Human-Induced Pluripotent Stem Cells and Herbal Small-Molecule Drugs for Treatment of Alzheimer’s Disease


Abstract
1. Introduction
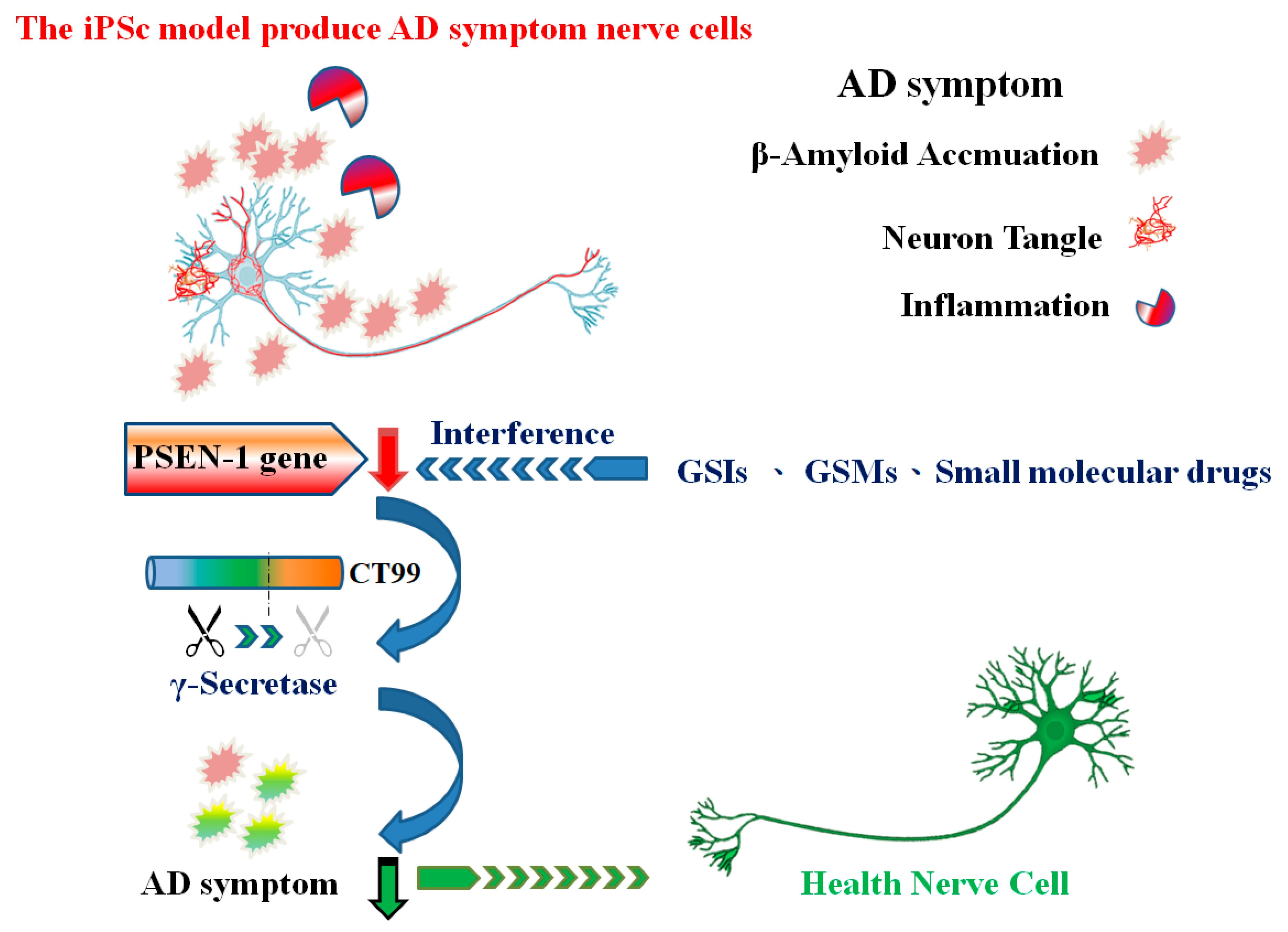
2. Pluripotent Stem Cells Induced by AD Gene Mutations
2.1. APP Gene Mutations
2.2. Trisomy 21 Gene Mutation
2.3. PSEN1/2 Gene Mutation
2.4. Microtubule-Associated Protein Tau Gene Mutation
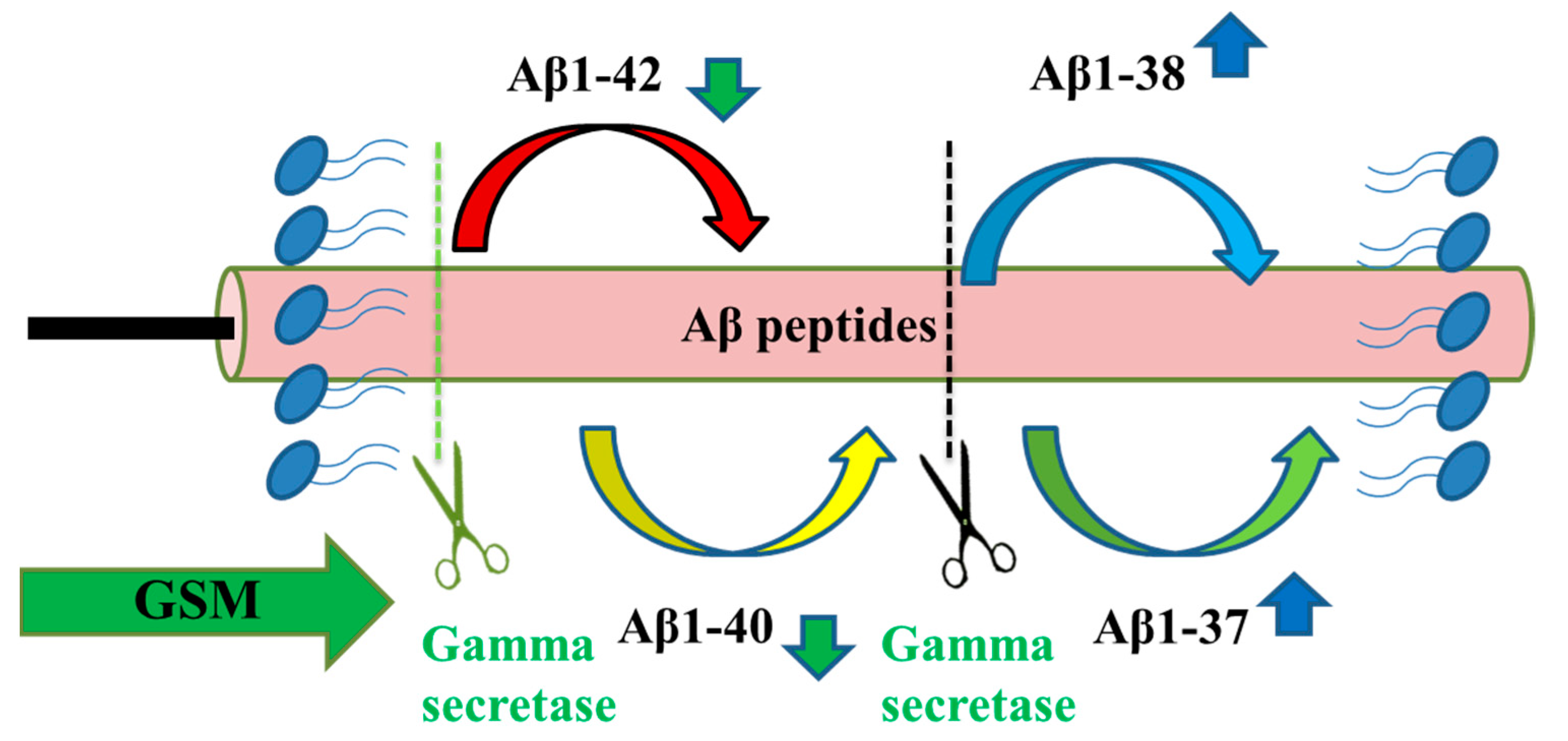
3. γ-secretase Inhibitors and γ-Secretase Modulators
3.1. γ-Secretase Inhibitors
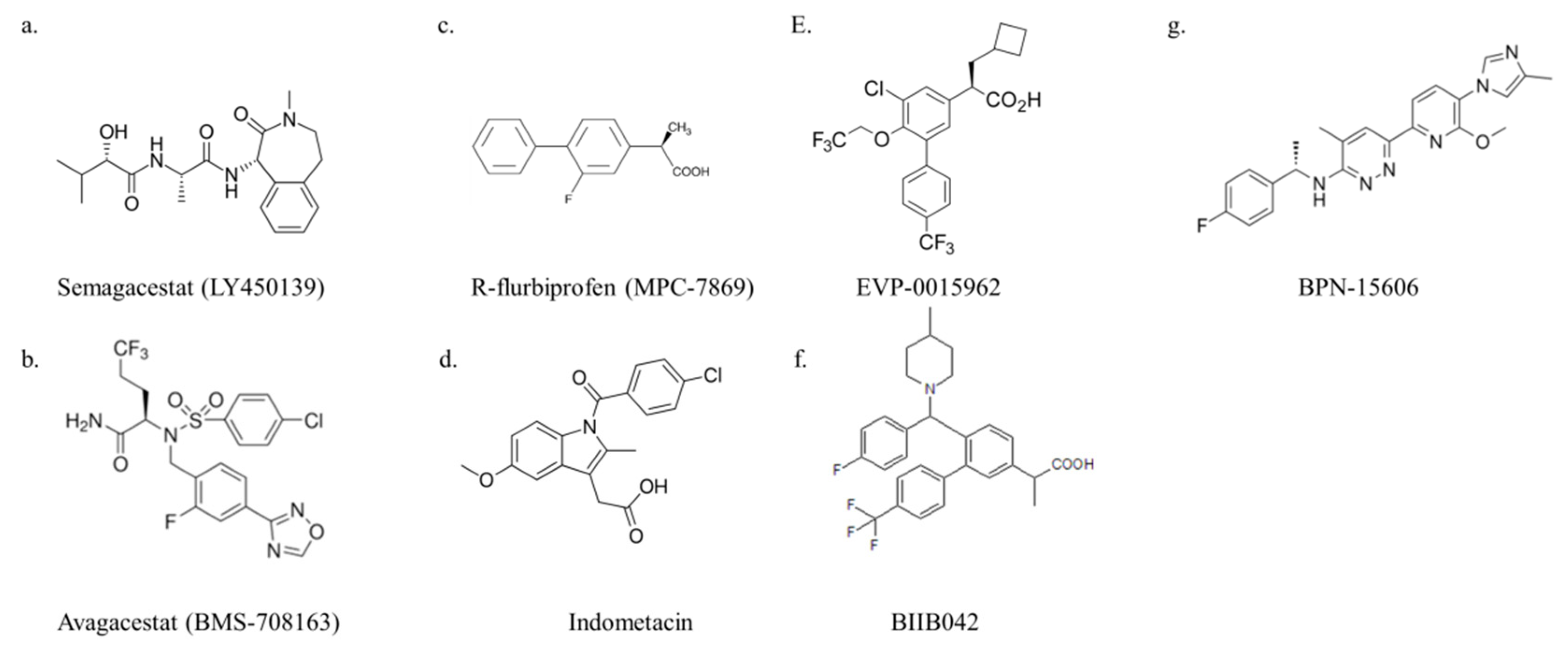
3.1.1. Semagacestat
3.1.2. Avagacestat
3.2. γ-secretase modulators
3.2.1. R-flurbiprofen (Tarenflurbil, MPC-7869)
3.2.2. Indomethacin
3.2.3. EVP-0015962
3.2.4. BIIB042
3.2.5. BPN-15606
4. Small-Molecular Combination GSM Treatments of Alzheimer’s Disease
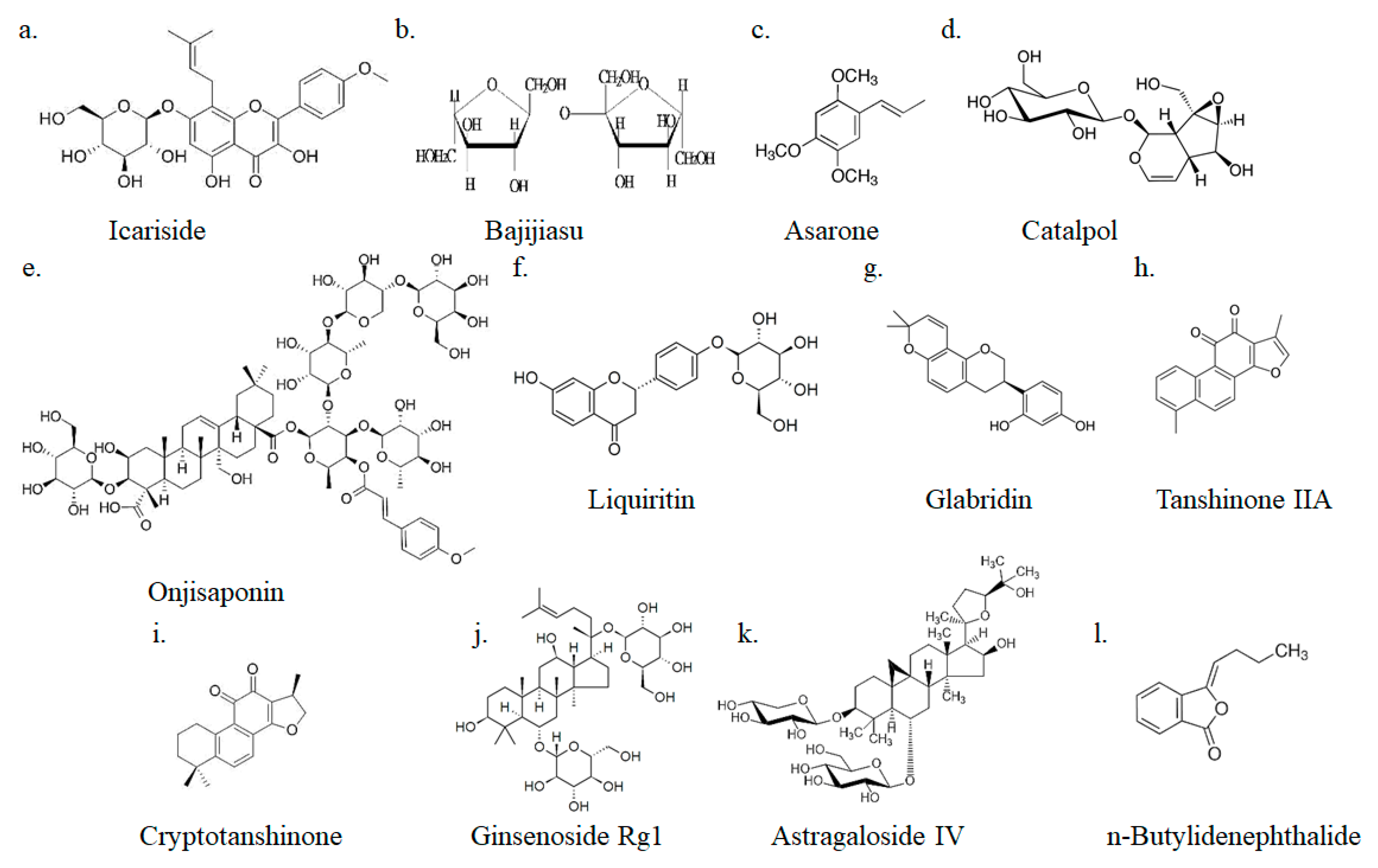
4.1. Icariside
4.2. Onjisaponin
4.3. Bajijiasu
4.4. Asarones
4.5. Catalpol
4.6. Liquiritin
4.7. Glabridin
4.8. Tanshinone IIA and Cryptotanshinone
4.9. Ginsenoside Rg1
4.10. Astragaloside IV
4.11. n-Butylidenephthalide
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAM | A disintegrin and metalloproteinase |
| APP | Amyloid precursor protein |
| AS-IV | Astragaloside IV |
| BACE | β-site beta-secretase |
| BDNF | Brain-derived neurotropic factor |
| cryo-EM | Cryo-electron microscopy |
| COX-2 | Cyclooxygenase-2 |
| CT | Cryptotanshinone |
| DLB | Dementia with Lewy bodies |
| ERK | Extracellular signal-regulated kinase |
| FTLD | Frontotemporal dementia |
| GSI | γ-secretase inhibitors |
| GSMs | γ-secretase modulators |
| GFAP | Glial fibrillary acidic protein |
| iNOS | Inducible nitric oxide synthase |
| MAPT | Microtubule-associated protein tau |
| MCI | Mild cognitive impairment |
| MDA | Malondialdehyde |
| NICD | Notch intracellular domains |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| NF-kBp65 | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| n-BP | n-Butylidenephthalide |
| OB | Onjisaponin B |
| PSEN-1 | Presenilin-1 |
| PSEN-2 | Presenilin-2 |
| PDAPP | PDGF promoter driven amyloid precursor protein |
| PPAR | Peroxisome proliferator-activated receptor |
| SCD | Subjective cognitive decline |
| TIIA | Tanshinone IIA |
| VaD | Vascular dementia |
| 8-OHdG | 8-hydroxy-2′deoxyguanosine |
References
- Van der Flier, W.M.; Scheltens, P. Amsterdam Dementia Cohort: Performing Research to Optimize Care. J. Alzheimers Dis. 2018, 62, 1091–1111. [Google Scholar] [CrossRef] [PubMed]
- Association, A.S. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Forstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.; Chen, M.K. Neuroimaging in Dementias. Semin Neurol. 2019, 39, 188–199. [Google Scholar] [CrossRef]
- Koedam, E.L.; Lauffer, V.; van der Vlies, A.E.; van der Flier, W.M.; Scheltens, P.; Pijnenburg, Y.A. Early-versus late-onset Alzheimer’s disease: more than age alone. J. Alzheimers Dis. 2010, 19, 1401–1408. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-onset Alzheimer’s disease: Nonamnestic subtypes and type 2 AD. Arch. Med. Res. 2012, 43, 677–685. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Westerink, R.H.; Ewing, A.G. The PC12 cell as model for neurosecretion. Acta Physiol (Oxf) 2008, 192, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kumar, M.; Zhu, Y. Protective Effect of Hyperforin on beta Amyloid Protein Induced Apoptosis in PC12 Cells and Colchicine Induced Alzheimer’s Disease: An Anti-oxidant and Anti-inflammatory Therapy. J. Oleo Sci. 2018, 67, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca(2+) entry. J. Mol. Med. (Berl) 2018, 96, 1061–1079. [Google Scholar] [CrossRef]
- Ng, J.; Kaur, H.; Collier, T.; Chang, K.; Brooks, A.E.S.; Allison, J.R.; Brimble, M.A.; Hickey, A.; Birch, N.P. Site-specific glycation of Abeta1-42 affects fibril formation and is neurotoxic. J. Biol. Chem. 2019, 294, 8806–8818. [Google Scholar] [CrossRef]
- Galante, D.; Corsaro, A.; Florio, T.; Vella, S.; Pagano, A.; Sbrana, F.; Vassalli, M.; Perico, A.; D’Arrigo, C. Differential toxicity, conformation and morphology of typical initial aggregation states of Abeta1-42 and Abetapy3-42 beta-amyloids. Int. J. Biochem. Cell Biol. 2012, 44, 2085–2093. [Google Scholar] [CrossRef]
- Arber, C.; Lovejoy, C.; Wray, S. Stem cell models of Alzheimer’s disease: Progress and challenges. Alzheimers Res. Ther. 2017, 9, 42. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Shimojo, D.; Onodera, K.; Doi-Torii, Y.; Ishihara, Y.; Hattori, C.; Miwa, Y.; Tanaka, S.; Okada, R.; Ohyama, M.; Shoji, M.; et al. Rapid, efficient, and simple motor neuron differentiation from human pluripotent stem cells. Mol. Brain 2015, 8, 79. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.M.; Wong, M.M.K.; Vowles, J.; Cowley, S.A.; Becker, E.B.E. A Simplified Method for Generating Purkinje Cells from Human-Induced Pluripotent Stem Cells. Cerebellum 2018, 17, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Crompton, L.A.; Byrne, M.L.; Taylor, H.; Kerrigan, T.L.; Bru-Mercier, G.; Badger, J.L.; Barbuti, P.A.; Jo, J.; Tyler, S.J.; Allen, S.J.; et al. Stepwise, non-adherent differentiation of human pluripotent stem cells to generate basal forebrain cholinergic neurons via hedgehog signaling. Stem Cell Res. 2013, 11, 1206–1221. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Srikanth, P.; Callahan, D.G.; Young-Pearse, T.L. Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS ONE 2014, 9, e105807. [Google Scholar] [CrossRef] [PubMed]
- Stanslowsky, N.; Haase, A.; Martin, U.; Naujock, M.; Leffler, A.; Dengler, R.; Wegner, F. Functional differentiation of midbrain neurons from human cord blood-derived induced pluripotent stem cells. Stem Cell Res. Ther. 2014, 5, 35. [Google Scholar] [CrossRef]
- Wang, S.; Wang, B.; Pan, N.; Fu, L.; Wang, C.; Song, G.; An, J.; Liu, Z.; Zhu, W.; Guan, Y.; et al. Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci. Rep. 2015, 5, 9232. [Google Scholar] [CrossRef]
- Callahan, P.M.; Bertrand, D.; Bertrand, S.; Plagenhoef, M.R.; Terry, A.V., Jr. Tropisetron sensitizes alpha7 containing nicotinic receptors to low levels of acetylcholine in vitro and improves memory-related task performance in young and aged animals. Neuropharmacology 2017, 117, 422–433. [Google Scholar] [CrossRef]
- Chien, M.Y.; Chuang, C.H.; Chern, C.M.; Liou, K.T.; Liu, D.Z.; Hou, Y.C.; Shen, Y.C. Salvianolic acid A alleviates ischemic brain injury through the inhibition of inflammation and apoptosis and the promotion of neurogenesis in mice. Free Radic. Biol. Med. 2016, 99, 508–519. [Google Scholar] [CrossRef]
- Fujiwara, N.; Shimizu, J.; Takai, K.; Arimitsu, N.; Saito, A.; Kono, T.; Umehara, T.; Ueda, Y.; Wakisaka, S.; Suzuki, T.; et al. Restoration of spatial memory dysfunction of human APP transgenic mice by transplantation of neuronal precursors derived from human iPS cells. Neurosci Lett. 2013, 557 Pt B, 129–134. [Google Scholar] [CrossRef]
- Tang, T.; Xiao, J.; Suh, C.Y.; Burroughs, A.; Cerminara, N.L.; Jia, L.; Marshall, S.P.; Wise, A.K.; Apps, R.; Sugihara, I.; et al. Heterogeneity of Purkinje cell simple spike-complex spike interactions: zebrin- and non-zebrin-related variations. J. Physiol. 2017, 595, 5341–5357. [Google Scholar] [CrossRef]
- Mavroudis, I.A.; Manani, M.G.; Petrides, F.; Petsoglou, K.; Njau, S.D.; Costa, V.G.; Baloyannis, S.J. Dendritic and spinal pathology of the Purkinje cells from the human cerebellar vermis in Alzheimer’s disease. Psychiatr Danub 2013, 25, 221–226. [Google Scholar] [PubMed]
- Jiang, Y.Q.; Wang, X.L.; Cao, X.H.; Ye, Z.Y.; Li, L.; Cai, W.Q. Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res. 2013, 1519, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tcw, J. Human iPSC application in Alzheimer’s disease and Tau-related neurodegenerative diseases. Neurosci Lett. 2019, 699, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, A.E.; Siegert, S.; Tsai, L.H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid beta Combination for Alzheimer’s Disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef]
- Oksanen, M.; Hyotylainen, I.; Voutilainen, J.; Puttonen, K.A.; Hamalainen, R.H.; Graff, C.; Lehtonen, S.; Koistinaho, J. Generation of a human induced pluripotent stem cell line (LL008 1.4) from a familial Alzheimer’s disease patient carrying a double KM670/671NL (Swedish) mutation in APP gene. Stem Cell Res. 2018, 31, 181–185. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Lehtonen, S.; Hoytylainen, I.; Voutilainen, J.; Sonninen, T.M.; Kuusisto, J.; Laakso, M.; Hamalainen, R.H.; Oksanen, M.; Koistinaho, J. Generation of a human induced pluripotent stem cell line from a patient with a rare A673T variant in amyloid precursor protein gene that reduces the risk for Alzheimer’s disease. Stem Cell Res. 2018, 30, 96–99. [Google Scholar] [CrossRef]
- Potter, H.; Granic, A.; Caneus, J. Role of Trisomy 21 Mosaicism in Sporadic and Familial Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 7–17. [Google Scholar] [CrossRef]
- Chang, C.Y.; Chen, S.M.; Lu, H.E.; Lai, S.M.; Lai, P.S.; Shen, P.W.; Chen, P.Y.; Shen, C.I.; Harn, H.J.; Lin, S.Z.; et al. N-butylidenephthalide attenuates Alzheimer’s disease-like cytopathy in Down syndrome induced pluripotent stem cell-derived neurons. Sci. Rep. 2015, 5, 8744. [Google Scholar] [CrossRef]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Okano, H.; Suzuki, N. [Modeling familial Alzheimer’s disease with induced pluripotent stem cells]. Rinsho Shinkeigaku 2012, 52, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Cheng, C.; Mair, W.; Almeida, S.; Fong, H.; Biswas, M.H.U.; Zhang, Z.; Huang, Y.; Temple, S.; Coppola, G.; et al. Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Rep. 2016, 7, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Leon, J.A.; Cabrera-Socorro, A.; Eggermont, K.; Swijsen, A.; Terryn, J.; Fazal, R.; Nami, F.; Ordovas, L.; Quiles, A.; Lluis, F.; et al. Generation of a human induced pluripotent stem cell-based model for tauopathies combining three microtubule-associated protein TAU mutations which displays several phenotypes linked to neurodegeneration. Alzheimers Dement. 2018, 14, 1261–1280. [Google Scholar] [CrossRef]
- Iovino, M.; Agathou, S.; Gonzalez-Rueda, A.; Del Castillo Velasco-Herrera, M.; Borroni, B.; Alberici, A.; Lynch, T.; O’Dowd, S.; Geti, I.; Gaffney, D.; et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 2015, 138, 3345–3359. [Google Scholar] [CrossRef]
- Maclean, G.A.; Menne, T.F.; Guo, G.; Sanchez, D.J.; Park, I.H.; Daley, G.Q.; Orkin, S.H. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 17567–17572. [Google Scholar] [CrossRef]
- Clarimon, J.; Djaldetti, R.; Lleo, A.; Guerreiro, R.J.; Molinuevo, J.L.; Paisan-Ruiz, C.; Gomez-Isla, T.; Blesa, R.; Singleton, A.; Hardy, J. Whole genome analysis in a consanguineous family with early onset Alzheimer’s disease. Neurobiol. Aging 2009, 30, 1986–1991. [Google Scholar] [CrossRef]
- Awada, A.A. Early and late-onset Alzheimer’s disease: What are the differences? J. Neurosci. Rural Pract. 2015, 6, 455–456. [Google Scholar] [CrossRef]
- Kehoe, P.; Williams, J.; Lovestone, S.; Wilcock, G.; Owen, M.J. Presenilin-1 polymorphism and Alzheimer’s disease. The UK Alzheimer’s Disease Collaborative Group. Lancet 1996, 347, 1185. [Google Scholar] [CrossRef]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/gamma-secretase. J. Alzheimers Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human gamma-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef]
- Haass, C.; De Strooper, B. The presenilins in Alzheimer’s disease--proteolysis holds the key. Science 1999, 286, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Kaether, C.; Haass, C.; Steiner, H. Assembly, trafficking and function of gamma-secretase. Neurodegener Dis. 2006, 3, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, S.K.; Singh, S.K. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Ables, J.L.; Breunig, J.J.; Eisch, A.J.; Rakic, P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci. 2011, 12, 269–283. [Google Scholar] [CrossRef]
- Mumm, J.S.; Kopan, R. Notch signaling: from the outside in. Dev. Biol. 2000, 228, 151–165. [Google Scholar] [CrossRef]
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 2012, 32, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Elvang, A.B.; Volbracht, C.; Pedersen, L.O.; Jensen, K.G.; Karlsson, J.J.; Larsen, S.A.; Mork, A.; Stensbol, T.B.; Bastlund, J.F. Differential effects of gamma-secretase and BACE1 inhibition on brain Abeta levels in vitro and in vivo. J. Neurochem. 2009, 110, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Barten, D.M.; Meredith, J.E., Jr.; Zaczek, R.; Houston, J.G.; Albright, C.F. Gamma-secretase inhibitors for Alzheimer’s disease: balancing efficacy and toxicity. Drugs R D 2006, 7, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Yanagida, K.; Kodama, T.S.; Takami, M.; Mizuta, N.; Oyama, H.; Nishitomi, K.; Chiu, Y.W.; Okamoto, T.; Ikeuchi, T.; et al. Semagacestat Is a Pseudo-Inhibitor of gamma-Secretase. Cell Rep. 2017, 21, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Gillman, K.W.; Starrett, J.E., Jr.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.E.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable gamma-Secretase Inhibitor. ACS Med. Chem. Lett. 2010, 1, 120–124. [Google Scholar] [CrossRef]
- Albright, C.F.; Dockens, R.C.; Meredith, J.E., Jr.; Olson, R.E.; Slemmon, R.; Lentz, K.A.; Wang, J.S.; Denton, R.R.; Pilcher, G.; Rhyne, P.W.; et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J. Pharmacol. Exp. Ther. 2013, 344, 686–695. [Google Scholar] [CrossRef]
- Coric, V.; Salloway, S.; van Dyck, C.H.; Dubois, B.; Andreasen, N.; Brody, M.; Curtis, C.; Soininen, H.; Thein, S.; Shiovitz, T.; et al. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar] [CrossRef]
- Ran, Y.; Cruz, P.E.; Ladd, T.B.; Fauq, A.H.; Jung, J.I.; Matthews, J.; Felsenstein, K.M.; Golde, T.E. gamma-Secretase processing and effects of gamma-secretase inhibitors and modulators on long Abeta peptides in cells. J. Biol. Chem. 2014, 289, 3276–3287. [Google Scholar] [CrossRef]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. gamma-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [PubMed]
- Woodling, N.S.; Colas, D.; Wang, Q.; Minhas, P.; Panchal, M.; Liang, X.; Mhatre, S.D.; Brown, H.; Ko, N.; Zagol-Ikapitte, I.; et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer’s disease model mice. Brain 2016, 139, 2063–2081. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Giardina, G.A. gamma-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr. Top. Med. Chem. 2011, 11, 1555–1570. [Google Scholar] [CrossRef]
- Xia, W.; Wong, S.T.; Hanlon, E.; Morin, P. gamma-Secretase modulator in Alzheimer’s disease: shifting the end. J. Alzheimers Dis. 2012, 31, 685–696. [Google Scholar] [CrossRef]
- Wong, L.R.; Ho, P.C. Role of serum albumin as a nanoparticulate carrier for nose-to-brain delivery of R-flurbiprofen: implications for the treatment of Alzheimer’s disease. J. Pharm. Pharmacol. 2018, 70, 59–69. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, X.; Zhang, X.; Liu, G.; Wang, Y.; Zhou, W.; Zhang, Y. A combination of indomethacin and atorvastatin ameliorates cognitive and pathological deterioration in PrP-hAbetaPPswe/PS1(DeltaE9) transgenic mice. J. Neuroimmunol. 2019, 330, 108–115. [Google Scholar] [CrossRef]
- De Jong, D.; Jansen, R.; Hoefnagels, W.; Jellesma-Eggenkamp, M.; Verbeek, M.; Borm, G.; Kremer, B. No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: A randomized controlled trial. PLoS ONE 2008, 3, e1475. [Google Scholar] [CrossRef]
- Hahn, S.; Bruning, T.; Ness, J.; Czirr, E.; Baches, S.; Gijsen, H.; Korth, C.; Pietrzik, C.U.; Bulic, B.; Weggen, S. Presenilin-1 but not amyloid precursor protein mutations present in mouse models of Alzheimer’s disease attenuate the response of cultured cells to gamma-secretase modulators regardless of their potency and structure. J. Neurochem. 2011, 116, 385–395. [Google Scholar] [CrossRef]
- Borgegard, T.; Jureus, A.; Olsson, F.; Rosqvist, S.; Sabirsh, A.; Rotticci, D.; Paulsen, K.; Klintenberg, R.; Yan, H.; Waldman, M.; et al. First and second generation gamma-secretase modulators (GSMs) modulate amyloid-beta (Abeta) peptide production through different mechanisms. J. Biol. Chem. 2012, 287, 11810–11819. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.L.; Rynearson, K.D.; Duddy, S.K.; Zhang, C.; Nguyen, P.D.; Becker, A.; Vo, U.; Masliah, D.; Monte, L.; Klee, J.B.; et al. Pharmacological and Toxicological Properties of the Potent Oral gamma-Secretase Modulator BPN-15606. J. Pharmacol. Exp. Ther. 2017, 362, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.; Felsenstein, K.M.; Hrdlicka, L.; Tu, Z.; Albayya, F.; Lee, W.; Hopp, S.; Miller, M.J.; Spaulding, D.; Yang, Z.; et al. Modulation of gamma-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Mol. Neurodegener 2012, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Talreja, T.; Xin, Z.; Cuervo, J.H.; Kumaravel, G.; Humora, M.J.; Xu, L.; Rohde, E.; Gan, L.; Jung, M.Y.; et al. Discovery of BIIB042, a Potent, Selective, and Orally Bioavailable gamma-Secretase Modulator. ACS Med. Chem. Lett. 2011, 2, 786–791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scannevin, R.H.; Chollate, S.; Brennan, M.S.; Snodgrass-Belt, P.A.; Peng, H.; Xu, L.; Jung, M.Y.; Bussiere, T.; Arastu, M.F.; Talreja, T.; et al. BIIB042, a novel gamma-secretase modulator, reduces amyloidogenic Abeta isoforms in primates and rodents and plaque pathology in a mouse model of Alzheimer’s disease. Neuropharmacology 2016, 103, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Prikhodko, O.; Rynearson, K.D.; Sekhon, T.; Mante, M.M.; Nguyen, P.D.; Rissman, R.A.; Tanzi, R.E.; Wagner, S.L. The GSM BPN-15606 as a Potential Candidate for Preventative Therapy in Alzheimer’s Disease. J. Alzheimers Dis. 2020. [Google Scholar] [CrossRef]
- Endres, K.; Deller, T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front. Mol. Neurosci. 2017, 10, 56. [Google Scholar] [CrossRef]
- Yan, L.; Deng, Y.; Gao, J.; Liu, Y.; Li, F.; Shi, J.; Gong, Q. Icariside II Effectively Reduces Spatial Learning and Memory Impairments in Alzheimer’s Disease Model Mice Targeting Beta-Amyloid Production. Front. Pharmacol. 2017, 8, 106. [Google Scholar] [CrossRef]
- Li, X.; Cui, J.; Yu, Y.; Li, W.; Hou, Y.; Wang, X.; Qin, D.; Zhao, C.; Yao, X.; Zhao, J.; et al. Traditional Chinese Nootropic Medicine Radix Polygalae and Its Active Constituent Onjisaponin B Reduce beta-Amyloid Production and Improve Cognitive Impairments. PLoS ONE 2016, 11, e0151147. [Google Scholar] [CrossRef]
- Singh, J.C.; Kakalij, R.M.; Kshirsagar, R.P.; Kumar, B.H.; Komakula, S.S.; Diwan, P.V. Cognitive effects of vanillic acid against streptozotocin-induced neurodegeneration in mice. Pharm. Biol. 2015, 53, 630–636. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Wei, Y.; Zhou, L.; Liu, L.; Yin, P.; Liu, Y.; Wu, S.; Li, J.; Lu, C. Onjisaponin B (OB) is Neuroprotective During Cognitive Loss Through Immune-mediated and SIRT1 Pathways. Curr. Neurovasc Res. 2018, 15, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wang, Y.; He, J.; Cai, T.; Wu, J.; Fang, J.; Zhang, R.; Guo, Z.; Guan, L.; Zhan, Q.; et al. Neuroprotective effects of bajijiasu against cognitive impairment induced by amyloid-beta in APP/PS1 mice. Oncotarget 2017, 8, 92621–92634. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, D.L.; Zhang, P.; Lin, L.; Shuai, O.; Zhang, H.M.; Liu, S.H.; Wang, J.Y. Protective effect of Bajijiasu against beta-amyloid-induced neurotoxicity in PC12 cells. Cell Mol. Neurobiol. 2013, 33, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.T.; Zhang, Y.; He, J.Y.; Luo, D.; Luo, Y.; Wang, Y.J.; Liu, W.; Wu, J.; Zhao, W.; Fang, J.; et al. Bajijiasu Ameliorates beta-Amyloid-Triggered Endoplasmic Reticulum Stress and Related Pathologies in an Alzheimer’s Disease Model. Cell Physiol. Biochem. 2018, 46, 107–117. [Google Scholar] [CrossRef]
- Mao, J.; Huang, S.; Liu, S.; Feng, X.L.; Yu, M.; Liu, J.; Sun, Y.E.; Chen, G.; Yu, Y.; Zhao, J.; et al. A herbal medicine for Alzheimer’s disease and its active constituents promote neural progenitor proliferation. Aging Cell 2015, 14, 784–796. [Google Scholar] [CrossRef]
- Yang, C.; Li, X.; Mo, Y.; Liu, S.; Zhao, L.; Ma, X.; Fang, Z.; Chen, J.; Chen, Y.; Yu, X.; et al. beta-Asarone Mitigates Amyloidosis and Downregulates RAGE in a Transgenic Mouse Model of Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 121–130. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Q.; Zhang, R.; Liu, S.; Xia, Z.; Hu, Y. Catalpol ameliorates beta amyloid-induced degeneration of cholinergic neurons by elevating brain-derived neurotrophic factors. Neuroscience 2009, 163, 1363–1372. [Google Scholar] [CrossRef]
- Huang, J.Z.; Wu, J.; Xiang, S.; Sheng, S.; Jiang, Y.; Yang, Z.; Hua, F. Catalpol preserves neural function and attenuates the pathology of Alzheimer’s disease in mice. Mol. Med. Rep. 2016, 13, 491–496. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Draper, H.H.; Hadley, M. Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 1990, 186, 421–431. [Google Scholar] [CrossRef]
- Jia, S.L.; Wu, X.L.; Li, X.X.; Dai, X.L.; Gao, Z.L.; Lu, Z.; Zheng, Q.S.; Sun, Y.X. Neuroprotective effects of liquiritin on cognitive deficits induced by soluble amyloid-beta1-42 oligomers injected into the hippocampus. J. Asian Nat. Prod. Res. 2016, 18, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Small, D.H.; Mok, S.S.; Bornstein, J.C. Alzheimer’s disease and Abeta toxicity: from top to bottom. Nat. Rev. Neurosci. 2001, 2, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.M.; Ao, M.Z.; Li, W.; Yu, L.J. Effect of glabridin from Glycyrrhiza glabra on learning and memory in mice. Planta Med. 2008, 74, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Li, C.G. Tanshinone IIA and Cryptotanshinone Prevent Mitochondrial Dysfunction in Hypoxia-Induced H9c2 Cells: Association to Mitochondrial ROS, Intracellular Nitric Oxide, and Calcium Levels. Evid Based Complement. Alternat. Med. 2013, 2013, 610694. [Google Scholar] [CrossRef]
- Maione, F.; Piccolo, M.; De Vita, S.; Chini, M.G.; Cristiano, C.; De Caro, C.; Lippiello, P.; Miniaci, M.C.; Santamaria, R.; Irace, C.; et al. Down regulation of pro-inflammatory pathways by tanshinone IIA and cryptotanshinone in a non-genetic mouse model of Alzheimer’s disease. Pharmacol Res. 2018, 129, 482–490. [Google Scholar] [CrossRef]
- Li, F.; Wu, X.; Li, J.; Niu, Q. Ginsenoside Rg1 ameliorates hippocampal long-term potentiation and memory in an Alzheimer’s disease model. Mol. Med. Rep. 2016, 13, 4904–4910. [Google Scholar] [CrossRef]
- Zhu, J.; Mu, X.; Zeng, J.; Xu, C.; Liu, J.; Zhang, M.; Li, C.; Chen, J.; Li, T.; Wang, Y. Ginsenoside Rg1 prevents cognitive impairment and hippocampus senescence in a rat model of D-galactose-induced aging. PLoS ONE 2014, 9, e101291. [Google Scholar] [CrossRef]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Hu, J.P.; Yu, S.; Li, B.K.; Cui, Y.; Ren, L.; Zhang, L.D. Astragaloside IV, a Natural PPARgamma Agonist, Reduces Abeta Production in Alzheimer’s Disease Through Inhibition of BACE1. Mol. Neurobiol. 2017, 54, 2939–2949. [Google Scholar] [CrossRef]
- Fu, R.H.; Harn, H.J.; Liu, S.P.; Chen, C.S.; Chang, W.L.; Chen, Y.M.; Huang, J.E.; Li, R.J.; Tsai, S.Y.; Hung, H.S.; et al. n-butylidenephthalide protects against dopaminergic neuron degeneration and alpha-synuclein accumulation in Caenorhabditis elegans models of Parkinson’s disease. PLoS ONE 2014, 9, e85305. [Google Scholar] [CrossRef]
- Hsueh, K.W.; Chiou, T.W.; Chiang, S.F.; Yamashita, T.; Abe, K.; Borlongan, C.V.; Sanberg, P.R.; Huang, A.Y.; Lin, S.Z.; Harn, H.J. Autophagic down-regulation in motor neurons remarkably prolongs the survival of ALS mice. Neuropharmacology 2016, 108, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Hasanein, P. Glabridin as a major active isoflavan from Glycyrrhiza glabra (licorice) reverses learning and memory deficits in diabetic rats. Acta Physiol. Hung. 2011, 98, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.C.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Sreenivasmurthy, S.G.; Liu, J.Y.; Song, J.X.; Yang, C.B.; Malampati, S.; Wang, Z.Y.; Huang, Y.Y.; Li, M. Neurogenic Traditional Chinese Medicine as a Promising Strategy for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 272. [Google Scholar] [CrossRef]
- Liu, S.J.; Yang, C.; Zhang, Y.; Su, R.Y.; Chen, J.L.; Jiao, M.M.; Chen, H.F.; Zheng, N.; Luo, S.; Chen, Y.B.; et al. Neuroprotective effect of beta-asarone against Alzheimer’s disease: regulation of synaptic plasticity by increased expression of SYP and GluR1. Drug Des. Devel Ther. 2016, 10, 1461–1469. [Google Scholar] [CrossRef]
- Chang, C.P.; Liu, Y.F.; Lin, H.J.; Hsu, C.C.; Cheng, B.C.; Liu, W.P.; Lin, M.T.; Hsu, S.F.; Chang, L.S.; Lin, K.C. Beneficial Effect of Astragaloside on Alzheimer’s Disease Condition Using Cultured Primary Cortical Cells Under beta-amyloid Exposure. Mol. Neurobiol. 2016, 53, 7329–7340. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Mutation of iPSCs | iPS Cells Produce Effect | References |
|---|---|---|
| Presenilin-1 ΔE9 mutation | Impair γ-secretase activity but do not disrupt γ-secretase-independent functions of PSEN1 | Minna Oksanen [42] |
| FAD patients with mutations in PSEN1 (A246E) and PSEN2 (N141I) | Increased toxic Aβ1-42 secretion | Takuya Yagi [41] |
| Amyloid precursor protein (APP) A673T mutation | Protective against Alzheimer’s disease and cognitive decline | Šárka Lehtonen [38] |
| APP double mutation (KM670/671NL) | Increase the total Aβ burden | Minna Oksanen [36] |
| Duplication of the amyloid precursor protein gene (APP(Dp)) | Increased Aβ 1–40, p-tau (Thr 231), and active glycogen synthase kinase-3β | Mason A. Israel [37] |
| Trisomy of chromosome 21 (Ts21) | Aβ aggregation and increased tau protein | Glenn A. Maclean [46] |
| A152T variant in MAPT | A152T-iPS cells-derived neurons showed accumulation, redistribution, and decreased solubility of tau. | M. Catarina Silva [43] |
| MAPT gene mutations (N279K, P301L, and E10+16) | Deficiencies in neurite outgrowth and upregulation of neurodegenerative pathways | Juan Antonio García-León [44] |
| Extracts | Extracted from/Chinese Name | Drug Doses & Experimental Model | Possible Molecular Mechanism Effect | Reference |
|---|---|---|---|---|
| Icariin II | Herba Epimedii/淫羊藿 | APP/PS1 transgenic mice were treated orally g icariside II 30 m/kg. | Effectively ameliorated cognitive function deficits, but also inhibited neuronal degeneration and reduced the formation of plaque burden. | [88] |
| Onjisaponin B | Radix polygalae/遠志 | APP/PS1 transgenic mice were treated orally 200 μL of onjisaponin B 1 mg/mL. | The Onjisaponin B suppresses Aβ production without direct inhibition of β-secretase and γ-secretase activities. | [89] |
| Bajijiasu | Morinda officinalis/巴戟天 | APP/PS1 transgenic mice were treated orally 20 mg and 80 mg bajijiasu/kg. | Reductions of Aβ deposition and senile plaques and have higher levels of neurotrophic factors and inhibitory function on neuroinflammation in the brains of APP/PS1 mice. | [92] |
| Asarones | Rhizoma Acori tatarinowii/石菖蒲 | Adult hippocampal neural progenitor cells (NPC) cultures treated with 1 μM asarones. | The Asarones enhanced NPC proliferation and neurogenesis in the hippocampi of transgenic AD model mice. | [95] |
| Catalpol | Rehmannia glutinosa/地黃 | APP/PS1 transgenic mice were treated orally catalpol 50 mg / kg. | Improves memory and protects the forebrain neurons through increasing BDNF expression. | [97] |
| Liquiritin | Glycyrrhizae radix/甘草 | Soluble amyloid-β1-42 oligomers injected into the hippocampus induced cognitive-deficit rats. Treated by liquiritin was orally 50~100 mg/kg. | The drugs improve Aβ1-42-induced spatial learning and memory impairment through inhibiting oxidative stress and neural apoptosis. | [101] |
| Glabridin | Glycyrrhiza glabra/洋甘草 | Oral glabridin 25 and 50 mg/kg treated for diabetic rats [112]. | It reversed learning and memory deficits of diabetic rats. | [103] |
| Tanshinone IIA | Salviae miltiorrhizae/丹参 | Oral 10mg/kg treated for nongenetic mouse model of β-amyloid-induced AD. | That TIIA and CT display anti-inflammatory and neuron-protective effects in a nongenetic mouse model of AD. | [105]. |
| Cryptotanshinone | ||||
| Ginsenoside Rg1 | Radix ginseng/人參 | Oral 20 mg/kg D-galactose-induced aging rat model. | It prevents cognitive impairment and hippocampus senescence of D-galactose-induced aging in a rat model. | [107] |
| Astragaloside IV | Astragalus membranaceus/黃耆 | APP/PS1 mice that were treated orally AS-IV 10 mg/kg. | AS-IV treatment increased PPARγ and reduced Aβ plaque formation in the brain. | [109] |
| n-Butylidenephthalide | Angelica sinensis/當歸 | Down syndrome - iPSCs were treated n-BP 10 μM for 3 days. | n-BP benefitted AD treatment by scavenging Aβ aggregates and neurofibrillary tangles. | [40] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wuli, W.; Tsai, S.-T.; Chiou, T.-W.; Harn, H.-J. Human-Induced Pluripotent Stem Cells and Herbal Small-Molecule Drugs for Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1327. https://doi.org/10.3390/ijms21041327
Wuli W, Tsai S-T, Chiou T-W, Harn H-J. Human-Induced Pluripotent Stem Cells and Herbal Small-Molecule Drugs for Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(4):1327. https://doi.org/10.3390/ijms21041327
Chicago/Turabian StyleWuli, Wei, Sheng-Tzung Tsai, Tzyy-Wen Chiou, and Horng-Jyh Harn. 2020. "Human-Induced Pluripotent Stem Cells and Herbal Small-Molecule Drugs for Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 4: 1327. https://doi.org/10.3390/ijms21041327
APA StyleWuli, W., Tsai, S.-T., Chiou, T.-W., & Harn, H.-J. (2020). Human-Induced Pluripotent Stem Cells and Herbal Small-Molecule Drugs for Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences, 21(4), 1327. https://doi.org/10.3390/ijms21041327