Bioinformatics Approaches to the Understanding of Molecular Mechanisms in Antimicrobial Resistance


Abstract
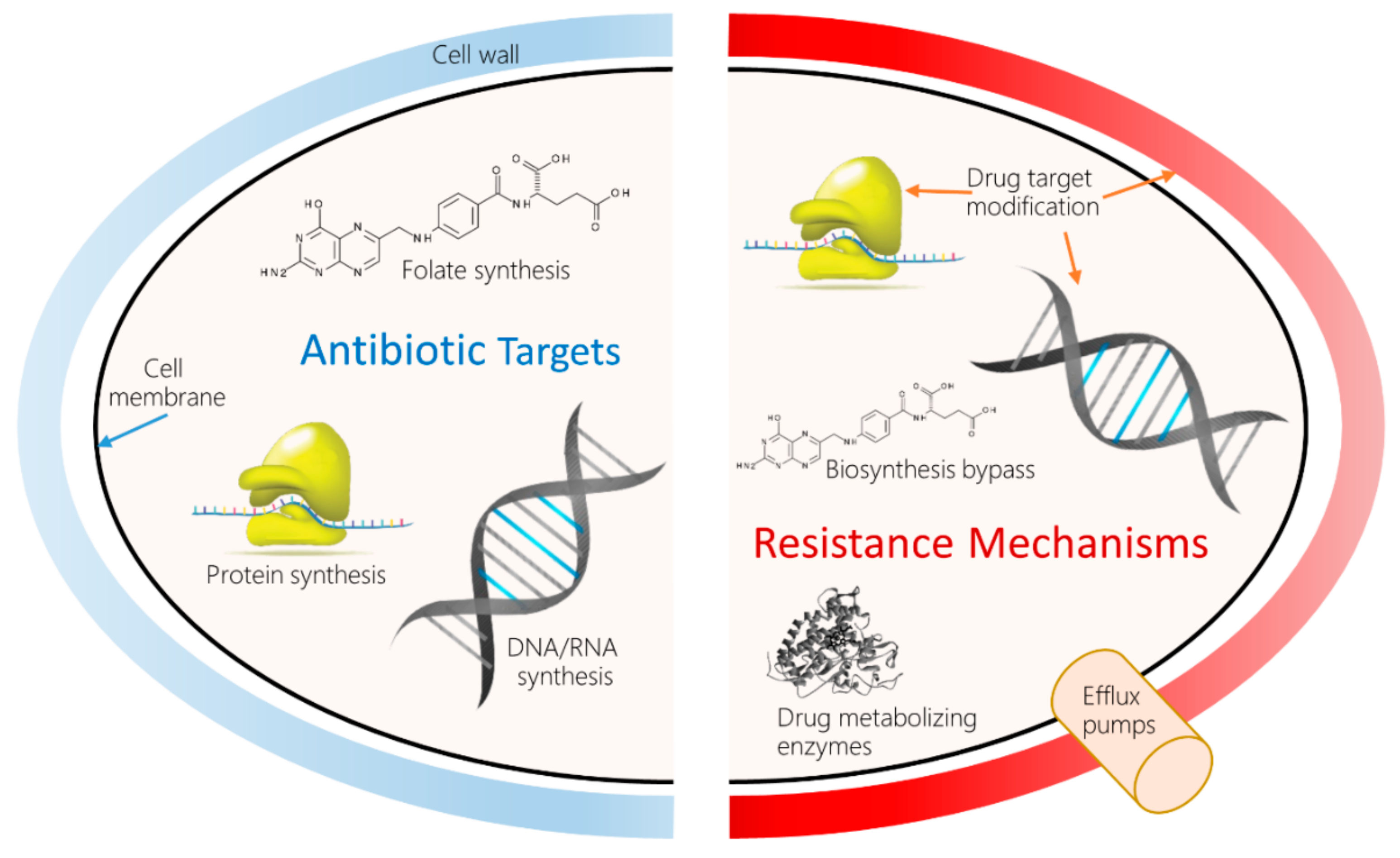
1. Introduction
2. Bioinformatics Approaches to the Analysis and Prediction of Antimicrobial Resistance
2.1. Approach 1: Identification of Known Genomic Signatures of AMR from WGS Data
2.2. Approach 2: Identification of AMR Signatures from Gene Expression Data
2.3. Approach 3: ARG Agnostic Identification of AMR Mechanisms via Pan-Genome Analysis
2.4. Approach 4: Identification of AMR Mechanisms from Metabolomics Data
3. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AB | antibiotic |
| AMR | antimicrobial resistance |
| ARG | antibiotic resistance gene |
| FBA | flux balance analysis |
| MIC | minimum inhibitory concentration |
| ML | machine learning |
| SNP | single nucleotide polymorphism |
| WGS | whole-genome sequencing |
References
- Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 17 February 2020).
- Hughes, D.; Andersson, D.I. Environmental and genetic modulation of the phenotypic expression of antibiotic resistance. FEMS Microbiol. Rev. 2017, 41, 374–391. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.C.; Price, J.R.; Cole, K.; Everitt, R.; Morgan, M.; Finney, J.; Kearns, A.M.; Pichon, B.; Young, B.; Wilson, D.J.; et al. Prediction of Staphylococcus aureus Antimicrobial Resistance by Whole-Genome Sequencing. J. Clin. Microbiol. 2014, 52, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Prediction of antibiotic resistance by gene expression profiles. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K. PhyResSE: A Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; de Cesare, M.; et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Ward, D.V.; Pasolli, E.; Tolio, T.; Zolfo, M.; Asnicar, F.; Truong, D.T.; Tett, A.; Morrow, A.L.; Segata, N. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 2016, 13, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Enke, T.; Chubukov, V.; Ricci, V.; Piddock, L.; Sauer, U. Metabolic constraints on the evolution of antibiotic resistance. Mol. Syst. Biol. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Mahé, P.; Tournoud, M. Predicting bacterial resistance from whole-genome sequences using k-mers and stability selection. BMC Bioinformatics 2018, 19, 383. [Google Scholar] [CrossRef] [PubMed]
- Kavvas, E.S.; Catoiu, E.; Mih, N.; Yurkovich, J.T.; Seif, Y.; Dillon, N.; Heckmann, D.; Anand, A.; Yang, L.; Nizet, V.; et al. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- The CRyPTIC Consortium and the 100,000 Genomes Project Prediction of Susceptibility to First-Line Tuberculosis Drugs by DNA Sequencing | NEJM. Available online: https://www.nejm.org/doi/full/10.1056/NEJMoa1800474 (accessed on 27 November 2019).
- Drouin, A.; Letarte, G.; Raymond, F.; Marchand, M.; Corbeil, J.; Laviolette, F. Interpretable genotype-to-phenotype classifiers with performance guarantees. Sci. Rep. 2019, 9, 4071. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Wright, S.N.; Hamblin, M.; McCloskey, D.; Alcantar, M.A.; Schrübbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661.e9. [Google Scholar] [CrossRef] [PubMed]
- Darnell, R.L.; Knottenbelt, M.K.; Todd Rose, F.O.; Monk, I.R.; Stinear, T.P.; Cook, G.M. Genomewide Profiling of the Enterococcus faecalis Transcriptional Response to Teixobactin Reveals CroRS as an Essential Regulator of Antimicrobial Tolerance. mSphere 2019, 4, e00228-19. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 14 January 2020).
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.E.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Method | Targeted Species | Number of AB | Type of Prediction; Category ** | Input Data | Feature Space |
|---|---|---|---|---|---|
| TypeWriter by Gordon et al., 2014 [3] | Staphylococcus aureus | 12 | Binary SvR classification; CA | WGS | 24 ARG and their mutations |
| By Suzuki et al., 2014 [4] | Escherichia coli | 11 | Regression, MIC; EA | Gene expression (Microarray) | 8 differentially expressed genes |
| PhyResSE by Feuerriegel et al., 2015 [5] | Mycobacterium tuberculosis | NS | Binary SvR classification; CA | WGS | 11 ARG and a catalog of SNPs defining 92 strains |
| Mykrobe by Bradley et al., 2015 [6] | S. aureus, M. tuberculosis | 12 | Binary SvR classification; CA | WGS | Curated list of known ARG and their mutations (n = NS) |
| PanPhlAn by Scholz et al., 2016 [7] | E. coli | NA | Binary SvR classification; PCU | Metagenomics, Meta-transcriptomics | 122 strains |
| PointFinder by Zankari et al., 2017 [8] | E. coli, Campylobacter jejuni, Salmonella enterica | 11 | Binary SvR classification; CA | WGS | 16 ARG and their mutations |
| By Zampieri et al., 2017 [9] | E. coli | 3 | NA; EA | WGS, Metabolomics | Metabolites, Mutations, 12 lineages |
| By Mahe and Tournoud, 2018 [10] | S. aureus, M. tuberculosis | 7 | Logistic regression; PCU | WGS | 1 to 8 genomic k-mers per AB (k = 31) |
| By Kavvas et al., 2018 [11] | M. tuberculosis | 13 | NA; PCU | WGS, protein structures | 1595 strains |
| CRyPTIC consortium, 2018 [12] | M. tuberculosis | 4 | Binary SvR classification; CA | WGS | 9 ARG and their mutations |
| By Drouin et al., 2019 [13] | A. baumannii E. faecium E. coli K. pneumoniae M. tuberculosis N. gonorrhoeae P. difficile P. aeruginosa S. enterica S. aureus S. haemolyticus S. pneumoniae | 56 | Multi-class classification; PCU | WGS | Genomic k-mers |
| By Yang et al., 2019 [14] | E. coli | 3 | NN regression; EA | Metabolomics | Metabolites, Metabolic networks, FBA |
| By Darnell et al., 2019 [15] | Enterococcus faecalis | 1 | NA; EA | Gene expression (RNA-seq) | 573 differentially expressed genes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Camp, P.-J.; Haslam, D.B.; Porollo, A. Bioinformatics Approaches to the Understanding of Molecular Mechanisms in Antimicrobial Resistance. Int. J. Mol. Sci. 2020, 21, 1363. https://doi.org/10.3390/ijms21041363
Van Camp P-J, Haslam DB, Porollo A. Bioinformatics Approaches to the Understanding of Molecular Mechanisms in Antimicrobial Resistance. International Journal of Molecular Sciences. 2020; 21(4):1363. https://doi.org/10.3390/ijms21041363
Chicago/Turabian StyleVan Camp, Pieter-Jan, David B. Haslam, and Aleksey Porollo. 2020. "Bioinformatics Approaches to the Understanding of Molecular Mechanisms in Antimicrobial Resistance" International Journal of Molecular Sciences 21, no. 4: 1363. https://doi.org/10.3390/ijms21041363
APA StyleVan Camp, P.-J., Haslam, D. B., & Porollo, A. (2020). Bioinformatics Approaches to the Understanding of Molecular Mechanisms in Antimicrobial Resistance. International Journal of Molecular Sciences, 21(4), 1363. https://doi.org/10.3390/ijms21041363