalic acid to 100 μL, buffered to pH 7.5 using Na2CO3 1M (106 μL) and added to 400 μL of DFO-NCS-huA33 solution in PBS (1.53 mg/mL, ≈10 μM). The radiolabelling mixture was agitated for 1 h at 37 °C. EDTA (50 mM solution in water) was added to reach a final concentration of 1 mM. ITLC analysis of the crude radiolabelling mixture (50 mM EDTA in water as a mobile phase) confirmed quantitative radiolabelling. The radiolabelling mixture was then applied to a PD10 column preconditioned with PBS. The resulting fractions were analysed by ITLC and the activity trapped on the column measured. The highest activity fractions were used to prepare doses for injections.
4.7. Cell Culture
Human colorectal cancer cell line SW1222 expressing the A33 antigen was obtained from Sigma-Aldrich and maintained in Minimum Essential Medium (GibcoTM, ThermoFisher, Waltham, MA, USA). Cell culture media were supplemented with 10% foetal calf serum, L-glutamine (2 mM), penicillin (100 units/mL), and streptomycin (0.1 mg/mL). Cell cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2. A trypsin EDTA solution was used to passage the cells (0.25% trypsin/0.53 mM EDTA in Hank’s Buffered Salt Solution without calcium and magnesium).
4.8. Immunoreactive Fraction
An immunoreactivity assay was performed according to a modified literature procedure, measuring binding of
68Ga-THP
Me-NCS-huA33 (5 and 10 equivalent batches) in excess antigen conditions, in which unmodified antibody would achieve quasi-quantitative target-binding [
18,
24]. Then, 10 μL of a radioimmunoconjugate solution in 0.4 ng/μL in PBS supplemented with 1% bovine serum albumin, ≈70 kBq, was added to a suspension of 2.0 × 10
7 SW1222 colorectal cancer cells in 100 μL of medium. The resulting mixture was agitated by pipetting and incubated for 1 h on ice. Cells were then centrifuged (600 RCF for 5 min) and the supernatant carefully removed. The cells were then washed three times by resuspension of the pellet in 1 mL of ice-cold PBS followed by centrifugation and removal of the supernatant. Radioactivity in the cell pellet, supernatant and washings was measured by γ-counting. After background correction of the data, the immunoreactive fraction was calculated by dividing the counts in the cell pellet by the sum of the counts in the cell pellet, media and three washings and multiplying by 100. The experiment was performed in triplicate.
4.9. In Vivo Radiolabelling Studies in Healthy Mice
All in vivo experiments performed at KCL were carried out in accordance with UK Home Office regulations governing animal experimentation and complied with guidelines on responsibility in the use of animals in bioscience research of the UK Research Councils and Medical Research Charities, under UK Home Office project and personal licences.
Experiments included both PET/CT imaging and ex vivo biodistribution. BALB/c mice (female, 7–9 weeks, from Charles River, Wilmington, MA, USA) were used for clearance experiments in healthy animals. PET and CT scans were carried out on a nanoScan
® PET/CT (Mediso Medical Imaging Systems, Budapest, Hungary) [
35] (PET: 1:5 coincidence, 5 ns time window, 400–600 KeV energy window, CT: 180 projection, 45 KVp). Respiration rate and bed temperature were monitored throughout all scans. PET/CT datasets were reconstructed using the Monte-Carlo based full 3D iterative algorithm Tera-Tomo (Mediso Medical Imaging Systems) [
36] using 1:3 coincidence and a voxel size of 0.21 × 0.21 × 0.21 mm
3. Images were analysed using VivoQuant software (inviCRO, London, UK).
Mice were anaesthetised with isoflurane (O2 flow rate: 1.0–1.5 L/min and isoflurane levels 2%–2.5%) and kept anaesthetised for the entire procedure so that dynamic PET scans could be performed. Activity was injected into the tail vein using an in-house made catheter (25 μL volume) or an insulin syringe (0.3 mL, Terumo, Tokyo, Japan), followed by injection of the chelator, or saline as a control, after a defined time interval. Relevant PET/CT scans were performed. Upon procedure completion, animals were sacrificed by neck dislocation while still anaesthetised, tissues harvested and weighed and ex vivo biodistribution assessed by γ-counting. Tubes containing different volumes of the radiotracer solution (corresponding to different %ID) were also counted as internal standards and the calibration curve obtained by linear regression of the data was used to convert measured CPM in %ID for every organ.
Ex vivo biodistribution was performed by collecting the whole organ in the case of tail, heart, lungs, liver, spleen, stomach and kidneys. The femur was used as representative of bone. Skin and fur were collected from the abdomen or the ears of the mouse and muscle was taken from the hind limb. Only part of the liver, small and large intestine was collected. All organs were washed in water to eliminate residual blood. Residual activity in the tail (due to any imperfections in IV injection) was subtracted from the total activity injected, and tail-corrected %ID was calculated for each organ. %ID/g was obtained by dividing the tail-corrected %ID of each organ by its weight.
4.10. THPMe vs. DFO Clearance in Healthy Mice
This experiment was performed at KCL. BALB/c mice were anaesthetised and injected with 68Ga-acetate (100 µL, ≈10 MBq) via a tail vein injection, as previously described. After 1 h, 50 µL of a clearance agent solution (24 nmol of THPMe or DFO, or saline as a control) was administered in the same way. At least one mouse per group was imaged before and after the second injection (PET from 20 to 45 min and from 80 to 105 min after injection of 68Ga-acetate). Mice were kept anaesthetised for the whole experiment, sacrificed at 130 min post 68Ga injection and ex vivo biodistribution performed.
4.11. Dose Finding Experiment
This experiment was performed at KCL. BALB/c mice were anaesthetised and injected with 68Ga-acetate (100 µL, 5–10 MBq) via a tail vein cannula. Then, 100 µL of the relevant THPMe solution (0, 1, 5 or 20 µg) was administered in the same way 5 min after the first injection. All animals were kept under anaesthesia for the duration of the experiment. Mice were culled 15 min after the second injection and biodistribution performed.
4.12. Xenograft Model
All experiments in xenograft models were performed at MSKCC and were conducted according to the guidelines approved by the Research Animal Resource Center and Institutional Animal Care and Use Committee at MSK. Athymic nude female mice (Athymic Nude-nu, 8–10 weeks old) were obtained from Charles River Laboratories (Wilmington, MA, USA). Animals were housed in ventilated cages, given food and water ad libitum, and were allowed to acclimatise for approximately 1 week prior to inoculation of tumour cells. SW1222 tumours were induced on the left shoulder by a subcutaneous injection of 5 × 106 cells suspended in 150 μL of a 1:1 mixture of fresh medium and BD Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). The xenografts were monitored daily until an ideal size for imaging and biodistribution was reached (≈100–150 mm3) in 2–3 weeks.
4.13. PET Imaging of SW1222 Xenograft Models
Mice were randomly assigned to the five different experimental groups (for each group n = 5). Administration of antibody/radiotracers was performed via tail vein injection on conscious mice after gentle warming with a heat lamp. Approximately 5 min prior to imaging, mice were anaesthetised by inhalation of 2% isoflurane/oxygen gas mixture and positioned on the bed of a MicroPET Focus scanner (Concorde MicroSystem Inc, Knoxville, TN, USA). A 1% isoflurane anaesthesia was maintained throughout the scan. Static scans of 10 min each were recorded at different time points after injections. An energy window of 350–700 keV and a coincidence timing window of 6 ns were used. Data were sorted into 2-dimensional histograms by Fourier re-binning, and transverse images were reconstructed by filtered back-projection (FBP) into 0.87 × 0.87 × 0.8 mm3 matrix. The imaging data were normalised to correct for non-uniformity of PET response, dead-time count losses, positron branching ratio and physical decay to the time of injection. No attenuation, scatter or partial-volume averaging correction was applied. Activity concentrations (%ID/g) were calculated from the counting rates in the reconstructed images by use of a system calibration factor derived from the imaging of a mouse-sized water-equivalent phantom containing 68Ga or 89Zr. Images were analysed using ASIPro VMTM software (Concorde MicroSystems Inc, Knoxville, TN, USA). Mice were sacrificed by CO2 asphyxiation. Details of the different experiments performed are described below.
4.14. Ex Vivo Biodistribution
For all 68Ga experiments, tissues were harvested and weighed as described above and ex vivo biodistribution assessed by γ-counting. Calibration of the γ-counter with known amounts of activity allowed determination of the decay-corrected CPM/MBq conversion coefficient, which was used to calculate % injected dose (%ID) in each organ.
4.15. Imaging with 89Zr-DFO-NCS-huA33 (Positive Control)
Mice (group 1) were injected with 89Zr-DFO-NCS-huA33 in saline (200 μL, ≈7 MBq, 88 μg) and imaged at 25 h after injection. At 26 h post injection, mice were sacrificed.
4.16. Pretargeting Experiments
Mice were injected with 100 μg of THPMe-NCS-huA33 in saline using either the 10 eq. or the 30 eq. batch (groups 2 and 4, respectively; five mice per group). After 24 h, mice were injected with 8–10 MBq of 68Ga-acetate (pH 7) at the same time. PET imaging of the five mice was then performed starting from 50 to 90 min after injection (10 min per mouse) and then repeated 1 h later. Mice were sacrificed 3 h after 68Ga-acetate injection and ex vivo biodistribution performed.
4.17. 68Ga-Only Control
Mice (group 3) were injected with 8–10 MBq of 68Ga-acetate (pH 7) and imaging performed as described for the pretargeting experiments, from 1 to 2 h following administration of the radioactivity. Mice were culled at 3 h post injection and ex vivo biodistribution evaluated.
4.18. 68Ga Followed by THPMe Blood Clearance
Mice (group 5) were injected with 10–11 MBq of 68Ga-acetate (pH 7). Imaging of two of the mice, one at 40 and one at 50 min after injection, was performed. A solution of THPMe in saline (3 mg/mL) was prepared and pH adjusted to 7 with Na2CO3. A 400 μg/mL solution was prepared by dilution of the stock solution in saline. At 1 h after gallium injection, 20 μg of THPMe (24 nmol, in 50 μL of saline) was also administered intravenously and all mice imaged between 20 and 60 min after THPMe injection. Mice were sacrificed 3 h after the 68Ga injection and the ex vivo biodistribution of the activity was assessed.
4.19. Power Calculations
Sample size for in vivo experiments was calculated in order to have a statistical power (1-β) of 80% and a type I error probability (α) of 5%. A minimal sample size of n = 3 was calculated, to detect a difference between a signal-to-noise ratio of 10 ± 2 (experimental group) and 5 ± 2 (control) when performing one-way analysis of variances (ANOVA) followed by up to two pairwise comparisons between groups (THPMe vs. DFO experiments). When three pairwise comparisons were considered (dose finding experiment) a minimal sample size of n = 4 was calculated.
4.20. Statistical Analysis of the Data
Data were analysed using GraphPad Prism, version 7.04 for Windows (GraphPad software). An unpaired, two-tailed Student’s t-test was generally used when comparing two groups. One-way ANOVA was used to analyse results where more than two groups were compared. In this case, a Tukey post-hoc test was employed to correct for multiple comparisons when performed between all the groups. A Dunnett post-hoc test was used for comparison between each of the experimental groups and the control group. Whenever variances of groups were found to be significantly different (according to an F test when comparing two groups, and to both Brown–Forsythe and Bartlett’s test when comparing more than two groups) the data were reanalysed using a Welch-adjusted t-test, which does not assume equal variances among groups.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}