Atomistic Insights of Calmodulin Gating of Complete Ion Channels


Abstract
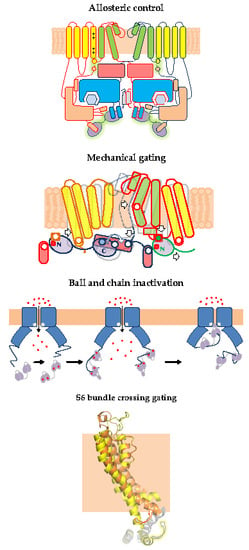
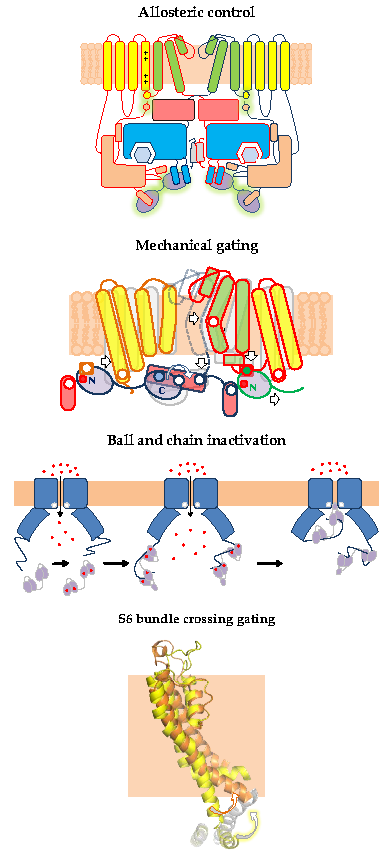{kind=link}
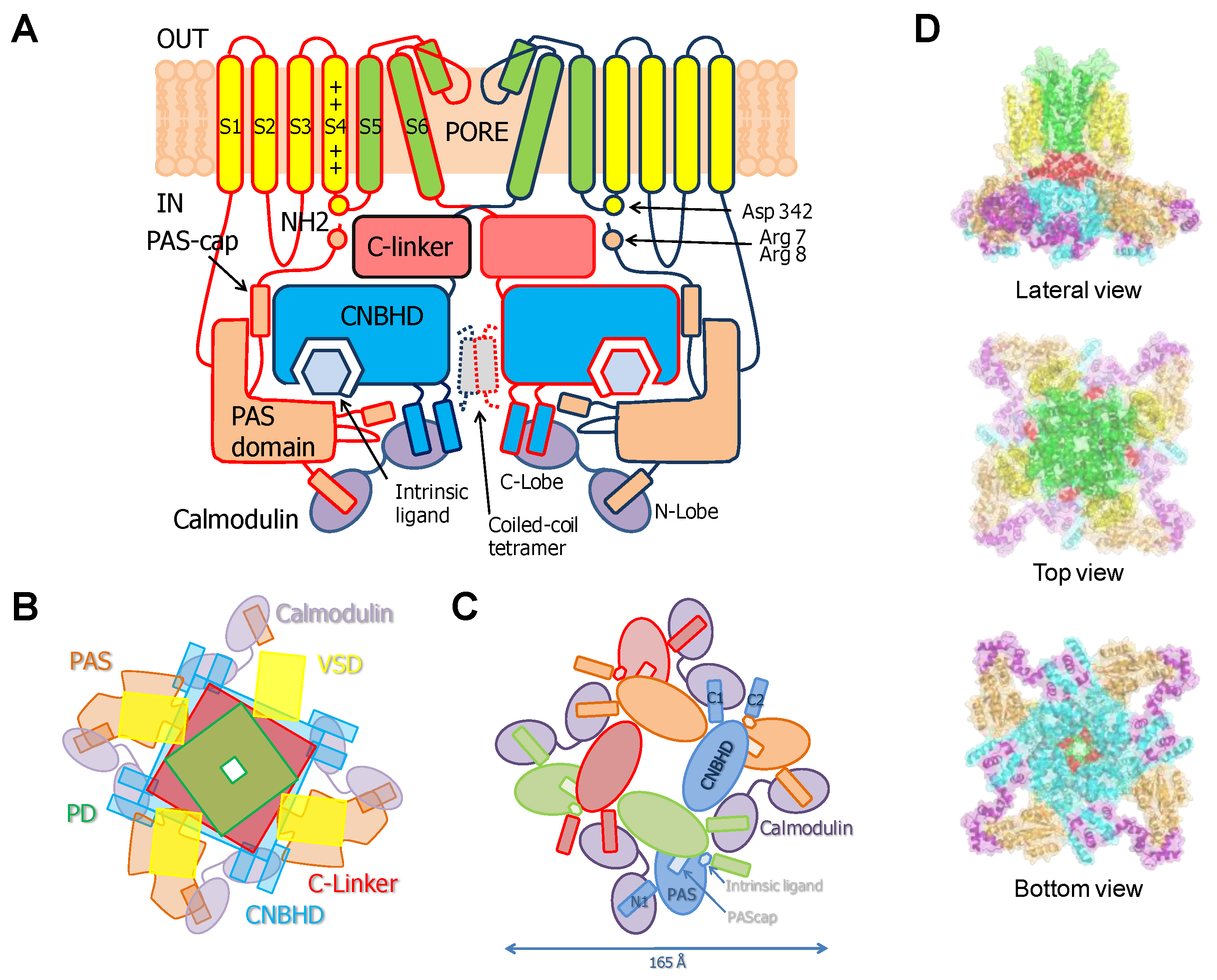{kind=link}
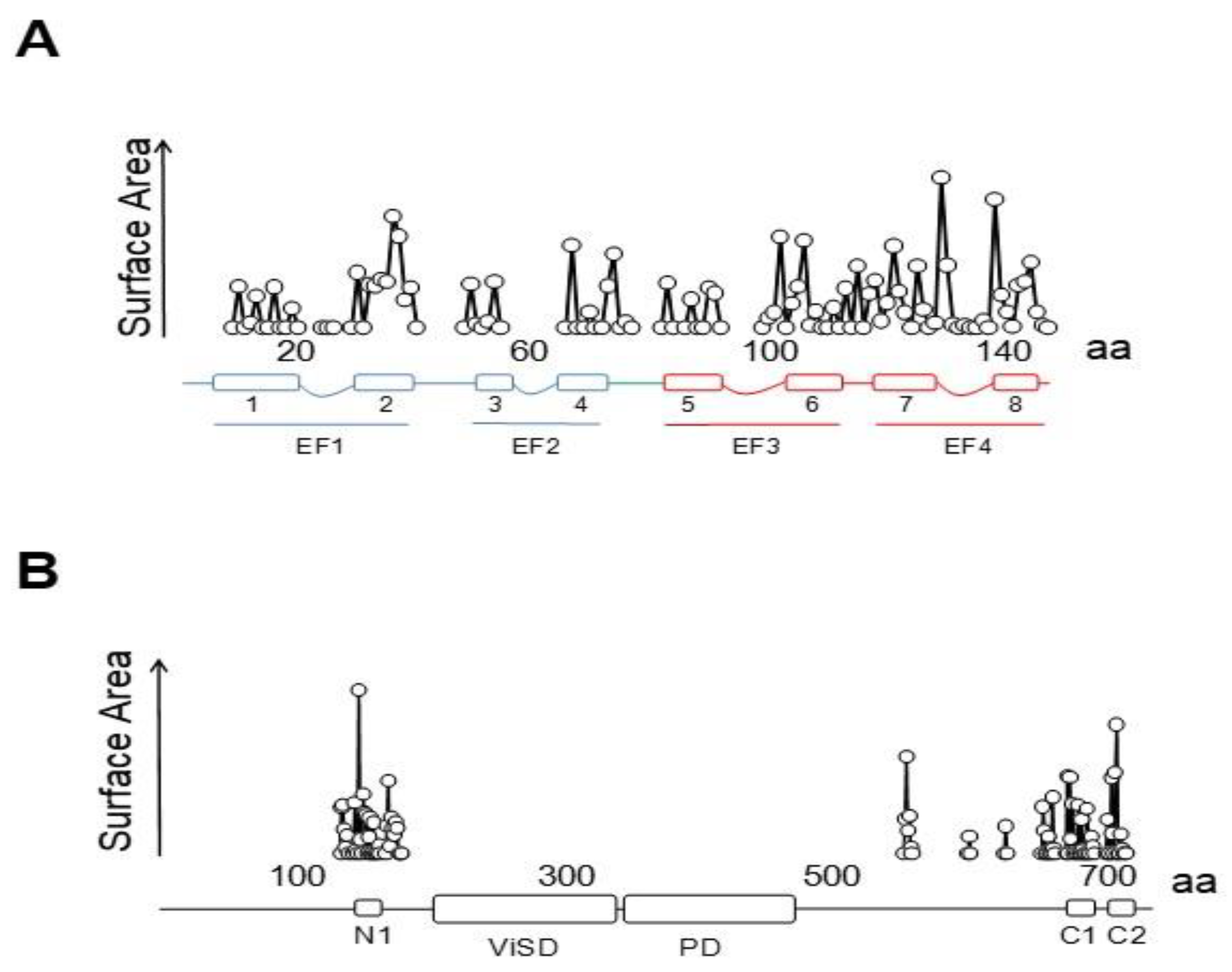{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
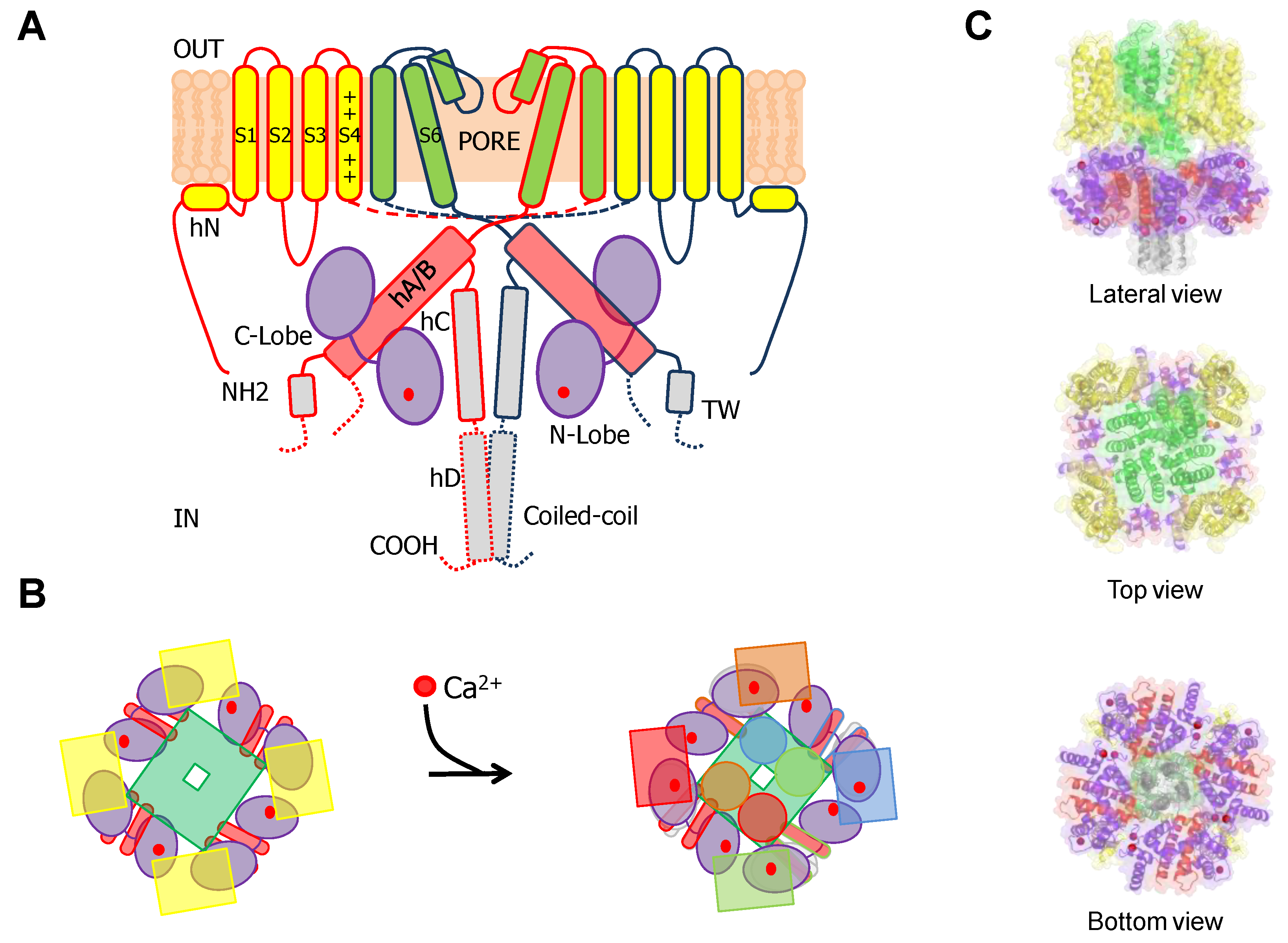{kind=link}
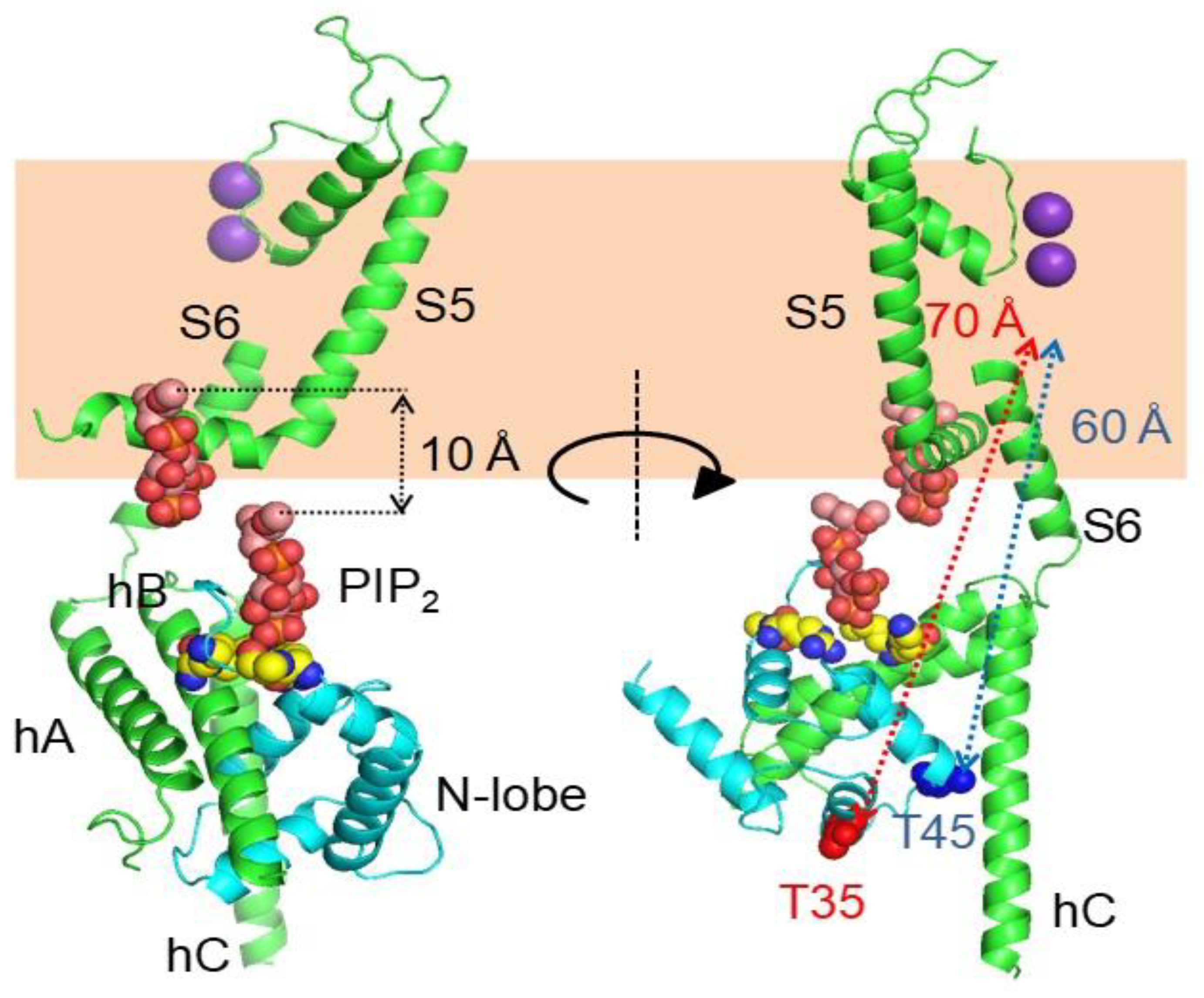{kind=link}
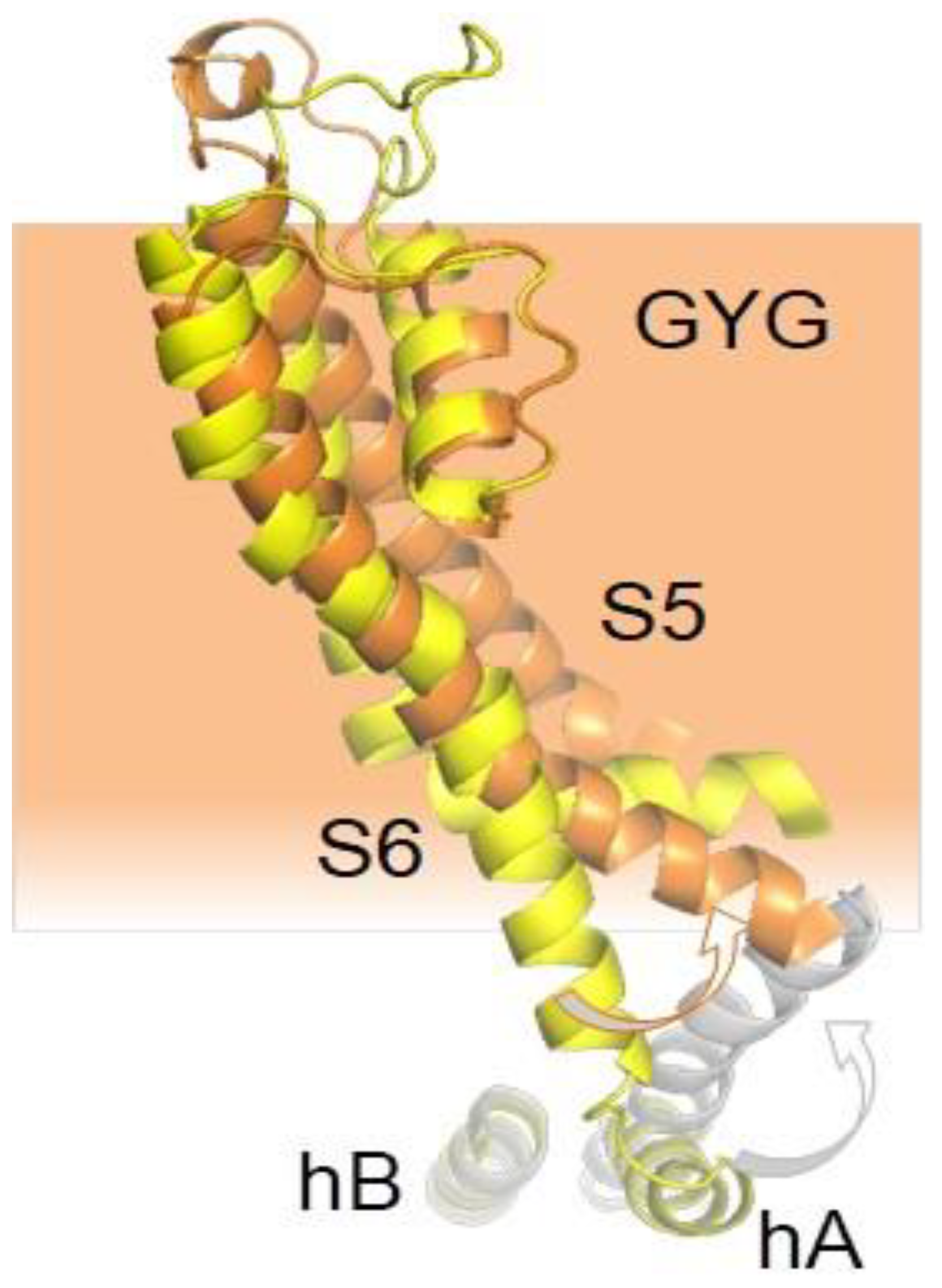{kind=link}
{kind=link}
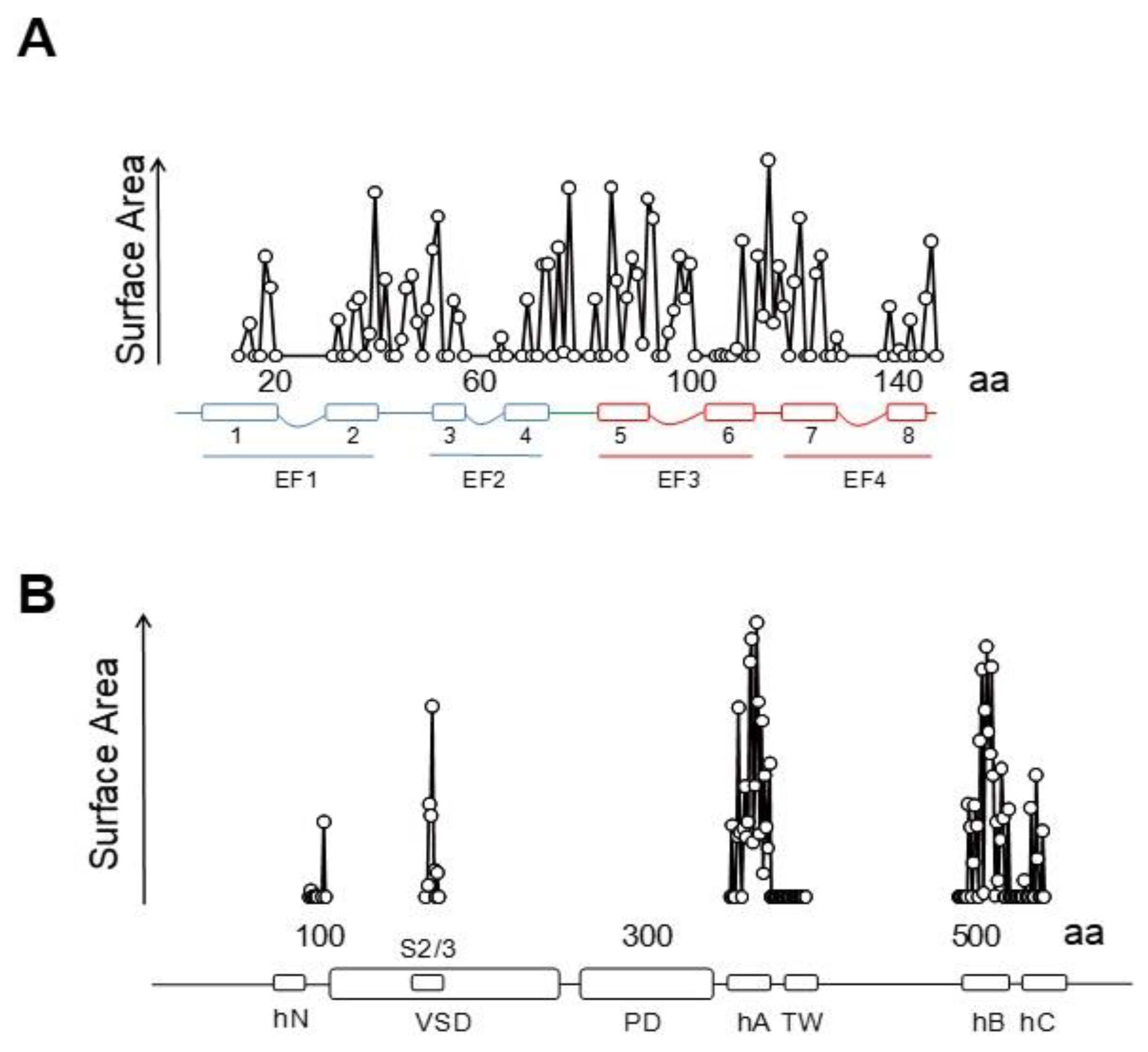{kind=link}
1. Gating of 6TM Ion Channels
2. Eag Channels
2.1. Allosteric Control of Eag Channel Gating by Calmodulin
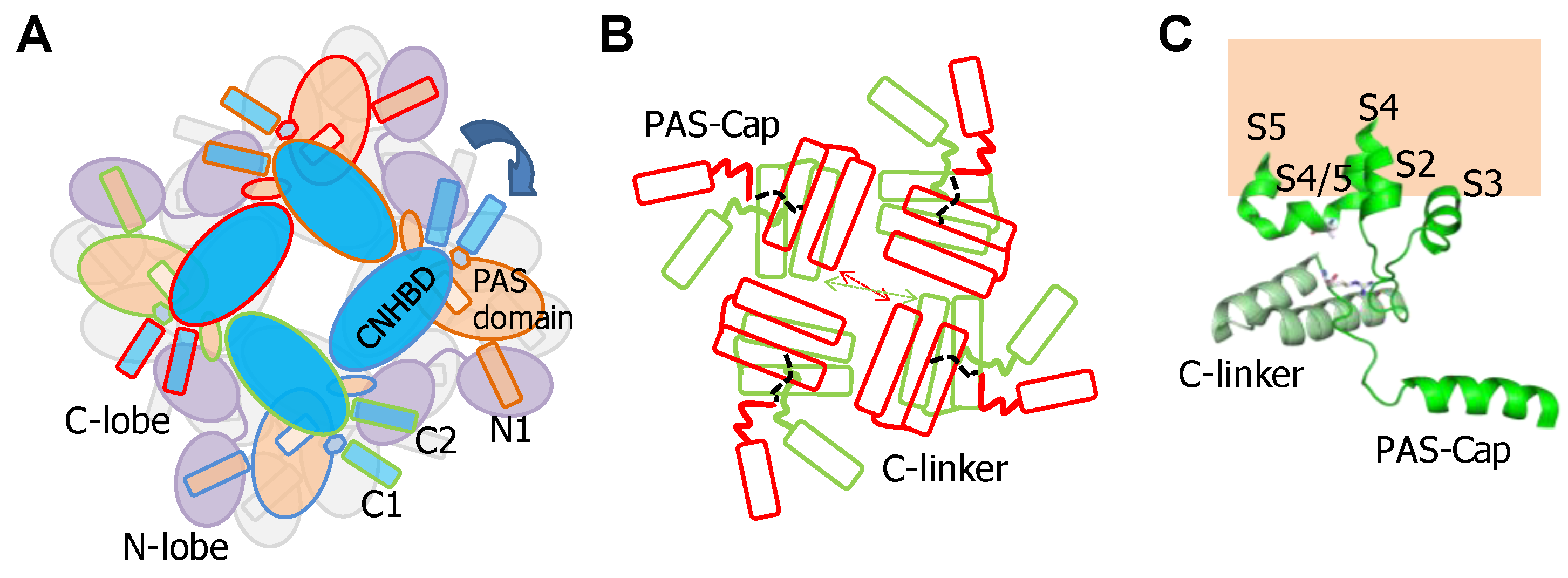
2.2. The PAS-Cap Domain of Eag Channels is Critical for Calmodulin-Mediated Gating
2.3. Interplay between PAS-Cap and the S4/5 Linker in Eag Channels
2.4. The PAS/CNBHD Complex of Eag Channels is not Compacted by Calmodulin
3. SK Channels
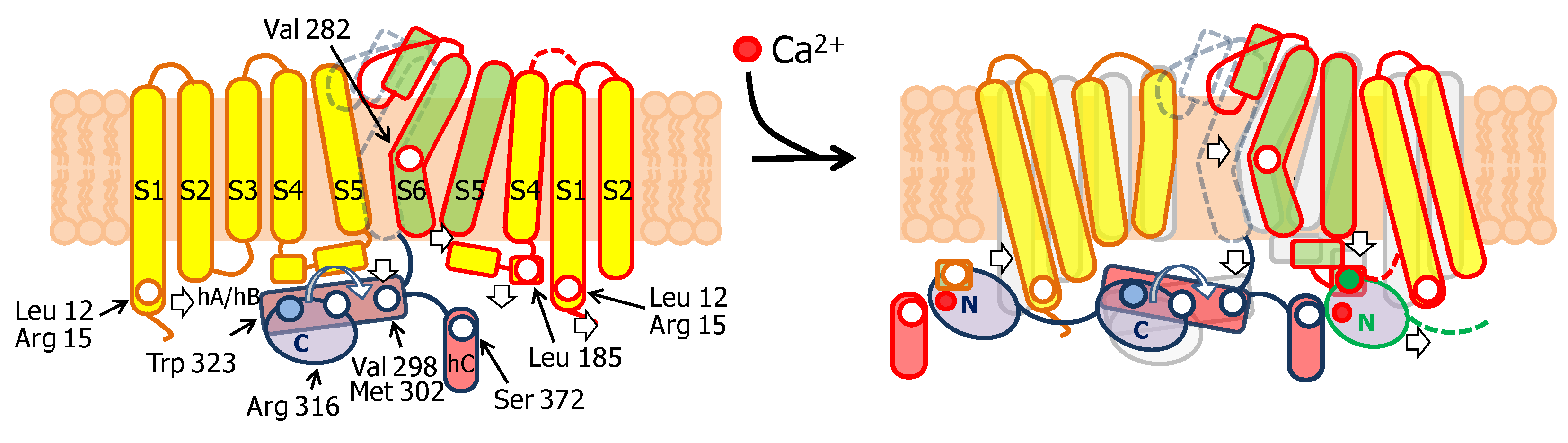
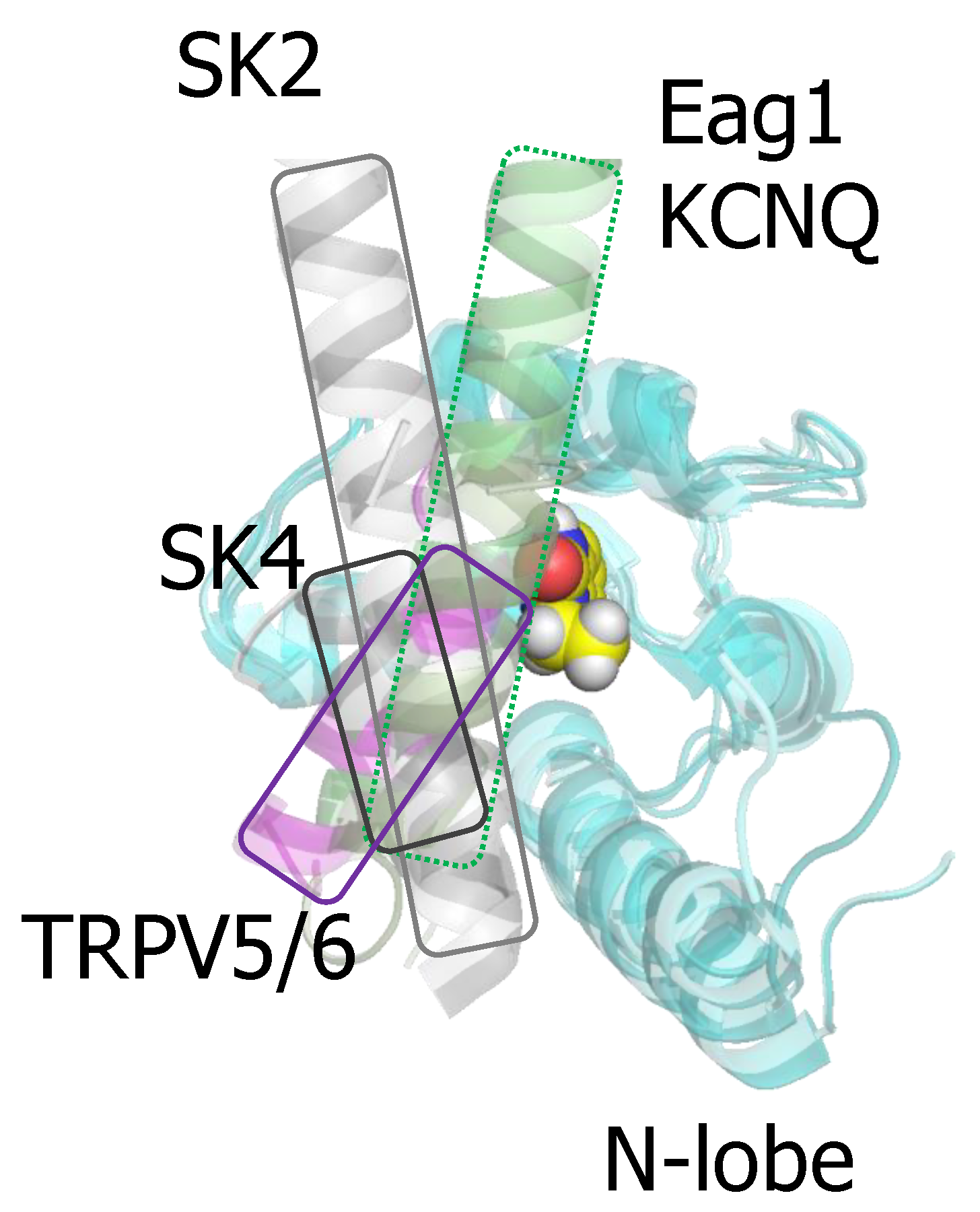
3.1. The N-Lobe Acts upon the S4/5 Linker and the ViSD to Mechanically Pull the Gate Open of SK4 Channels
3.2. The N-lobe Mediates Gating on SK Channels
3.3. Some Clinically Relevant Drugs Stabilize the Interaction with the N-lobe in SK Channels
3.4. The Interaction between SK Channels and Calmodulin is Regulated by CK2-Mediated Phosphorylation of the Lobe Linker
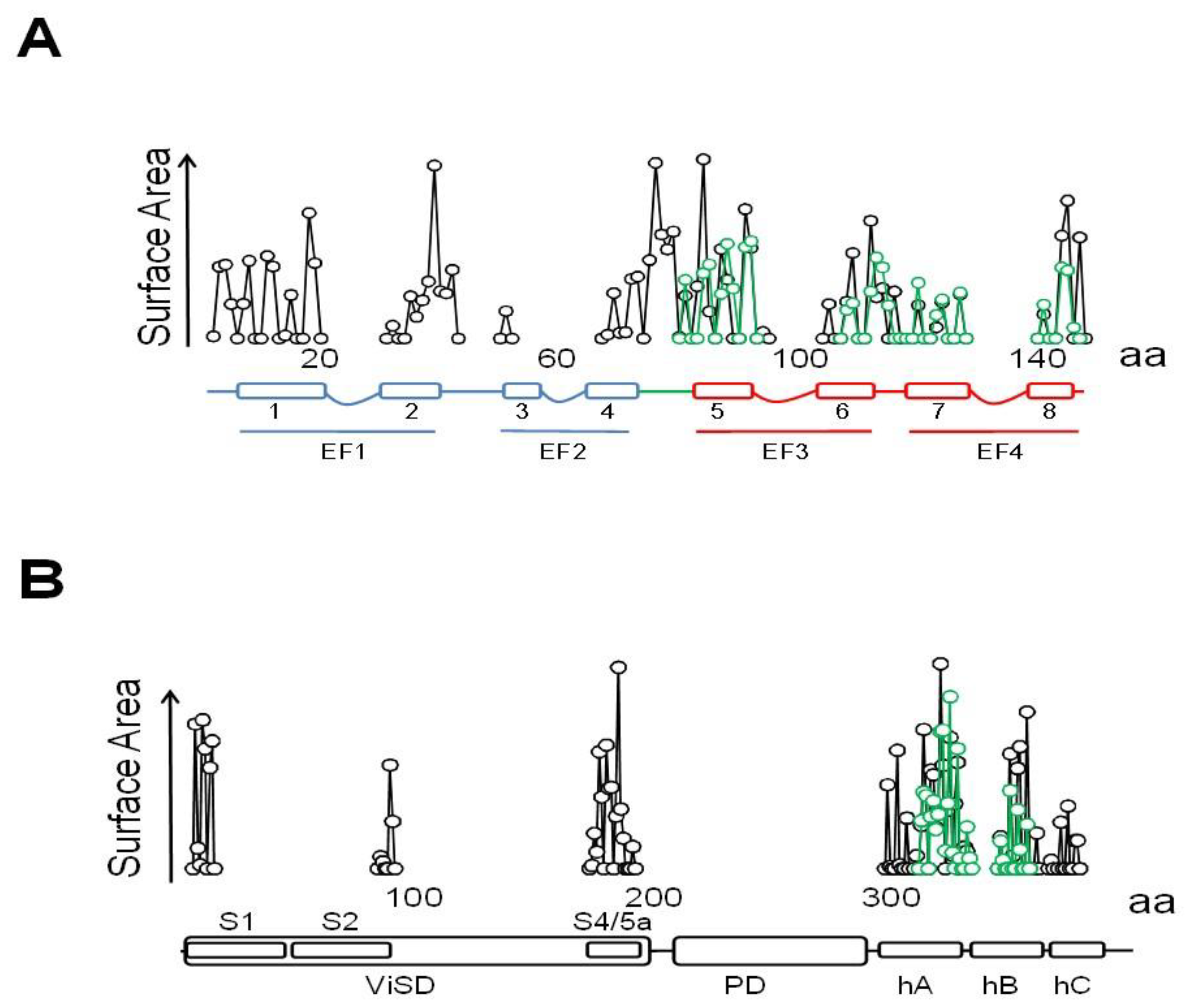
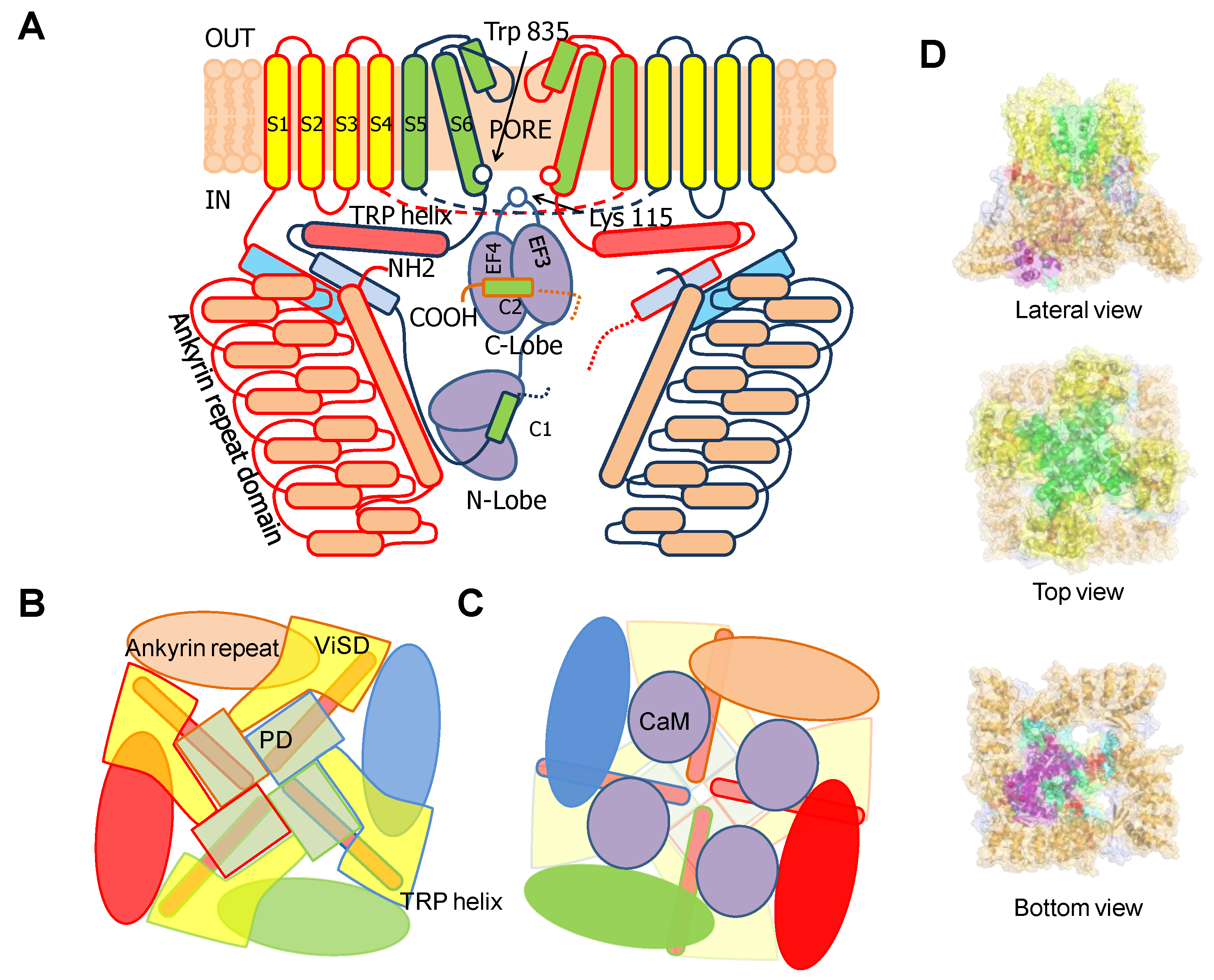
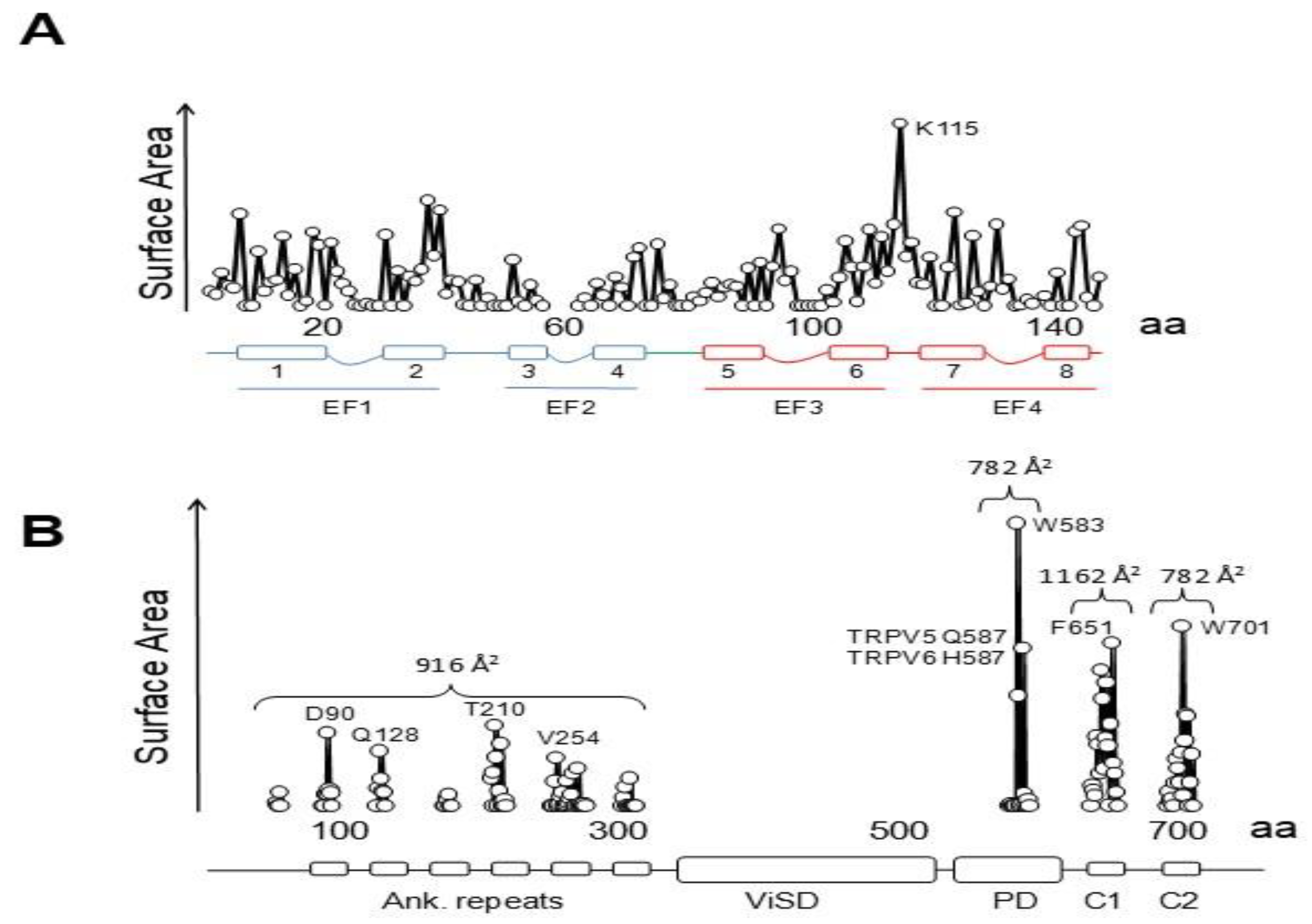
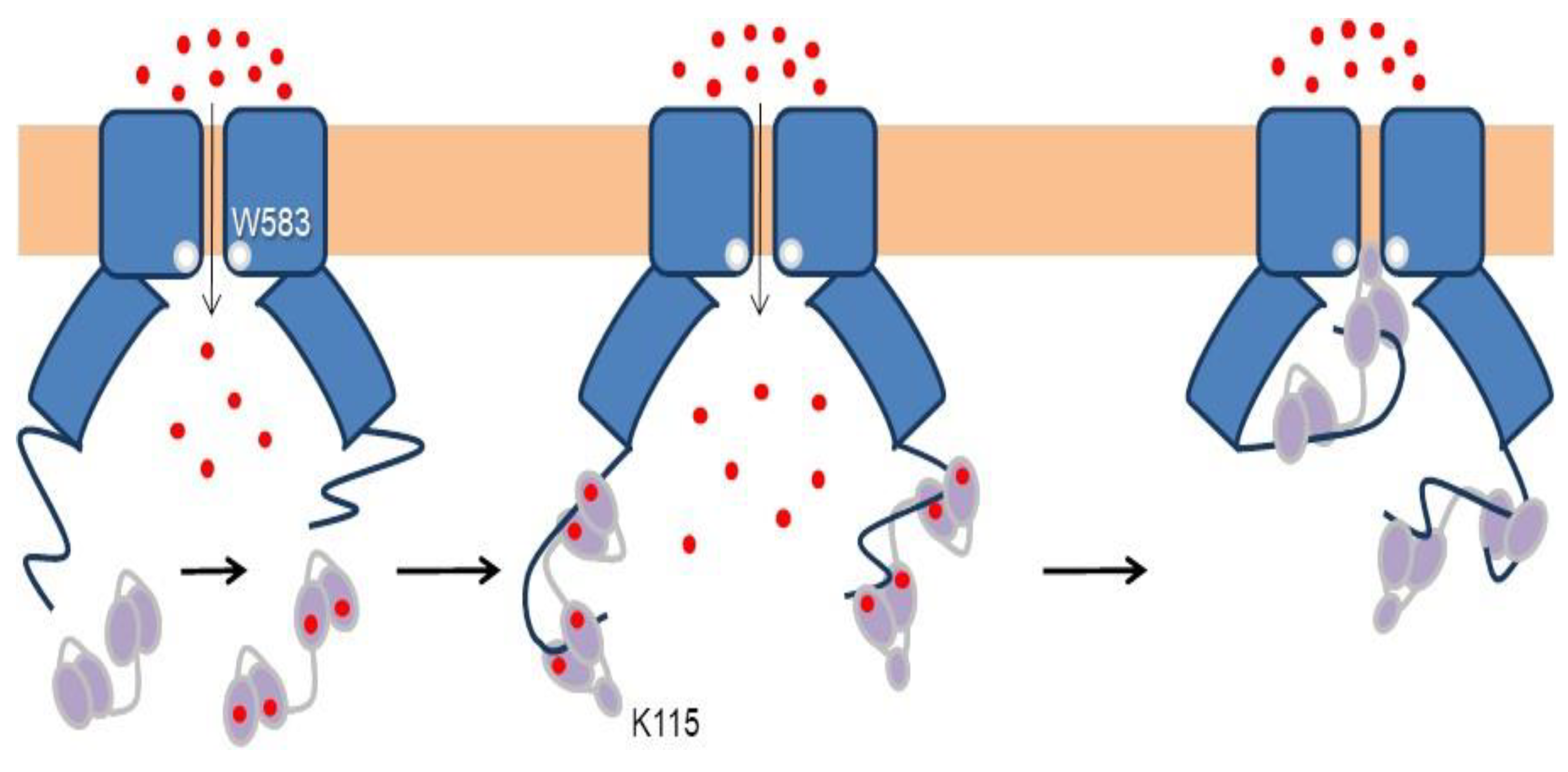
4. TRPV5/6 Channels
4.1. A Ball and Chain Mechanism for Holo-CaM-Dependent Inactivation of TRPV5 and TRPV6 Channels
4.2. Variable Stoichiometry of the Calmodulin-TRPV5/6 Complexes
5. KCNQ channels
5.1. KCNQ Channels Can be Potentiated or Inhibited by Calcium
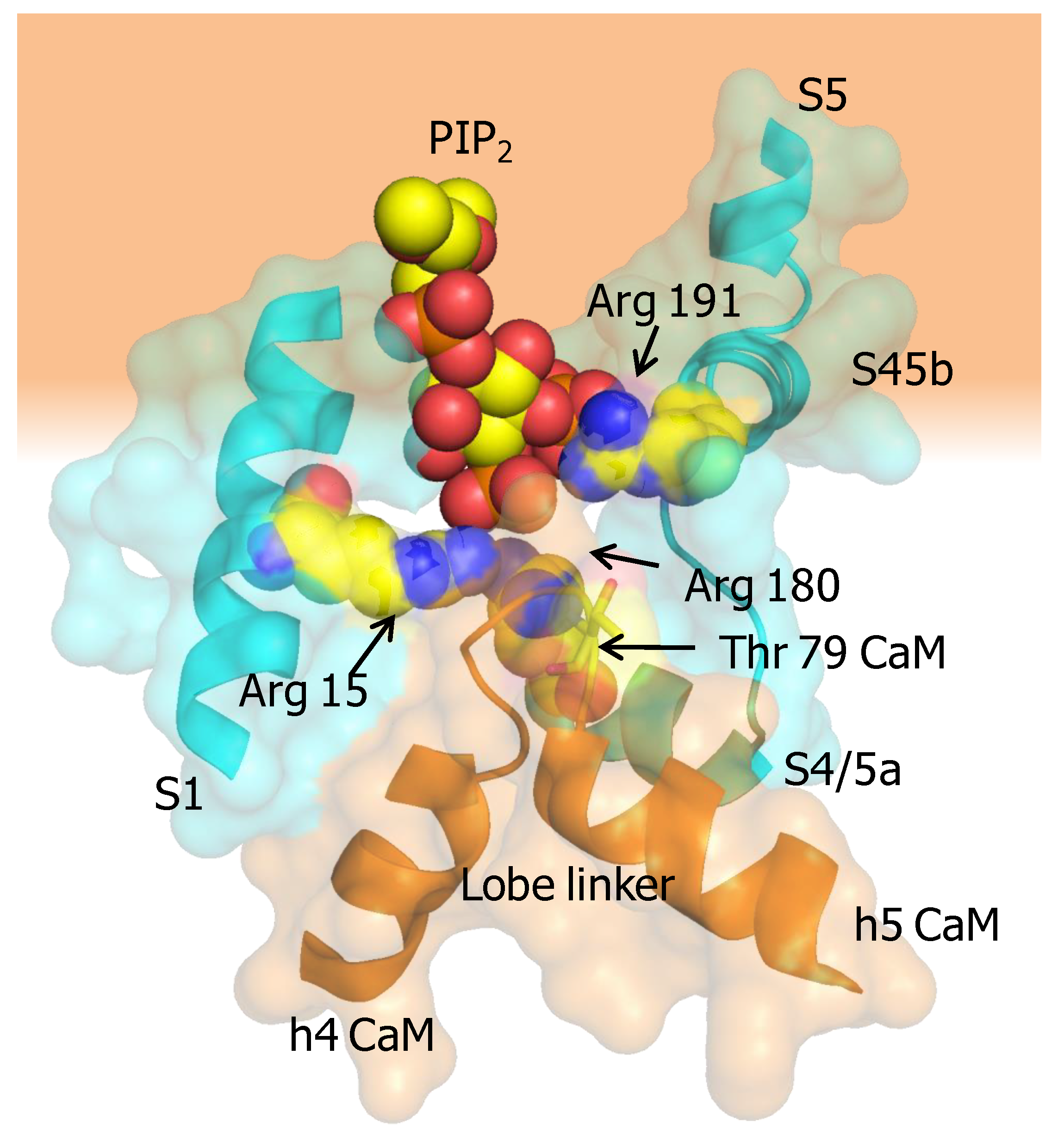
5.2. The PIP2 Site Delineated by CaM and Helices B/C Linker is Far from the Membrane in the Cryo-EM Structure of KCNQ1
5.3. Calmodulin may Act Directly upon the S6 Gate of KCNQ Channels
5.4. Calmodulin May Alter PIP2 Binding to KCNQ Channels by Two Mechanisms
5.5. The Interaction between KCNQ2 Channels and Calmodulin is Regulated by CK2-Mediated Phosphorylation of the Lobe Linker
6. Summary
| Eag1 | SK | TRPV5/6 | KCNQ | ||
| CaM Docking | N-lobe | N1 (holo) [23,24] | S4-S5 (holo) [45] | C1 (holo) [79,80,81] | hB (holo) [117] |
| C-lobe | C1, C2 (holo) [23] | hA (apo) [45] | C2 (holo) [79,80,81] | hA (apo) [117] | |
| Ca2+ signaling | C-lobe [30] | N-lobe [45] | EF3/4 linker [79,80,81] | C-lobe [85,97,121] | |
| Residence/motility | N-lobe | Dynamic [25] | Dynamic [45] | Dynamic [75] | Static [85,97,121] |
| C-lobe | Dynamic [25] | Static [45] | Dynamic [75] | Partially static [85,97,121] | |
| Mechanism | Allosteric(S4/5, PAS cap) [20] | Mechanical (ViSD, S4/5a) [45] | Direct pore [75] | Indirect (PIP2, S6 gate) [97,121] | |
| EC50 Ca2+ | 70–100 nM [25,29] | 440 nM [50] | 90 nM [67] | ? | |
| Ca2+ Effect | Inhibition [25,29] | Activation [39] | Blockade [79,80,81] | ? |
7. Outlook
Author Contributions
Funding
Conflicts of Interest
Note
References
- Urrutia, J.; Aguado, A.; Muguruza-Montero, A.; Nunez, E.; Malo, C.; Casis, O.; Villarroel, A. The Crossroad of Ion Channels and Calmodulin in Disease. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Yazdani, M.; Zhang, G.; Cui, J.; Chen, J. Hydrophobic gating in BK channels. Nat. Commun. 2018, 9, 3408. [Google Scholar] [CrossRef] [PubMed]
- Hite, R.K.; Tao, X.; MacKinnon, R. Structural basis for gating the high-conductance Ca2+-activated K+ channel. Nature 2017, 541, 52–57. [Google Scholar] [CrossRef]
- Tao, X.; Hite, R.K.; MacKinnon, R. Cryo-EM structure of the open high-conductance Ca2+-activated K+ channel. Nature 2017, 541, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Klesse, G.; Stansfeld, P.J.; Tucker, S.J.; Sansom, M.S.P. A heuristic derived from analysis of the ion channel structural proteome permits the rapid identification of hydrophobic gates. Proc. Natl. Acad. Sci USA 2019, 116, 13989–1399. [Google Scholar] [CrossRef]
- Thouta, S.; Sokolov, S.; Abe, Y.; Clark, S.J.; Cheng, Y.M.; Claydon, T.W. Proline scan of the HERG channel S6 helix reveals the location of the intracellular pore gate. Biophys. J. 2014, 106, 1057–1069. [Google Scholar] [CrossRef]
- Tomczak, A.P.; Fernandez-Trillo, J.; Bharill, S.; Papp, F.; Panyi, G.; Stuhmer, W.; Isacoff, E.Y.; Pardo, L.A. A new mechanism of voltage-dependent gating exposed by KV10.1 channels interrupted between voltage sensor and pore. J. Gen. Physiol 2017, 149, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.M.; Yarov-Yarovoy, V.; Agarwal, G.; Roux, B.; Barth, P.; Kohout, S.; Tombola, F.; Isacoff, E.Y. Closing in on the resting state of the Shaker K+ channel. Neuron 2007, 56, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K+ Channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef]
- Santos, J.S.; Grigoriev, S.M.; Montal, M. Molecular template for a voltage sensor in a novel K+ channel. III. Functional reconstitution of a sensorless pore module from a prokaryotic Kv channel. J Gen. Physiol 2008, 132, 651–666. [Google Scholar] [CrossRef]
- Vargas, E.; Yarov-Yarovoy, V.; Khalili-Araghi, F.; Catterall, W.A.; Klein, M.L.; Tarek, M.; Lindahl, E.; Schulten, K.; Perozo, E.; Bezanilla, F.; et al. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J. Gen. Physiol 2012, 140, 587–594. [Google Scholar] [CrossRef]
- Gonzalez, C.; Contreras, G.F.; Peyser, A.; Larsson, P.; Neely, A.; Latorre, R. Voltage sensor of ion channels and enzymes. Biophys. Rev. 2012, 4, 1–15. [Google Scholar] [CrossRef]
- Gandhi, C.S.; Clark, E.; Loots, E.; Pralle, A.; Isacoff, E.Y. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron 2003, 40, 515–525. [Google Scholar] [CrossRef]
- Lorinczi, E.; Gomez-Posada, J.C.; de la Pena, P.; Tomczak, A.P.; Fernandez-Trillo, J.; Leipscher, U.; Stuhmer, W.; Barros, F.; Pardo, L.A. Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat Commun 2015, 6, 6672. [Google Scholar] [CrossRef]
- Barros, F.; Pardo, L.A.; Dominguez, P.; Sierra, L.M.; de la Pena, P. New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- De la Pena, P.; Dominguez, P.; Barros, F. Gating mechanism of Kv11.1 (hERG) K+ channels without covalent connection between voltage sensor and pore domains. Pflugers Arch. 2018, 470, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; Stuhmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Bauer, C.K.; Schwarz, J.R. Ether-a-go-go K+ channels: Effective modulators of neuronal excitability. J Physiol 2018, 596, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; del Camino, D.; Sanchez, A.; Alves, F.; Bruggemann, A.; Beckh, S.; Stuhmer, W. Oncogenic potential of EAG K+ channels. Embo. J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef]
- Whicher, J.R.; MacKinnon, R. Regulation of Eag1 gating by its intracellular domains. Elife 2019, 8. [Google Scholar] [CrossRef]
- Jenke, M.; Sanchez, A.; Monje, F.; Stuhmer, W.; Weseloh, R.M.; Pardo, L.A. C-terminal domains implicated in the functional surface expression of potassium channels. Embo. J. 2003, 22, 395–403. [Google Scholar] [CrossRef]
- Brelidze, T.I.; Carlson, A.E.; Sankaran, B.; Zagotta, W.N. Structure of the carboxy-terminal region of a KCNH channel. Nature 2012, 481, 530–533. [Google Scholar] [CrossRef]
- Whicher, J.R.; MacKinnon, R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 2016, 353, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Marques-Carvalho, M.J.; Oppermann, J.; Munoz, E.; Fernandes, A.S.; Gabant, G.; Cadene, M.; Heinemann, S.H.; Schonherr, R.; Morais-Cabral, J.H. Molecular Insights into the Mechanism of Calmodulin Inhibition of the EAG1 Potassium Channel. Structure. 2016, 24, 1742–1754. [Google Scholar] [CrossRef]
- Schönherr, R.; Lober, K.; Heinemann, S.H. Inhibition of human ether a go-go potassium channels by Ca2+/calmodulin. EMBO J. 2000, 19, 3263–3271. [Google Scholar] [CrossRef]
- Ziechner, U.; Schonherr, R.; Born, A.K.; Gavrilova-Ruch, O.; Glaser, R.W.; Malesevic, M.; Kullertz, G.; Heinemann, S.H. Inhibition of human ether a go-go potassium channels by Ca2+/calmodulin binding to the cytosolic N- and C-termini. FEBS J. 2006, 273, 1074–1086. [Google Scholar] [CrossRef]
- Sobolev, V.; Sorokine, A.; Prilusky, J.; Abola, E.E.; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999, 15, 327–332. [Google Scholar] [CrossRef]
- Goncalves, J.T.; Stuhmer, W. Calmodulin interaction with hEAG1 visualized by FRET microscopy. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Stansfeld, C.E.; Roper, J.; Ludwig, J.; Weseloh, R.M.; Marsh, S.J.; Brown, D.A.; Pongs, O. Elevation of intracellular calcium by muscarinic receptor activation induces a block of voltage-activated rat ether-a-go-go channels in a stably transfected cell line. Proc. Natl. Acad. Sci. USA 1996, 93, 9910–9914. [Google Scholar] [CrossRef] [PubMed]
- Lorinczi, E.; Helliwell, M.; Finch, A.; Stansfeld, P.J.; Davies, N.W.; Mahaut-Smith, M.; Muskett, F.W.; Mitcheson, J.S. Calmodulin Regulates Human Ether a Go-Go 1 (hEAG1) Potassium Channels through Interactions of the Eag Domain with the Cyclic Nucleotide Binding Homology Domain. J. Biol. Chem. 2016, 291, 17907–17918. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gayen, S.; Chen, A.S.; Huang, Q.; Raida, M.; Kang, C. NMR solution structure of the N-terminal domain of hERG and its interaction with the S4-S5 linker. Biochem. Biophys. Res. Commun. 2010, 403, 126–132. [Google Scholar] [CrossRef]
- Muskett, F.W.; Thouta, S.; Thomson, S.J.; Bowen, A.; Stansfeld, P.J.; Mitcheson, J.S. Mechanistic insight into human ether-a-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 2011, 286, 6184–6191. [Google Scholar] [CrossRef]
- Terlau, H.; Heinemann, S.H.; Stuhmer, W.; Pongs, O.; Ludwig, J. Amino terminal-dependent gating of the potassium channel rat eag is compensated by a mutation in the S4 segment. J. Physiol. 1997, 502, 537–543. [Google Scholar] [CrossRef]
- Rodriguez-Castaneda, F.; Maestre-Martinez, M.; Coudevylle, N.; Dimova, K.; Junge, H.; Lipstein, N.; Lee, D.; Becker, S.; Brose, N.; Jahn, O.; et al. Modular architecture of Munc13/calmodulin complexes: Dual regulation by Ca2+ and possible function in short-term synaptic plasticity. EMBO J. 2010, 29, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, A.; Taglialatela, M.; Bernardo-Seisdedos, G.; Alaimo, A.; Agirre, J.; Alberdi, A.; Gomis-Perez, C.; Soldovieri, M.V.; Ambrosino, P.; Malo, C.; et al. The ever changing moods of calmodulin: How structural plasticity entails transductional adaptability. J. Mol. Biol. 2014, 426, 2717–2735. [Google Scholar] [CrossRef]
- Kohler, M.; Hirschberg, B.; Bond, C.T.; Kinzie, J.M.; Marrion, N.V.; Maylie, J.; Adelman, J.P. Small-conductance, calcium-activated potassium channels from mammalian brain. Science 1996, 273, 1709–1714. [Google Scholar] [CrossRef]
- Joiner, W.J.; Wang, L.Y.; Tang, M.D.; Kaczmarek, L.K. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA 1997, 94, 11013–11018. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.M.; Fakler, B.; Rivard, A.; Wayman, G.; Johnson-Pais, T.; Keen, J.E.; Ishii, T.; Hirschberg, B.; Bond, C.T.; Lutsenko, S.; et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 1998, 395, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Fanger, C.M.; Ghanshani, S.; Logsdon, N.J.; Rauer, H.; Kalman, K.; Zhou, J.; Beckingham, K.; Chandy, K.G.; Cahalan, M.D.; Aiyar, J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J. Biol. Chem. 1999, 274, 5746–5754. [Google Scholar] [CrossRef]
- Adelman, J.P.; Maylie, J.; Sah, P. Small-conductance Ca2+-activated K+ channels: Form and function. Annu. Rev. Physiol. 2012, 74, 245–269. [Google Scholar] [CrossRef]
- Feske, S.; Concepcion, A.R.; Coetzee, W.A. Eye on ion channels in immune cells. Sci Signal. 2019, 12. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef]
- Hirschberg, B.; Maylie, J.; Adelman, J.P.; Marrion, N.V. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol 1998, 111, 565–581. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Rivard, A.F.; Bachinger, H.P.; Adelman, J.P. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 2001, 410, 1120–1124. [Google Scholar] [CrossRef]
- Lee, C.H.; MacKinnon, R. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 2018, 360, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Joiner, W.J.; Khanna, R.; Schlichter, L.C.; Kaczmarek, L.K. Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 2001, 276, 37980–37985. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Ngo-Anh, T.J.; Bruening-Wright, A.; Maylie, J.; Adelman, J.P. Small conductance Ca2+-activated K+ channels and calmodulin - Cell surface expression and gating. J. Biol. Chem. 2003, 278, 25940–25946. [Google Scholar] [CrossRef]
- Alaimo, A.; Alberdi, A.; Gomis-Perez, C.; Fernandez-Orth, J.; Bernardo-Seisdedos, G.; Malo, C.; Millet, O.; Areso, P.; Villarroel, A. Pivoting between Calmodulin Lobes Triggered by Calcium in the Kv7.2/Calmodulin Complex. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Zhang, M.; Pascal, J.M.; Zhang, J.F. Unstructured to structured transition of an intrinsically disordered protein peptide in coupling Ca2+-sensing and SK channel activation. Proc. Natl. Acad. Sci. USA 2013, 110, 4828–4833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pascal, J.M.; Schumann, M.; Armen, R.S.; Zhang, J.F. Identification of the functional binding pocket for compounds targeting small-conductance Ca(2)(+)-activated potassium channels. Nat. Commun. 2012, 3, 1021. [Google Scholar] [CrossRef]
- Zhang, M.; Meng, X.Y.; Cui, M.; Pascal, J.M.; Logothetis, D.E.; Zhang, J.F. Selective phosphorylation modulates the PIP2 sensitivity of the CaM-SK channel complex. Nat. Chem. Biol. 2014, 10, 753–759. [Google Scholar] [CrossRef]
- Halling, D.B.; Kenrick, S.A.; Riggs, A.F.; Aldrich, R.W. Calcium-dependent stoichiometries of the KCa2.2 (SK) intracellular domain/calmodulin complex in solution. J. Gen. Physiol 2014, 143, 231–252. [Google Scholar] [CrossRef]
- Simoes, M.; Garneau, L.; Klein, H.; Banderali, U.; Hobeila, F.; Roux, B.; Parent, L.; Sauve, R. Cysteine mutagenesis and computer modeling of the S6 region of an intermediate conductance IKCa channel. J. Gen. Physiol. 2002, 120, 99–116. [Google Scholar] [CrossRef]
- Klein, H.; Garneau, L.; Banderali, U.; Simoes, M.; Parent, L.; Sauve, R. Structural determinants of the closed KCa3.1 channel pore in relation to channel gating: Results from a substituted cysteine accessibility analysis. J. Gen. Physiol. 2007, 129, 299–315. [Google Scholar] [CrossRef]
- Shim, H.; Brown, B.M.; Singh, L.; Singh, V.; Fettinger, J.C.; Yarov-Yarovoy, V.; Wulff, H. The Trials and Tribulations of Structure Assisted Design of KCa Channel Activators. Front. Pharmacol. 2019, 10, 972. [Google Scholar] [CrossRef]
- Nam, Y.W.; Baskoylu, S.N.; Gazgalis, D.; Orfali, R.; Cui, M.; Hart, A.C.; Zhang, M. A V-to-F substitution in SK2 channels causes Ca2+ hypersensitivity and improves locomotion in a C. elegans ALS model. Sci. Rep. 2018, 8, 10749. [Google Scholar] [CrossRef]
- Alaimo, A.; Gomez-Posada, J.C.; Aivar, P.; Etxeberria, A.; Rodriguez-Alfaro, J.A.; Areso, P.; Villarroel, A. Calmodulin activation limits the rate of KCNQ2 K+ channel exit from the endoplasmic reticulum. J. Biol. Chem. 2009, 284, 20668–20675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Abrams, C.; Wang, L.; Gizzi, A.; He, L.; Lin, R.; Chen, Y.; Loll, P.J.; Pascal, J.M.; Zhang, J.F. Structural basis for calmodulin as a dynamic calcium sensor. Structure 2012, 20, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.W.; Orfali, R.; Liu, T.; Yu, K.; Cui, M.; Wulff, H.; Zhang, M. Structural insights into the potency of SK channel positive modulators. Sci. Rep. 2017, 7, 17178. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.T.; Alexandrou, A.J.; Torella, R.; Knafels, J.; Hobbs, J.; Taylor, T.; Loucif, A.; Konopacka, A.; Bell, S.; Stevens, E.B.; et al. An Intracellular Allosteric Modulator Binding Pocket in SK2 Ion Channels Is Shared by Multiple Chemotypes. Structure. 2018, 26, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.J.; Dreixler, J.C.; Couey, J.J.; Houamed, K.M. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur. J. Pharmacol 2002, 449, 47–54. [Google Scholar] [CrossRef]
- Dimitriadi, M.; Kye, M.J.; Kalloo, G.; Yersak, J.M.; Sahin, M.; Hart, A.C. The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J. Neurosci. 2013, 33, 6557–6562. [Google Scholar] [CrossRef]
- Allen, D.; Fakler, B.; Maylie, J.; Adelman, J.P. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J. Neurosci. 2007, 27, 2369–2376. [Google Scholar] [CrossRef]
- Arrigoni, G.; Marin, O.; Pagano, M.A.; Settimo, L.; Paolin, B.; Meggio, F.; Pinna, L.A. Phosphorylation of calmodulin fragments by protein kinase CK2. Mechanistic aspects and structural consequences. Biochemistry 2004, 43, 12788–12798. [Google Scholar] [CrossRef]
- Zhang, M.; Meng, X.Y.; Zhang, J.F.; Cui, M.; Logothetis, D.E. Molecular overlap in the regulation of SK channels by small molecules and phosphoinositides. Sci. Adv. 2015, 1, e1500008. [Google Scholar] [CrossRef]
- Van Goor, M.K.C.; Hoenderop, J.G.J.; van der Wijst, J. TRP channels in calcium homeostasis: From hormonal control to structure-function relationship of TRPV5 and TRPV6. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Prenen, J.; Vennekens, R.; Hoenderop, J.G.; Bindels, R.J.; Droogmans, G. Modulation of the epithelial calcium channel, ECaC, by intracellular Ca2+. Cell Calcium. 2001, 29, 417–428. [Google Scholar] [CrossRef]
- Nijenhuis, T.; Hoenderop, J.G.; Bindels, R.J. TRPV5 and TRPV6 in Ca2+ (re)absorption: Regulating Ca2+ entry at the gate. Pflugers Arch. 2005, 451, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Alaimo, A.; Rubert, J. The Pivotal Role of TRP Channels in Homeostasis and Diseases throughout the Gastrointestinal Tract. Int. J. Mol. Sci. 2019, 20, doi103390/ijms20215277. [Google Scholar] [CrossRef] [PubMed]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef]
- Lambers, T.T.; Weidema, A.F.; Nilius, B.; Hoenderop, J.G.; Bindels, R.J. Regulation of the mouse epithelial Ca2+ channel TRPV6 by the Ca2+-sensor calmodulin. J. Biol. Chem. 2004, 279, 28855–28861. [Google Scholar] [CrossRef] [PubMed]
- De Groot, T.; Kovalevskaya, N.V.; Verkaart, S.; Schilderink, N.; Felici, M.; van der Hagen, E.A.; Bindels, R.J.; Vuister, G.W.; Hoenderop, J.G. Molecular mechanisms of calmodulin action on TRPV5 and modulation by parathyroid hormone. Mol. Cell Biol. 2011, 31, 2845–2853. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Weidema, F.; Prenen, J.; Hoenderop, J.G.; Vennekens, R.; Hoefs, S.; Droogmans, G.; Bindels, R.J. The carboxyl terminus of the epithelial Ca2+ channel ECaC1 is involved in Ca2+-dependent inactivation. Pflugers Arch. 2003, 445, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Hofbauer, M.; Kahr, H.; Fritsch, R.; Muik, M.; Kepplinger, K.; Hack, M.E.; Moritz, S.; Schindl, R.; Groschner, K.; et al. Dynamic but not constitutive association of calmodulin with rat TRPV6 channels enables fine tuning of Ca2+-dependent inactivation. J. Physiol. 2006, 577, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Zakharian, E.; Borbiro, I.; Rohacs, T. Interplay between calmodulin and phosphatidylinositol 4,5-bisphosphate in Ca2+-induced inactivation of transient receptor potential vanilloid 6 channels. J. Biol. Chem. 2013, 288, 5278–5290. [Google Scholar] [CrossRef]
- Tran, Q.K.; Black, D.J.; Persechini, A. Intracellular coupling via limiting calmodulin. J. Biol. Chem. 2003, 278, 24247–24250. [Google Scholar] [CrossRef]
- Gregorio-Teruel, L.; Valente, P.; Liu, B.; Fernandez-Ballester, G.; Qin, F.; Ferrer-Montiel, A. The Integrity of the TRP Domain Is Pivotal for Correct TRPV1 Channel Gating. Biophys. J. 2015, 109, 529–541. [Google Scholar] [CrossRef]
- Van der Wijst, J.; Leunissen, E.H.; Blanchard, M.G.; Venselaar, H.; Verkaart, S.; Paulsen, C.E.; Bindels, R.J.; Hoenderop, J.G. A Gate Hinge Controls the Epithelial Calcium Channel TRPV5. Sci. Rep. 2017, 7, 45489. [Google Scholar] [CrossRef]
- Hughes, T.E.T.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V.; et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 4198. [Google Scholar] [CrossRef]
- Singh, A.K.; McGoldrick, L.L.; Twomey, E.C.; Sobolevsky, A.I. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci. Adv. 2018. [Google Scholar] [CrossRef]
- Dang, S.; van Goor, M.K.; Asarnow, D.; Wang, Y.; Julius, D.; Cheng, Y.; van der Wijst, J. Structural insight into TRPV5 channel function and modulation. Proc. Natl. Acad. Sci. USA 2019, 116, 8869–8878. [Google Scholar] [CrossRef]
- Hoshi, T.; Zagotta, W.N.; Aldrich, R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990, 250, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Morais-Cabral, J.H.; Mann, S.; MacKinnon, R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 2001, 411, 657–661. [Google Scholar] [CrossRef]
- Swain, S.M.; Sahoo, N.; Dennhardt, S.; Schonherr, R.; Heinemann, S.H. Ca2+/calmodulin regulates Kvbeta1.1-mediated inactivation of voltage-gated K+ channels. Sci. Rep. 2015, 5, 15509. [Google Scholar] [CrossRef]
- Chang, A.; Abderemane-Ali, F.; Hura, G.L.; Rossen, N.D.; Gate, R.E.; Minor, D.L., Jr. A Calmodulin C-Lobe Ca2+-Dependent Switch Governs Kv7 Channel Function. Neuron 2018, 97, 836–852. [Google Scholar] [CrossRef]
- Maljevic, S.; Wuttke, T.V.; Seebohm, G.; Lerche, H. Kv7 channelopathies. Pflugers Arch. 2010, 460, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Marrion, N.V. Control of M-current. Annu. Rev. Physiol. 1997, 59, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Tokimasa, T. Intracellular Ca2+-ions inactivate K+-current in bullfrog sympathetic neurons. Brain Res. 1985, 337, 386–391. [Google Scholar] [CrossRef]
- Kirkwood, A.; Simmons, M.A.; Mather, R.J.; Lisman, J. Muscarinic suppression of the M-current is mediated by a rise in internal Ca2+ concentration. Neuron 1991, 6, 1009–1014. [Google Scholar] [CrossRef]
- Ikeda, S.R.; Kammermeier, P.J. M current mystery messenger revealed? Neuron 2002, 35, 411–412. [Google Scholar] [CrossRef][Green Version]
- Suh, B.C.; Hille, B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 2002, 35, 507–520. [Google Scholar] [CrossRef]
- Zaydman, M.A.; Cui, J. PIP2 regulation of KCNQ channels: Biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Front Physiol 2014, 5, 195. [Google Scholar] [CrossRef]
- Marrion, N.V.; Zucker, R.S.; Marsh, S.J.; Adams, P.R. Modulation of M-current by intracellular Ca2+. Neuron 1991, 6, 533–545. [Google Scholar] [CrossRef]
- Selyanko, A.A.; Brown, D.A. Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 1996, 16, 151–162. [Google Scholar] [CrossRef]
- Gomez-Posada, J.C.; Aivar, P.; Alberdi, A.; Alaimo, A.; Etxeberria, A.; Fernandez-Orth, J.; Zamalloa, T.; Roura-Ferrer, M.; Villace, P.; Areso, P.; et al. Kv7 Channels Can Function without Constitutive Calmodulin Tethering. PLoS ONE 2011, e25508, 1055. [Google Scholar] [CrossRef]
- Shamgar, L.; Ma, L.J.; Schmitt, N.; Haitin, Y.; Peretz, A.; Wiener, R.; Hirsch, J.; Pongs, O.; Attali, B. Calmodulin is essential for cardiac IKs channel gating and assembly - Impaired function in long-QT mutations. Circ. Res. 2006, 98, 1055–1063. [Google Scholar] [CrossRef]
- Tobelaim, W.S.; Dvir, M.; Lebel, G.; Cui, M.; Buki, T.; Peretz, A.; Marom, M.; Haitin, Y.; Logothetis, D.E.; Hirsch, J.A.; et al. Competition of calcified calmodulin N lobe and PIP2 to an LQT mutation site in Kv7.1 channel. Proc. Natl. Acad. Sci. USA 2017, 114, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; MacKinnon, R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 2017, 169, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Yus-Nájera, E.; Santana-Castro, I.; Villarroel, A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 2002, 277, 28545–28553. [Google Scholar] [CrossRef]
- Gomis-Perez, C.; Alaimo, A.; Fernandez-Orth, J.; Alberdi, A.; Aivar-Mateo, P.; Bernardo-Seisdedos, G.; Malo, C.; Areso, P.; Felipe, A.; Villarroel, A. Unconventional calmodulin anchoring site within the AB module of Kv7.2 channels. J. Cell Sci. 2015, 128, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Wiener, R.; Haitin, Y.; Shamgar, L.; Fernandez-Alonso, M.C.; Martos, A.; Chomsky-Hecht, O.; Rivas, G.; Attali, B.; Hirsch, J.A. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J. Biol. Chem. 2008, 283, 5815–5830. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Nunziato, D.A.; Pitt, G.S. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 2006, 98, 1048–1054. [Google Scholar] [CrossRef]
- Etxeberria, A.; Aivar, P.; Rodriguez-Alfaro, J.A.; Alaimo, A.; Villace, P.; Gomez-Posada, J.C.; Areso, P.; Villarroel, A. Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB J. 2008, 22, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Cavaretta, J.P.; Sherer, K.R.; Lee, K.Y.; Kim, E.H.; Issema, R.S.; Chung, H.J. Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Liu, W.; Devaux, J.J. Calmodulin orchestrates the heteromeric assembly and the trafficking of KCNQ2/3 (Kv7.2/3) channels in neurons. Mol. Cell Neurosci. 2014, 58, 40–52. [Google Scholar] [CrossRef]
- Gamper, N.; Li, Y.; Shapiro, M.S. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol. Biol. Cell 2005, 16, 3538–3551. [Google Scholar] [CrossRef]
- Ambrosino, P.; Alaimo, A.; Bartollino, S.; Manocchio, L.; De, M.M.; Mosca, I.; Gomis-Perez, C.; Alberdi, A.; Scambia, G.; Lesca, G.; et al. Epilepsy-causing mutations in Kv7.2 C-terminus affect binding and functional modulation by calmodulin. Biochim. Biophys. Acta 2015, 1852, 1856–1866. [Google Scholar] [CrossRef]
- Alberdi, A.; Gomis-Perez, C.; Bernardo-Seisdedos, G.; Alaimo, A.; Malo, C.; Aldaregia, J.; Lopez-Robles, C.; Areso, P.; Butz, E.; Wahl-Schott, C.; et al. Uncoupling PIP2-calmodulin regulation of Kv7.2 channels by an assembly de-stabilizing epileptogenic mutation. J. Cell Sci. 2015, 128, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Perez, C.; Soldovieri, M.V.; Malo, C.; Ambrosino, P.; Taglialatela, M.; Areso, P.; Villarroel, A. Differential Regulation of PI(4,5)P2 Sensitivity of Kv7.2 and Kv7.3 Channels by Calmodulin. Front Mol. Neurosci. 2017, 10, 117. [Google Scholar] [CrossRef]
- Archer, C.R.; Enslow, B.T.; Taylor, A.B.; De, l.R.V.; Bhattacharya, A.; Shapiro, M.S. A mutually induced conformational fit underlies Ca2+-directed interactions between calmodulin and the proximal C terminus of KCNQ4 K+ channels. J. Biol. Chem. 2019, 294, 6094–6112. [Google Scholar] [CrossRef]
- Sihn, C.R.; Kim, H.J.; Woltz, R.L.; Yarov-Yarovoy, V.; Yang, P.C.; Xu, J.; Clancy, C.E.; Zhang, X.D.; Chiamvimonvat, N.; Yamoah, E.N. Mechanisms of Calmodulin Regulation of Different Isoforms of Kv7.4 K+ Channels. J. Biol. Chem. 2016, 291, 2499–2509. [Google Scholar] [CrossRef]
- Zaydman, M.A.; Silva, J.R.; Delaloye, K.; Li, Y.; Liang, H.; Larsson, H.P.; Shi, J.; Cui, J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. USA 2013, 110, 13180–13185. [Google Scholar] [CrossRef]
- Choveau, F.S.; De, l.R.V.; Bierbower, S.M.; Hernandez, C.C.; Shapiro, M.S. Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates KCNQ3 K+ channels by interacting with four cytoplasmic channel domains. J. Biol. Chem. 2018, 293, 19411–19428. [Google Scholar] [CrossRef] [PubMed]
- Tobelaim, W.S.; Dvir, M.; Lebel, G.; Cui, M.; Buki, T.; Peretz, A.; Marom, M.; Haitin, Y.; Logothetis, D.E.; Hirsch, J.A.; et al. Ca2+-Calmodulin and PIP2 interactions at the proximal C-terminus of Kv7 channels. Channels 2017, 11, 686–695. [Google Scholar] [CrossRef]
- Mruk, K.; Shandilya, S.M.; Blaustein, R.O.; Schiffer, C.A.; Kobertz, W.R. Structural insights into neuronal K+ channel-calmodulin complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 13579–13583. [Google Scholar] [CrossRef]
- Alaimo, A.; Villarroel, A. Calmodulin: A Multitasking Protein in Kv7.2 Potassium Channel Functions. Biomolecules. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Sachyani, D.; Dvir, M.; Strulovich, R.; Tria, G.; Tobelaim, W.; Peretz, A.; Pongs, O.; Svergun, D.; Attali, B.; Hirsch, J.A. Structural Basis of a Kv7.1 Potassium Channel Gating Module: Studies of the Intracellular C-Terminal Domain in Complex with Calmodulin. Structure 2014, 22, 1582–1594. [Google Scholar] [CrossRef]
- Alaimo, A.; Alberdi, A.; Gomis-Perez, C.; Fernandez-Orth, J.; Gomez-Posada, J.C.; Areso, P.; Villarroel, A. Cooperativity between calmodulin-binding sites in Kv7.2 channels. J. Cell Sci. 2013, 126, 244–253. [Google Scholar] [CrossRef]
- Alaimo, A.; Nunez, E.; Aivar, P.; Fernandez-Orth, J.; Gomis-Perez, C.; Bernardo-Seisdedos, G.; Malo, C.; Villarroel, A. Calmodulin confers calcium sensitivity to the stability of the distal intracellular assembly domain of Kv7.2 channels. Sci. Rep. 2017, 7, 13425. [Google Scholar] [CrossRef] [PubMed]
- Bonache, M.A.; Alaimo, A.; Malo, C.; Millet, O.; Villarroel, A.; Gonzalez-Muniz, R. Clicked bis-PEG-peptide conjugates for studying calmodulin-Kv7.2 channel binding. Org. Biomol. Chem. 2014, 12, 8877–8887. [Google Scholar] [CrossRef]
- Bernardo-Seisdedos, G.; Nunez, E.; Gomis, C.; Malo, C.; Villarroel, A.; Millet, O. Structural basis and energy landscape for the Ca2+ gating and calmodulation of the Kv7.2 K+ channel. Proc. Natl. Acad. Sci. USA 2018, 115, 2395–2400. [Google Scholar] [CrossRef]
- Gourgy-Hacohen, O.; Kornilov, P.; Pittel, I.; Peretz, A.; Attali, B.; Paas, Y. Capturing distinct KCNQ2 channel resting states by metal ion bridges in the voltage-sensor domain. J. Gen. Physiol 2014, 144, 513–527. [Google Scholar] [CrossRef]
- Strulovich, R.; Tobelaim, W.S.; Attali, B.; Hirsch, J.A. Structural Insights into the M-Channel Proximal C-Terminus/Calmodulin Complex. Biochemistry 2016, 55, 5353–5365. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, A.; Kang, S.; Smith, I.M.; Greene, D.L.; Langeberg, L.K.; Scott, J.D.; Hoshi, N. Coordinated signal integration at the M-type potassium channel upon muscarinic stimulation. EMBO J. 2012, 31, 3147–3156. [Google Scholar] [CrossRef] [PubMed]
- Bildl, W.; Strassmaier, T.; Thurm, H.; Andersen, J.; Eble, S.; Oliver, D.; Knipper, M.; Mann, M.; Schulte, U.; Adelman, J.P.; et al. Protein kinase CK2 is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron 2004, 43, 847–858. [Google Scholar] [CrossRef]
- Kang, S.; Xu, M.; Cooper, E.C.; Hoshi, N. Channel anchored protein kinase CK2 and protein phosphatase 1 reciprocally regulate KCNQ2-containing M-channels via phosphorylation of calmodulin. J. Biol. Chem. 2014, 289, 11536–11544. [Google Scholar] [CrossRef] [PubMed]
- Benaim, G.; Villalobo, A. Phosphorylation of calmodulin. Functional implications. Eur. J. Biochem. 2002, 269, 3619–3631. [Google Scholar] [CrossRef]
- Villalobo, A. The multifunctional role of phospho-calmodulin in pathophysiological processes. Biochem. J. 2018, 475, 4011–4023. [Google Scholar] [CrossRef]
















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez, E.; Muguruza-Montero, A.; Villarroel, A. Atomistic Insights of Calmodulin Gating of Complete Ion Channels. Int. J. Mol. Sci. 2020, 21, 1285. https://doi.org/10.3390/ijms21041285
Núñez E, Muguruza-Montero A, Villarroel A. Atomistic Insights of Calmodulin Gating of Complete Ion Channels. International Journal of Molecular Sciences. 2020; 21(4):1285. https://doi.org/10.3390/ijms21041285
Chicago/Turabian StyleNúñez, Eider, Arantza Muguruza-Montero, and Alvaro Villarroel. 2020. "Atomistic Insights of Calmodulin Gating of Complete Ion Channels" International Journal of Molecular Sciences 21, no. 4: 1285. https://doi.org/10.3390/ijms21041285
APA StyleNúñez, E., Muguruza-Montero, A., & Villarroel, A. (2020). Atomistic Insights of Calmodulin Gating of Complete Ion Channels. International Journal of Molecular Sciences, 21(4), 1285. https://doi.org/10.3390/ijms21041285