Label-Free Comparative Proteomic Analysis Combined with Laser-Capture Microdissection Suggests Important Roles of Stress Responses in the Black Layer of Maize Kernels

 , ,
, , 
 , and
, and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Observation of Black Layer Development in Maize Kernels
2.2. Isolation of Black Layer Cells
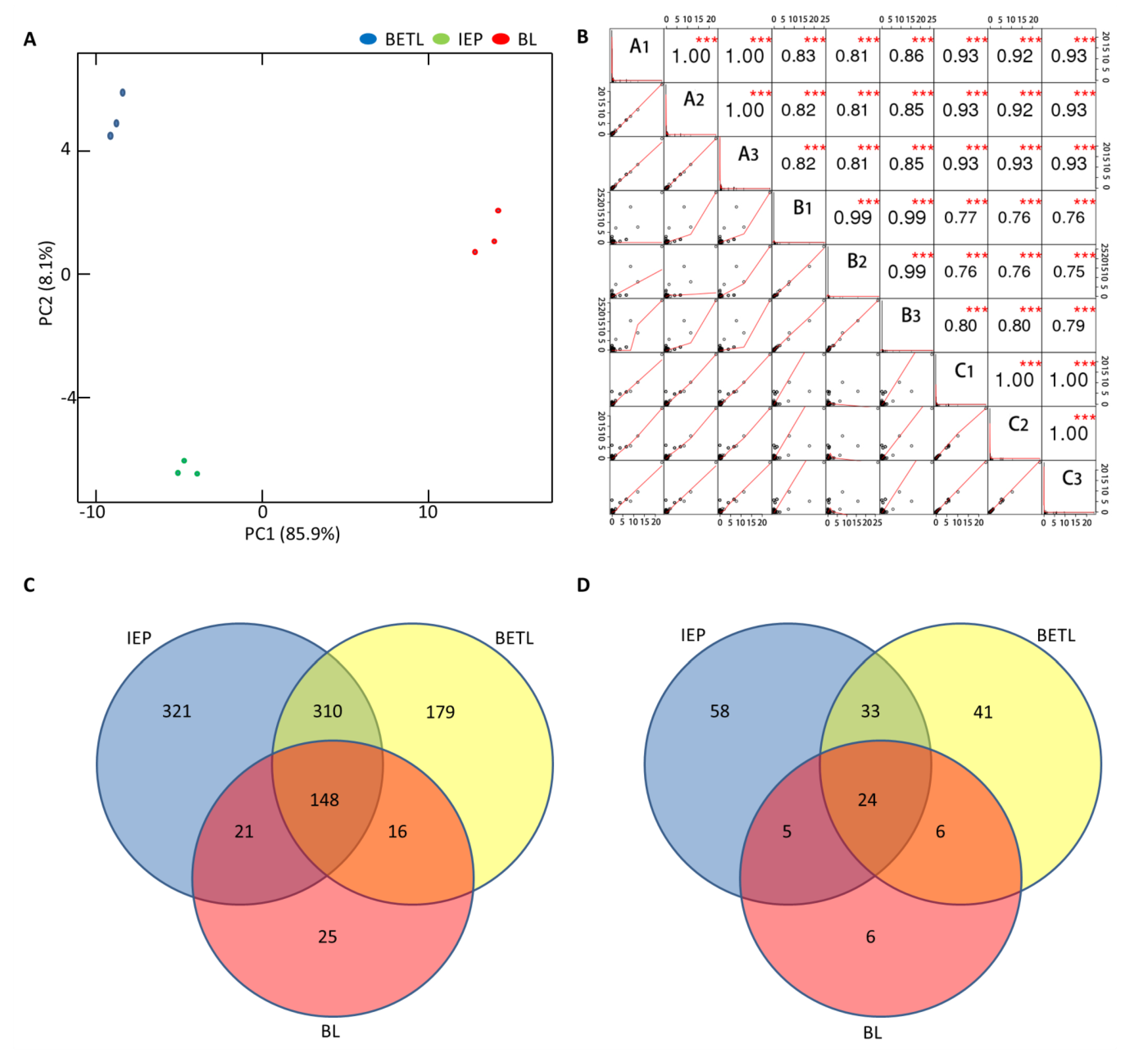
2.3. Proteomic Analysis of the Black Layer Cells
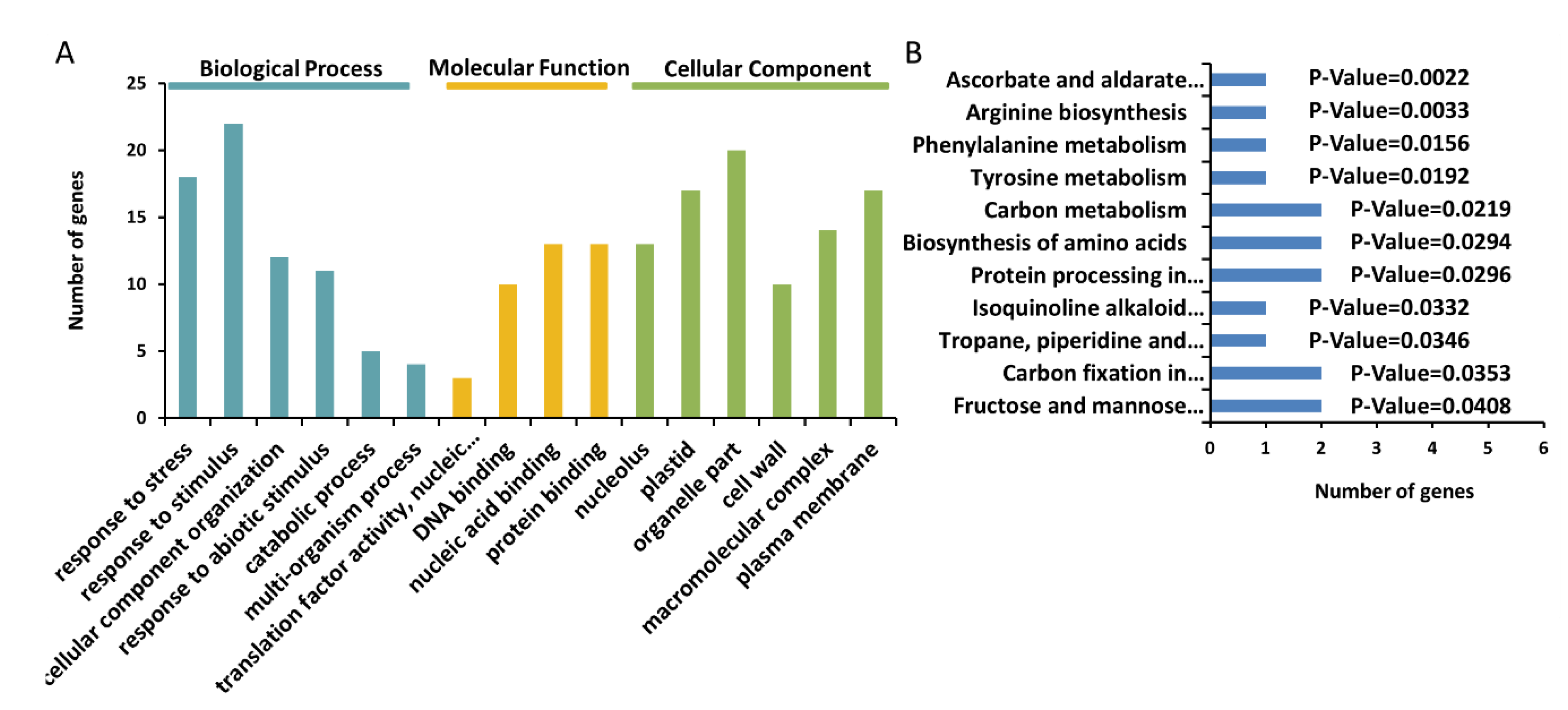
2.4. Gene ontology Annotation Function and Kyoto Encyclopedia of Genes and Genomes Analysis of Proteins Identified from BL
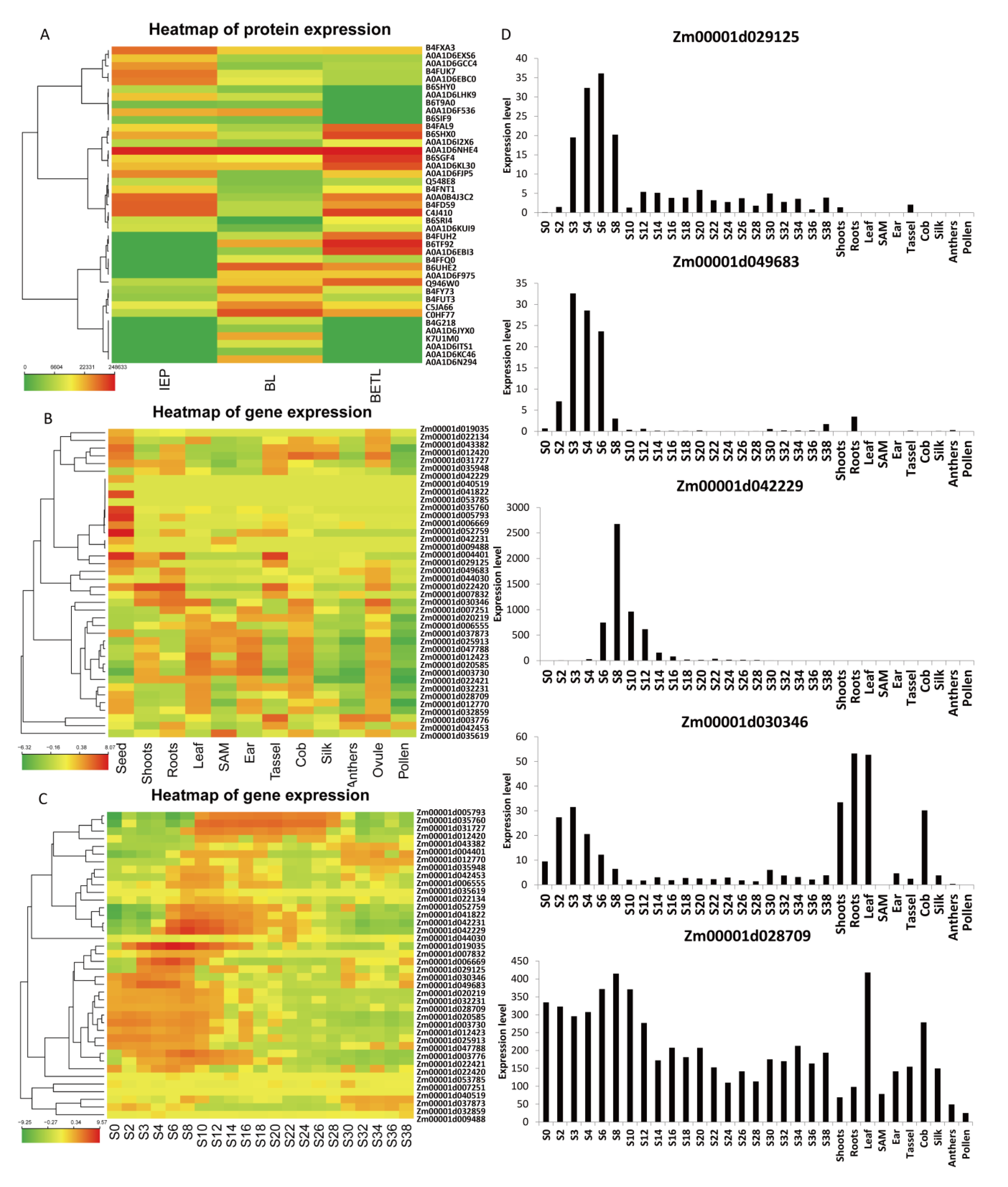
2.5. Gene Expression of BL Accumulated Proteins
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Light and Scanning Electron Microscopy
4.3. Laser Capture Microdissection
4.4. Protein Extraction
4.5. Proteomics Analysis
4.6. Expression Analysis of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BL | black layer |
| DAP | days after pollination |
| BETL | basal endosperm transfer layer |
| IEP | inner epidermis cells of the pedicel |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| P-C | placento-chalazal |
| iP-C | integumental P-C |
| nP-C | nucellar P-C |
| PCD | program cell death process |
| LCM | laser-capture microdissection |
| PBS | phosphate buffered saline |
| OCT | optimum cutting temperature |
| FDR | false discovery rate |
| iBAQ | Intensity-based absolute quantification |
| PCA | principal component analysis |
| L-GulL | L-gulono-1,4-lactone |
| Hsp21 | heat shock protein21 |
| BAP2 | basal layer-type antifungal protein |
| MALDI-TOF MS | Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry profiling |
References
- Joazeiro, C.A.P.; Wing, S.S.; Huang, H.; Leverson, J.D.; Hunter, T.; Liu, Y.-C. The Tyrosine Kinase Negative Regulator c-Cbl as a RING-Type, E2-Dependent Ubiquitin-Protein Ligase. Science 1999, 286, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Panison, F.; Sangoi, L.; Kolling, D.F.; Coelho, C.M.M.; Durli, M.M. Harvest time and agronomic performance of maize hybrids with contrasting growth cycles. Acta Sci. Agron. 2016, 38, 219. [Google Scholar] [CrossRef]
- Gu, R.; Li, L.; Liang, X.; Wang, Y.; Fan, T.; Wang, Y.; Wang, J. The ideal harvest time for seeds of hybrid maize (Zea mays L.) XY335 and ZD958 produced in multiple environments. Sci. Rep. 2017, 7, 17537. [Google Scholar] [CrossRef] [PubMed]
- Daynard, T.B.; Duncan, W.G. The Black Layer and Grain Maturity in Corn. Crop Sci. 1969, 9, 473. [Google Scholar] [CrossRef]
- Rench, W.E.; Shaw, R.H. Black Layer Development in Corn. Agron. J. 1971, 63, 303–305. [Google Scholar] [CrossRef]
- Daynard, T.B. Relationships Among Black Layer Formation, Grain Moisture Percentage, and Heat Unit Accumulation in Corn1. Agron. J. 1972, 64, 716. [Google Scholar] [CrossRef]
- Carter, M.W.; Poneleit, C.G. Black Layer Maturity and Filling Period Variation Among Inbred Lines of Corn (Zea mays L.). Crop Sci. 1973, 13, 436. [Google Scholar] [CrossRef]
- Hunter, J.L.; TeKrony, D.M.; Miles, D.F.; Egli, D.B. Corn Seed Maturity Indicators and their Relationship to Uptake of Carbon-14 Assimilate. Crop Sci. 1991, 31, 1309. [Google Scholar] [CrossRef]
- Gunn, R.B.; Christensen, R. Maturity Relationships Among Early to Late Hybrids of Corn (Zea mays L.). Crop Sci. 1965, 4. [Google Scholar]
- Esau, K. Anatomy of Seed Plants, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1977. [Google Scholar]
- Kladnik, A.; Chamusco, K.; Dermastia, M.; Chourey, P. Evidence of Programmed Cell Death in Post-Phloem Transport Cells of the Maternal Pedicel Tissue in Developing Caryopsis of Maize. Plant Physiol. 2004, 136, 3572–3581. [Google Scholar] [CrossRef]
- Felker, F.C.; Shannon, J.C. Movement of C-labeled Assimilates into Kernels of Zea mays L. Plant Physiol. 1980, 65, 7. [Google Scholar] [CrossRef] [PubMed]
- Schel, J.H.N.; Kieft, H.; Lammeren, A.A.M.V. Interactions between embryo and endosperm during early developmental stages of maize caryopses (Zea mays). Can. J. Bot. 1984, 62, 2842–2853. [Google Scholar] [CrossRef]
- Thorne, J.H. Phloem Unloading of C and N Assimilates in Developing Seeds. Plant Physiol. 1985, 36, 27. [Google Scholar] [CrossRef]
- Lowe, J.; Nelson, O.E. Miniature seed-A study in the development of a defective caryopsis in maize. Genetics 1946, 31, 11. [Google Scholar]
- Oeljeklaus, S.; Meyer, H.E.; Warscheid, B. Advancements in plant proteomics using quantitative mass spectrometry. J. Proteom. 2009, 72, 545–554. [Google Scholar] [CrossRef]
- Méchin, V.; Thévenot, C.; Le Guilloux, M.; Prioul, J.-L.; Damerval, C. Developmental Analysis of Maize Endosperm Proteome Suggests a Pivotal Role for Pyruvate Orthophosphate Dikinase. Plant Physiol. 2007, 143, 1203–1219. [Google Scholar] [CrossRef]
- Huang, H.; Møller, I.M.; Song, S.-Q. Proteomics of desiccation tolerance during development and germination of maize embryos. J. Proteom. 2012, 75, 1247–1262. [Google Scholar] [CrossRef]
- Jin, X.; Fu, Z.; Ding, D.; Li, W.; Liu, Z.; Tang, J. Proteomic Identification of Genes Associated with Maize Grain-Filling Rate. PLoS ONE 2013, 8, e59353. [Google Scholar] [CrossRef]
- Sabelli, P.A.; Liu, Y.; Dante, R.A.; Lizarraga, L.E.; Nguyen, H.N.; Brown, S.W.; Klingler, J.P.; Yu, J.; LaBrant, E.; Layton, T.M.; et al. Control of cell proliferation, endoreduplication, cell size, and cell death by the retinoblastoma-related pathway in maize endosperm. Proc. Natl. Acad. Sci. USA 2013, 110, E1827–E1836. [Google Scholar] [CrossRef]
- Balsamo, G.M.; de Mello, C.S.; Arisi, A.C.M. Proteome Comparison of Grains from Two Maize Genotypes, with Colorless Kernel Pericarp (P1-ww) and Red Kernel Pericarp (P1-rr). Food Biotechnol. 2016, 30, 110–122. [Google Scholar] [CrossRef]
- Yu, T.; Li, G.; Dong, S.; Liu, P.; Zhang, J.; Zhao, B. Proteomic analysis of maize grain development using iTRAQ reveals temporal programs of diverse metabolic processes. BMC Plant Biol. 2016, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, Y.; Wang, Q.; Du, C.; Xiong, W.; Li, X.; Zhu, S.; Li, Y. iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize. Int. J. Mol. Sci. 2017, 18, 1840. [Google Scholar] [CrossRef] [PubMed]
- Silva-Sanchez, C.; Chen, S.; Zhu, N.; Li, Q.-B.; Chourey, P.S. Proteomic comparison of basal endosperm in maize miniature1 mutant and its wild-type Mn1. Front. Plant Sci. 2013, 4, 211. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, G.; Wang, J.; Du, Y.; Yao, D.; Shuai, B.; Han, L.; Tang, Y.; Song, R. Comprehensive proteomic analysis of developing protein bodies in maize (Zea mays) endosperm provides novel insights into its biogenesis. J. Exp. Biol. 2016, 67, 6323–6335. [Google Scholar] [CrossRef]
- Albrethsen, J. The first decade of MALDI protein profiling: A lesson in translational biomarker research. J. Proteom. 2011, 74, 765–773. [Google Scholar] [CrossRef]
- Mehta, A.; Silva, L.P. MALDI-TOF MS profiling approach: How much can we get from it? Front. Plant Sci. 2015, 6, 184. [Google Scholar] [CrossRef]
- Olsen, O.-A. ENDOSPERM DEVELOPMENT: Cellularization and Cell Fate Specification. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 2001, 52, 233–267. [Google Scholar] [CrossRef]
- Sabelli, P.A.; Larkins, B.A. The Development of Endosperm in Grasses. Plant Physiol. 2009, 149, 14–26. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic Transcriptome Landscape of Maize Embryo and Endosperm Development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef]
- Wittmann-Liebold, B.; Graack, H.-R.; Pohl, T. Two-dimensional gel electrophoresis as tool for proteomics studies in combination with protein identification by mass spectrometry. PROTEOMICS 2006, 6, 4688–4703. [Google Scholar] [CrossRef]
- Ruedi Aebersold; Matthias Mann Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.R.; Abbatiello, S.E.; Carr, S.A. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinformatics 2012, 13, S9. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Köcher, T.; Pichler, P.; Schutzbier, M.; Stingl, C.; Kaul, A.; Teucher, N.; Hasenfuss, G.; Penninger, J.M.; Mechtler, K. High Precision Quantitative Proteomics Using iTRAQ on an LTQ Orbitrap: A New Mass Spectrometric Method Combining the Benefits of All. J. Proteome Res. 2009, 8, 4743–4752. [Google Scholar] [CrossRef]
- Ge, F.; Hu, H.; Huang, X. Metabolomic and Proteomic Analysis of Maize Embryonic Callus induced from immature embryo. Sci. Rep. 2017, 7, 1004. [Google Scholar] [CrossRef]
- Liu, S.; Zenda, T.; Dong, A.; Yang, Y.; Liu, X.; Wang, Y.; Li, J.; Tao, Y.; Duan, H. Comparative Proteomic and Morpho-Physiological Analyses of Maize Wild-Type Vp16 and Mutant vp16 Germinating Seed Responses to PEG-Induced Drought Stress. Int. J. Mol. Sci. 2019, 20, 5586. [Google Scholar] [CrossRef]
- Jiang, Z.; Jin, F.; Shan, X. iTRAQ-Based Proteomic Analysis Reveals Several Strategies to Cope with Drought Stress in MaizeSeedlings. Int. J. Mol. Sci. 2019, 20, 5956. [Google Scholar] [CrossRef]
- Yu, G.; Lv, Y.; Shen, L.; Wang, Y.; Qing, Y.; Wu, N.; Li, Y.; Huang, H.; Zhang, N.; Liu, Y.; et al. The Proteomic Analysis of Maize Endosperm Protein Enriched by Phos-tagtm Reveals the Phosphorylation of Brittle-2 Subunit of ADP-Glc Pyrophosphorylase in Starch Biosynthesis Process. Int. J. Mol. Sci. 2019, 20, 986. [Google Scholar] [CrossRef]
- Prioul, J.L.; Méchin, V.; Lessard, P.; Thévenot, C.; Grimmer, M.; Chateau-Joubert, S.; Coates, S.; Hartings, H.; Kloiber-Maitz, M.; Murigneux, A.; et al. A joint transcriptomic, proteomic and metabolic analysis of maize endosperm development and starch filling. Plant Biotechnol. J. 2008, 6, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Tong, Z.; Yang, Q.; Sun, Y.; Jin, X.; Peng, C.; Guo, A.; Wang, X. Proteomic analysis of phytase transgenic and non-transgenic maize seeds. Sci. Rep. 2017, 7, 9246. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Ding, H.; Zhang, J.; Wang, W. Proteomic Analysis of Starch Biosynthesis in Maize Seeds. Starch - Stärke 2019, 71. [Google Scholar] [CrossRef]
- Bonello, J.-F.; Sevilla-Lecoq, S.; Berne, A.; Risueño, M.-C.; Dumas, C.; Rogowsky, P.M. Esr proteins are secreted by the cells of the embryo surrounding region. J. Exp. Biol. 2002, 53, 1559–1568. [Google Scholar] [CrossRef][Green Version]
- Sharma, V.K.; Ramirez, J.; Fletcher, J.C. The ArabidopsisCLV3-like (CLE) genes are expressed in diverse tissues and encode secreted proteins. Plant Mol. Biol. 2003, 51, 415–425. [Google Scholar] [CrossRef]
- Bate, N.J.; Niu, X.; Wang, Y.; Reimann, K.S.; Helentjaris, T.G. An Invertase Inhibitor from Maize Localizes to the Embryo Surrounding Region during Early Kernel Development. Plant Physiol. 2004, 134, 246–254. [Google Scholar] [CrossRef]
- Balandín, M.; Royo, J.; Gómez, E.; Muniz, L.M.; Molina, A.; Hueros, G. A protective role for the embryo surrounding region of the maize endosperm, as evidenced by the characterisation of ZmESR-6, a defensin gene specifically expressed in this region. Plant Mol. Biol. 2005, 58, 269–282. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Liu, J.; Park, C.H.; He, F.; Nagano, M. The RhoGAP SPIN6 Associates with SPL11 and OsRac1 and Negatively Regulates Programmed Cell Death and Innate Immunity in Rice. PLoS Pathog. 2015, 11, e1004629. [Google Scholar] [CrossRef]
- Thao, N.P.; Chen, L.; Nakashima, A.; Hara, S. RAR1 and HSP90 Form a Complex with Rac/Rop GTPase and Function in Innate-Immune Responses in Rice. Plant Cell 2007, 19, 4035–4045. [Google Scholar] [CrossRef]
- Pnueli, L.; Liang, H.; Rozenberg, M.; Mittler, R. Growth suppression, altered stomatal responses, and augmented induction of heat shock proteins in cytosolic ascorbate peroxidase (Apx1)-deficient Arabidopsis plants. Plant J. 2003, 34, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Dubey, R.S. Ascorbate peroxidase from rice seedlings: Properties of enzyme isoforms, effects of stresses and protective roles of osmolytes. Plant Sci. 2004, 167, 541–550. [Google Scholar] [CrossRef]
- Davletova, S.; Rizhsky, L.; Liang, H.; Shengqiang, Z.; Oliver, D.J.; Coutu, J.; Shulaev, V.; Schlauch, K.; Mittler, R. Cytosolic Ascorbate Peroxidase 1 Is a Central Component of the Reactive Oxygen Gene Network of Arabidopsis. Plant Cell 2005, 17, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Suzuki, N.; Rizhsky, L.; Hegie, A.; Koussevitzky, S.; Mittler, R. Double Mutants Deficient in Cytosolic and Thylakoid Ascorbate Peroxidase Reveal a Complex Mode of Interaction between Reactive Oxygen Species, Plant Development, and Response to Abiotic Stresses. Plant Physiol. 2007, 144, 1777–1785. [Google Scholar] [CrossRef]
- Lu, R. High throughput virus-induced gene silencing implicates heat shock protein 90 in plant disease resistance. EMBO J. 2003, 22, 5690–5699. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Neta-Sharir, I.; Isaacson, T.; Lurie, S.; Weiss, D. Dual Role for Tomato Heat Shock Protein 21: Protecting Photosystem II from Oxidative Stress and Promoting Color Changes during Fruit Maturation. Plant Cell 2005, 17, 1829–1838. [Google Scholar] [CrossRef]
- Nishizawa, A.; Yabuta, Y.; Yoshida, E.; Maruta, T.; Yoshimura, K.; Shigeoka, S. Arabidopsis heat shock transcription factor A2 as a key regulator in response to several types of environmental stress. Plant J. 2006, 48, 535–547. [Google Scholar] [CrossRef]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.-D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- Leprince, O.; Hendry, G.A.F.; McKersie, B.D. The mechanisms of desiccation tolerance in developing seeds. Seed Sci. Res. 1993, 3, 231–246. [Google Scholar] [CrossRef]
- Oliver, M.J. Desiccation tolerance in vegetative plant cells. Physiol. Plant. 1996, 97, 779–787. [Google Scholar] [CrossRef]
- Angelovici, R.; Galili, G.; Fernie, A.R.; Fait, A. Seed desiccation: A bridge between maturation and germination. Trends Plant Sci. 2010, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Serna, A.; Maitz, M.; O’Connell, T.; Santandrea, G.; Thevissen, K.; Tienens, K.; Hueros, G.; Faleri, C.; Cai, G.; Lottspeich, F.; et al. Maize endosperm secretes a novel antifungal protein into adjacent maternal tissue: Maize basal endosperm antifungal protein. Plant J. 2001, 25, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Linnestad, C.; Doan, D.N.P.; Brown, R.C.; Lemmon, B.E.; Meyer, D.J.; Jung, R.; Olsen, O.-A. Nucellain, a Barley Homolog of the Dicot Vacuolar-Processing Protease, Is Localized in Nucellar Cell Walls. Plant Physiol. 1998, 118, 1169–1180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, Y.; Li, H.; Bhatti, S.; Zhou, S.; Yang, Y.; Fish, T.; Thannhauser, T.W. Development of a laser capture microscope-based single-cell-type proteomics tool for studying proteomes of individual cell layers of plant roots. Hortic. Res. 2016, 3, 16026. [Google Scholar] [CrossRef]
- Nakazono, M.; Qiu, F.; Borsuk, L.A.; Schnable, P.S. Laser-Capture Microdissection, a Tool for the Global Analysis of Gene Expression in Specific Plant Cell Types: Identification of Genes Expressed Differentially in Epidermal Cells or Vascular Tissues of Maize. Plant Cell 2003, 15, 583–596. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Combination of FASP and StageTip-Based Fractionation Allows In-Depth Analysis of the Hippocampal Membrane Proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef]
- Whitaker, J.R.; Granum, P.E. An absolute method for protein determination based on difference in absorbance at 235 and 280 nm. Anal. Biochem. 1980, 109, 156–159. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Huang, R.; Xu, Z.; Zhang, Y.; Li, L.; Fu, J.; Wang, G.; Wang, J.; Du, X.; Gu, R. Label-Free Comparative Proteomic Analysis Combined with Laser-Capture Microdissection Suggests Important Roles of Stress Responses in the Black Layer of Maize Kernels. Int. J. Mol. Sci. 2020, 21, 1369. https://doi.org/10.3390/ijms21041369
Chen Q, Huang R, Xu Z, Zhang Y, Li L, Fu J, Wang G, Wang J, Du X, Gu R. Label-Free Comparative Proteomic Analysis Combined with Laser-Capture Microdissection Suggests Important Roles of Stress Responses in the Black Layer of Maize Kernels. International Journal of Molecular Sciences. 2020; 21(4):1369. https://doi.org/10.3390/ijms21041369
Chicago/Turabian StyleChen, Quanquan, Ran Huang, Zhenxiang Xu, Yaxin Zhang, Li Li, Junjie Fu, Guoying Wang, Jianhua Wang, Xuemei Du, and Riliang Gu. 2020. "Label-Free Comparative Proteomic Analysis Combined with Laser-Capture Microdissection Suggests Important Roles of Stress Responses in the Black Layer of Maize Kernels" International Journal of Molecular Sciences 21, no. 4: 1369. https://doi.org/10.3390/ijms21041369
APA StyleChen, Q., Huang, R., Xu, Z., Zhang, Y., Li, L., Fu, J., Wang, G., Wang, J., Du, X., & Gu, R. (2020). Label-Free Comparative Proteomic Analysis Combined with Laser-Capture Microdissection Suggests Important Roles of Stress Responses in the Black Layer of Maize Kernels. International Journal of Molecular Sciences, 21(4), 1369. https://doi.org/10.3390/ijms21041369