The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective

 , and
, and
Abstract
1. Introduction
2. Strategy of Literature Review and Findings
3. miRNAs in VC: Positive and Negative VC Regulators
3.1. Negative VC-Regulating miRNAs
3.2. Positive VC-Regulating miRNAs
3.3. MiRNAs with Controversial Influences on VC
3.4. Non-Coding RNA and VC
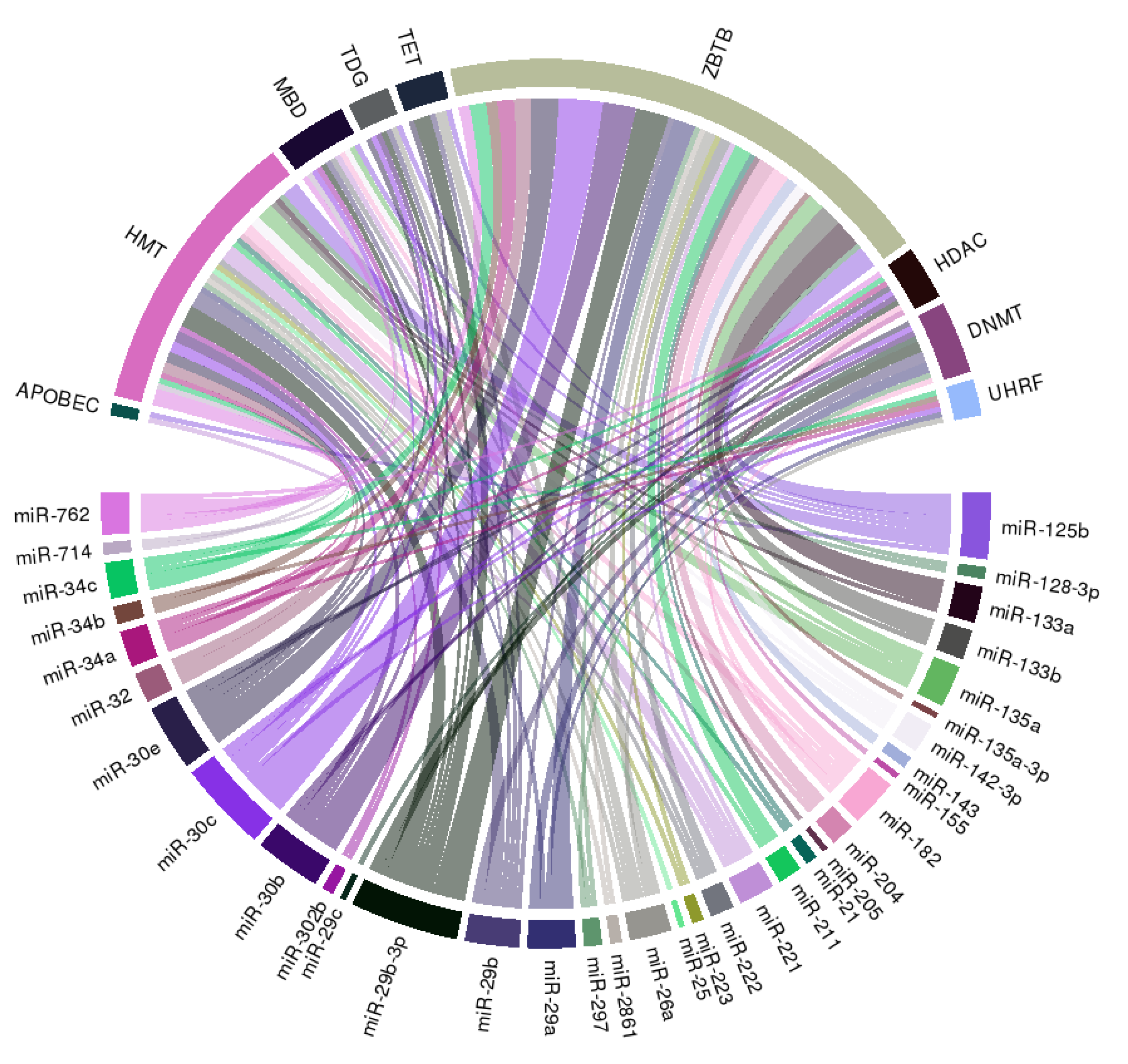
3.5. An Integrative Perspective: Other Targets of Literature-Identified miRNAs in VC
4. DNA Methylation in VC
5. Histone Modification in VC
6. Chromatin Changes in VC
7. The Cross-Talk between Epigenetic Mechanisms in VC
8. Discrepancies between Calcified and Non-Calcified Tissues or Cells: Potential for Uncovering Clinical Biomarkers
9. Conclusion and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3’-UTR | 3’-untranslated region |
| ALP | alkaline phosphatase |
| BMP | bone morphogenetic protein |
| CAC | coronary artery calcification |
| CKD | chronic kidney disease |
| DM | diabetes mellitus |
| DNMT | DNA methyltransferase |
| ESRD | end-stage renal disease |
| GSK | glycogen synthase kinase |
| HDAC | histone deacetylase |
| HMT | histone methyltransferase |
| HP | high phosphate |
| IGF | insulin-like growth factor |
| IS | indoxyl sulfate |
| KLF | Kruppel-like factor |
| LDLR | low density lipoprotein receptor |
| lncRNA | long non-coding RNA |
| MBD | methyl-CpG binding domain |
| Mecp | methyl-CpG binding protein |
| miRNA | microRNA |
| MSC | mesenchymal stem cell |
| mTOR | mammalian target of rapamycin |
| MV | matrix vesicle |
| NFAT | nuclear factor of activated T cells |
| NF-κB | nuclear factor-κB |
| PPAR-γ | peroxisome proliferator-activated receptor-γ |
| PRMT | protein arginine N-methyltransferase |
| PTEN | phosphatase and tensin homolog |
| qPCR | quantitative polymerase chain reaction |
| RUNX2 | Runt-related transcription factor 2 |
| SATB | special AT-rich sequence-binding protein |
| SIRT | sirtuin |
| SMA | smooth muscle actin |
| TGF | transforming growth factor |
| VC | vascular calcification |
| VSMC | vascular smooth muscle cell |
References
- Alluri, K.; Joshi, P.H.; Henry, T.S.; Blumenthal, R.S.; Nasir, K.; Blaha, M.J. Scoring of coronary artery calcium scans: History, assumptions, current limitations, and future directions. Atherosclerosis 2015, 239, 109–117. [Google Scholar] [CrossRef]
- McCullough, P.A.; Agrawal, V.; Danielewicz, E.; Abela, G.S. Accelerated Atherosclerotic Calcification and Mönckeberg’s Sclerosis: A Continuum of Advanced Vascular Pathology in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1585–1598. [Google Scholar] [CrossRef]
- Doherty, T.M.; Fitzpatrick, L.A.; Inoue, D.; Qiao, J.H.; Fishbein, M.C.; Detrano, R.C.; Shah, P.K.; Rajavashisth, T.B. Molecular, Endocrine, and Genetic Mechanisms of Arterial Calcification. Endocr. Rev. 2004, 25, 629–672. [Google Scholar] [CrossRef]
- Chen, N.X.; Moe, S.M. Pathophysiology of Vascular Calcification. Curr. Osteoporos. Rep. 2015, 13, 372–380. [Google Scholar] [CrossRef]
- de Oca, A.M.; Madueño, J.A.; Martinez-Moreno, J.M.; Guerrero, F.; Muñoz-Castañeda, J.; Rodriguez-Ortiz, M.E.; Mendoza, F.J.; Almaden, Y.; Lopez, I.; Rodriguez, M.; et al. High-phosphate-induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J. Bone Miner. Res. 2010, 25, 1996–2005. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Sales, V.M.; Ferguson-Smith, A.C.; Patti, M.E. Epigenetic Mechanisms of Transmission of Metabolic Disease across Generations. Cell Metab. 2017, 25, 559–571. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Wu, S.S.; Lin, X.; Yuan, L.Q.; Liao, E.Y. The Role of Epigenetics in Arterial Calcification. Biomed. Res. Int. 2015, 2015, 320849. [Google Scholar] [CrossRef]
- Nanoudis, S.; Pikilidou, M.; Yavropoulou, M.; Zebekakis, P. The Role of MicroRNAs in Arterial Stiffness and Arterial Calcification. An Update and Review of the Literature. Front. Genet. 2017, 8, 209. [Google Scholar] [CrossRef]
- Kwon, D.H.; Kim, Y.K.; Kook, H. New Aspects of Vascular Calcification: Histone Deacetylases and Beyond. J. Korean Med. Sci. 2017, 32, 1738–1748. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kook, H. Diverse roles of noncoding RNAs in vascular calcification. Arch. Pharm. Res. 2019, 42, 244–251. [Google Scholar] [CrossRef]
- Goettsch, C.; Rauner, M.; Pacyna, N.; Hempel, U.; Bornstein, S.R.; Hofbauer, L.C. miR-125b Regulates Calcification of Vascular Smooth Muscle Cells. Am. J. Pathol. 2011, 179, 1594–1600. [Google Scholar] [CrossRef]
- Du, Y.; Gao, C.; Liu, Z.; Wang, L.; Liu, B.; He, F.; Zhang, T.; Wang, Y.; Wang, X.; Xu, M.; et al. Upregulation of a Disintegrin and Metalloproteinase with Thrombospondin Motifs-7 by miR-29 Repression Mediates Vascular Smooth Muscle Calcification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2580–2588. [Google Scholar] [CrossRef]
- Cui, R.R.; Li, S.J.; Liu, L.J.; Yi, L.; Liang, Q.H.; Zhu, X.; Liu, G.Y.; Liu, Y.; Wu, S.S.; Liao, X.B.; et al. MicroRNA-204 regulates vascular smooth muscle cell calcification in vitro and in vivo. Cardiovasc. Res. 2012, 96, 320–329. [Google Scholar] [CrossRef]
- Balderman, J.A.; Lee, H.Y.; Mahoney, C.E.; Handy, D.E.; White, K.; Annis, S.; Lebeche, D.; Hajjar, R.J.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 decreases microRNA-30b and microRNA-30c to promote vascular smooth muscle cell calcification. J. Am. Heart Assoc. 2012, 1, e003905. [Google Scholar] [CrossRef]
- Gui, T.; Zhou, G.; Sun, Y.; Shimokado, A.; Itoh, S.; Oikawa, K.; Muragami, Y. MicroRNAs that target Ca2+ transporters are involved in vascular smooth muscle cell calcification. Lab. Investig. 2012, 92, 1250. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Metzinger-Le Meuth, V.; Hénaut, L.; Djelouat, M.S.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: Evidence for the involvement of miR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef]
- Liao, X.B.; Zhang, Z.Y.; Yuan, K.; Liu, Y.; Feng, X.; Cui, R.R.; Hu, Y.R.; Yuan, Z.S.; Gu, L.; Li, S.J.; et al. MiR-133a Modulates Osteogenic Differentiation of Vascular Smooth Muscle Cells. Endocrinology 2013, 154, 3344–3352. [Google Scholar] [CrossRef]
- Mackenzie, N.C.; Staines, K.A.; Zhu, D.; Genever, P.; Macrae, V.E. miRNA-221 and miRNA-222 synergistically function to promote vascular calcification. Cell Biochem. Funct. 2014, 32, 209–216. [Google Scholar] [CrossRef]
- Qiao, W.; Chen, L.; Zhang, M. MicroRNA-205 Regulates the Calcification and Osteoblastic Differentiation of Vascular Smooth Muscle Cells. Cell. Physiol. Biochem. 2014, 33, 1945–1953. [Google Scholar] [CrossRef]
- Wen, P.; Cao, H.; Fang, L.; Ye, H.; Zhou, Y.; Jiang, L.; Su, W.; Xu, H.; He, W.; Dai, C.; et al. miR-125b/Ets1 axis regulates transdifferentiation and calcification of vascular smooth muscle cells in a high-phosphate environment. Exp. Cell. Res. 2014, 322, 302–312. [Google Scholar] [CrossRef]
- M’Baya-Moutoula, E.; Louvet, L.; Metzinger-Le Meuth, V.; Massy, Z.A.; Metzinger, L. High inorganic phosphate concentration inhibits osteoclastogenesis by modulating miR-223. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2202–2212. [Google Scholar] [CrossRef]
- Xia, Z.Y.; Hu, Y.; Xie, P.L.; Tang, S.Y.; Luo, X.H.; Liao, E.Y.; Chen, Y.; Xie, H. Runx2/miR-3960/miR-2861 Positive Feedback Loop Is Responsible for Osteogenic Transdifferentiation of Vascular Smooth Muscle Cells. Biomed. Res. Int. 2015, 2015, 624037. [Google Scholar] [CrossRef]
- Ding, W.; Li, J.; Singh, J.; Alif, R.; Vazquez-Padron, R.I.; Gomes, S.A.; Hare, J.M.; Shehadeh, L.A. miR-30e targets IGF2-regulated osteogenesis in bone marrow-derived mesenchymal stem cells, aortic smooth muscle cells, and ApoE−/− mice. Cardiovasc. Res. 2015, 106, 131–142. [Google Scholar] [CrossRef]
- Sudo, R.; Sato, F.; Azechi, T.; Wachi, H. MiR-29-mediated elastin down-regulation contributes to inorganic phosphorus-induced osteoblastic differentiation in vascular smooth muscle cells. Genes Cells 2015, 20, 1077–1087. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Chen, N.X.; O’Neill, K.; McClintick, J.N.; Moe, S.M.; Janga, S.C. Differential miRNA Expression in Cells and Matrix Vesicles in Vascular Smooth Muscle Cells from Rats with Kidney Disease. PLoS ONE 2015, 10, e0131589. [Google Scholar] [CrossRef]
- Louvet, L.; Metzinger, L.; Büchel, J.; Steppan, S.; Massy, Z.A. Magnesium Attenuates Phosphate-Induced Deregulation of a MicroRNA Signature and Prevents Modulation of Smad1 and Osterix during the Course of Vascular Calcification. Biomed. Res. Int. 2016, 2016, 7419524. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, L.; Cong, G.; Ren, L.; Hao, L. MicroRNA-34b/c inhibits aldosterone-induced vascular smooth muscle cell calcification via a SATB2/Runx2 pathway. Cell Tissue Res. 2016, 366, 733–746. [Google Scholar] [CrossRef]
- Lin, L.; He, Y.; Xi, B.L.; Zheng, H.C.; Chen, Q.; Li, J.; Hu, Y.; Ye, M.H.; Chen, P.; Qu, Y. MiR-135a Suppresses Calcification in Senescent VSMCs by Regulating KLF4/STAT3 Pathway. Curr. Vasc. Pharmacol. 2016, 14, 211–218. [Google Scholar] [CrossRef][Green Version]
- Zheng, S.; Zhang, S.; Song, Y.; Guo, W.; Zhai, W.; Qiu, X.; Li, J. MicroRNA-297a regulates vascular calcification by targeting fibroblast growth factor 23. Iran. J. Basic Med. Sci. 2016, 19, 1331–1336. [Google Scholar]
- Panizo, S.; Naves-Díaz, M.; Carrillo-López, N.; Martínez-Arias, L.; Fernández-Martín, J.L.; Ruiz-Torres, M.P.; Cannata-Andia, J.B.; Rodriguez, I. MicroRNAs 29b, 133b, and 211 Regulate Vascular Smooth Muscle Calcification Mediated by High Phosphorus. J. Am. Soc. Nephrol. 2016, 27, 824–834. [Google Scholar] [CrossRef]
- Chao, C.T.; Liu, Y.P.; Su, S.F.; Yeh, H.Y.; Chen, H.Y.; Lee, P.J.; Chen, W.J.; Lee, Y.M.; Huang, J.W.; Chiang, C.K.; et al. Circulating MicroRNA-125b Predicts the Presence and Progression of Uremic Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1402–1414. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Z.; Yang, H.; Lin, Q.; Han, C.; Qin, X. The Involvement of miR-29b-3p in Arterial Calcification by Targeting Matrix Metalloproteinase-2. Biomed. Res. Int. 2017, 2017, 6713606. [Google Scholar] [CrossRef]
- Wu, W.; Shang, Y.; Dai, S.; Yi, F.; Wang, X. MiR-26a regulates vascular smooth muscle cell calcification in vitro through targeting CTGF. Bratisl. Lek. Listy 2017, 118, 499–503. [Google Scholar] [CrossRef]
- Li, J.; Xing, G.; Zhang, L.; Shang, J.; Li, Y.; Li, C.; Tian, F.; Yang, X. Satb1 promotes osteoclastogenesis by recruiting CBP to upregulate miR-223 expression in chronic kidney disease-mineral and bone disorder. Pharmazie 2017, 72, 680–686. [Google Scholar]
- Liu, J.; Xiao, X.; Shen, Y.; Chen, L.; Xu, C.; Zhao, H.; Wu, Y.; Zhang, Q.; Zhong, J.; Tang, Z.; et al. MicroRNA-32 promotes calcification in vascular smooth muscle cells: Implications as a novel marker for coronary artery calcification. PLoS ONE 2017, 12, e0174138. [Google Scholar] [CrossRef]
- Sun, W.I.; Wang, N.; Xu, Y. Impact of miR-302b on Calcium-phosphorus Metabolism and Vascular Calcification of Rats with Chronic Renal Failure by Regulating BMP-2/Runx2/Osterix Signaling Pathway. Arch. Med. Res. 2018, 49, 164–171. [Google Scholar] [CrossRef]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; et al. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2079–2090. [Google Scholar] [CrossRef]
- Han, H.; Yang, S.; Liang, Y.; Zeng, P.; Liu, L.; Yang, X.; Duan, Y.; Han, J.; Chen, Y. Teniposide regulates the phenotype switching of vascular smooth muscle cells in a miR-21-dependent manner. Biochem. Biophys. Res. Commun. 2018, 506, 1040–1046. [Google Scholar] [CrossRef]
- Lin, X.; Xu, F.; Cui, R.R.; Xiong, D.; Zhong, J.Y.; Zhu, T.; Li, F.; Wu, F.; Xie, X.B.; Mao, M.Z.; et al. Arterial Calcification Is Regulated Via an miR-204/DNMT3a Regulatory Circuit Both In Vitro and in Female Mice. Endocrinology 2018, 159, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, W.; Yang, H.; Lin, Q.; Qin, X. The miR-182/SORT1 axis regulates vascular smooth muscle cell calcification in vitro and in vivo. Exp. Cell Res. 2018, 362, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, M.; Skafi, N.; Fayyad-Kazan, M.; Kobeissy, F.; Hamade, E.; Mebarek, S.; Habib, A.; Borghol, N.; Zeidan, A.; Magne, D.; et al. Characterization and assessment of potential microRNAs involved in phosphate-induced aortic calcification. J. Cell. Physiol. 2018, 233, 4056–4067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/β-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kétszeri, M.; Kirsch, A.; Frauscher, B.; Moschovaki-Filippidou, F.; Mooslechner, A.A.; Kirsch, A.H.; Schabhuettl, C.; Aringer, I.; Artinger, K.; Pregartner, G.; et al. MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis 2019, 280, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Bao, S.; Guo, W.; Diao, Z.; Wang, L.; Han, X.; Guo, W.; Liu, W. Bone marrow mesenchymal stem cell–derived exosomes alleviate high phosphorus-induced vascular smooth muscle cells calcification by modifying microRNA profiles. Funct. Integr. Genomics 2019, 19, 633–643. [Google Scholar] [CrossRef]
- Xu, T.H.; Qiu, X.B.; Sheng, Z.T.; Han, Y.R.; Wang, J.; Tian, B.Y.; Yao, L. Restoration of microRNA-30b expression alleviates vascular calcification through the mTOR signaling pathway and autophagy. J. Cell. Physiol. 2019, 234, 14306–14318. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, X.Z.; Li, F.; Ji, Q.R. MiR-128-3p accelerates cardiovascular calcification and insulin resistance through ISL1-dependent Wnt pathway in type 2 diabetes mellitus rats. J. Cell. Physiol. 2019, 234, 4997–5010. [Google Scholar] [CrossRef]
- Cavallari, C.; Dellepiane, S.; Fonsato, V.; Medica, D.; Marengo, M.; Migliori, M.; Quercia, A.D.; Pitino, A.; Formica, M.; Panichi, V.; et al. Online Hemodiafiltration Inhibits Inflammation-Related Endothelial Dysfunction and Vascular Calcification of Uremic Patients Modulating miR-223 Expression in Plasma Extracellular Vesicles. J. Immunol. 2019, 202, 2372–2383. [Google Scholar] [CrossRef]
- Wang, C.; Xu, W.; An, J.; Liang, M.; Li, Y.; Zhang, F.; Tong, Q.; Huang, K. Poly(ADP-ribose) polymerase 1 accelerates vascular calcification by upregulating Runx2. Nat. Commun. 2019, 10, 1203. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, G.; Wei, T.; Yang, Z.; Tan, W.; Mo, Z.; Liu, J.; Li, D.; Wei, Y.; Zhang, L.; et al. MicroRNA-25 Protects Smooth Muscle Cells against Corticosterone-Induced Apoptosis. Oxid. Med. Cell. Longev. 2019, 2019, 2691514. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wei, D.; Chen, Y.; Chen, L.; Bian, Y.; Shen, Y.; Chen, J.; Pan, Y. Association of astragaloside IV-inhibited autophagy and mineralization in vascular smooth muscle cells with lncRNA H19 and DUSP5-mediated ERK signaling. Toxicol. Appl. Pharmacol. 2019, 364, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Azechi, T.; Kanehira, D.; Kobayashi, T.; Sudo, R.; Nishimura, A.; Sato, F.; Wachi, H. Trichostatin A, an HDAC Class I/II Inhibitor, Promotes Pi-Induced Vascular Calcification Via Up-Regulation of the Expression of Alkaline Phosphatase. J. Atheroscler. Thromb. 2013, 20, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Eom, G.H.; Ko, J.H.; Shin, S.; Joung, H.; Choe, N.; Nam, Y.S.; Min, H.K.; Kook, T.; Yoon, S.; et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 2016, 7, 10492. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Daoudi, M.; Rosa, M.; Vinod, M.; Louvet, L.; Copin, C.; Fanchon, M.; Vanhoutte, J.; Derudas, B.; Belloy, L.; et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ. Res. 2017, 121, 19–30. [Google Scholar] [CrossRef]
- Abend, A.; Shkedi, O.; Fertouk, M.; Caspi, L.H.; Kehat, I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 2017, 18, 1166–1185. [Google Scholar] [CrossRef]
- Kurozumi, A.; Nakano, K.; Yamagata, K.; Okada, Y.; Nakayamada, S.; Tanaka, Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 2019, 124, 53–61. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.; Zhang, H.; Ning, Y.; Song, N.; Hu, J.; Cai, J.; Shi, Y.; Ding, X.; Zhang, X. Amelioration of Uremic Toxin Indoxyl Sulfate-Induced Osteoblastic Calcification by SET Domain Containing Lysine Methyltransferase 7/9 Protein. Nephron 2019, 141, 287–294. [Google Scholar] [CrossRef]
- Azechi, T.; Sato, F.; Sudo, R.; Wachi, H. 5-aza-2’-Deoxycytidine, a DNA Methyltransferase Inhibitor, Facilitates the Inorganic Phosphorus-Induced Mineralization of Vascular Smooth Muscle Cells. J. Atheroscler. Thromb. 2014, 21, 463–476. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int. J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef]
- Xie, S.A.; Zhang, T.; Wang, J.; Zhao, F.; Zhang, Y.P.; Yao, W.J.; Hur, S.S.; Yeh, Y.T.; Pang, W.; Zheng, L.S.; et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: Role of DNA methyltransferase 1. Biomaterials 2018, 155, 203–216. [Google Scholar] [CrossRef]
- Ramachandran, B.; Stabley, J.N.; Cheng, S.L.; Behrmann, A.S.; Gay, A.; Li, L.; Mead, M.; Kozlitina, J.; Lemoff, A.; Mirzaei, H.; et al. A GTPase-activating protein-binding protein (G3BP1)/antiviral protein relay conveys arteriosclerotic Wnt signals in aortic smooth muscle cells. J. Biol. Chem. 2018, 293, 7942–7968. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, Y.; Zhang, Y.; Bi, X.; Nie, L.; Liu, C.; Xiong, J.; He, T.; Xu, X.; Yu, Y.; et al. High phosphate-induced downregulation of PPARγ contributes to CKD-associated vascular calcification. J. Mol. Cell. Cardiol. 2018, 114, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, F.; Xu, F.; Cui, R.R.; Xiong, D.; Zhong, J.Y.; Zhu, T.; Shan, S.K.; Wu, F.; Xie, X.B.; et al. Aberration methylation of miR-34b was involved in regulating vascular calcification by targeting Notch1. Aging 2019, 11, 3182. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.L.; Ramachandran, B.; Behrmann, A.; Shao, J.S.; Mead, M.; Smith, C.; Krchma, K.; Bello Arredondo, Y.; Kovacs, A.; Kapoor, K.; et al. Vascular smooth muscle LRP6 limits arteriosclerotic calcification in diabetic LDLR−/− mice by restraining noncanonical Wnt signals. Circ. Res. 2015, 117, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Yamamoto, M.; Ogino, T.; Kobuchi, H.; Ohmoto, N.; Aoyama, E.; Oka, T.; Nakanishi, T.; Inoue, K.; Sasaki, J. Necrotic and apoptotic cells serve as nuclei for calcification on osteoblastic differentiation of human mesenchymal stem cells in vitro. Cell. Biochem. Funct. 2014, 32, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Gilham, D.; Tsujikawa, L.M.; Sarsons, C.D.; Halliday, C.; Wasiak, S.; Stotz, S.C.; Jahagirdar, R.; Sweeney, M.; Johansson, J.O.; Wong, N.C.W.; et al. Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis 2019, 280, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tang, Y.; Wang, Y.; Li, G.; Wang, L.; Li, Y. Label-free quantitative proteomics identifies Smarca4 is involved in vascular calcification. Ren. Fail. 2019, 41, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, W.; Ouyang, X.; Dai, Y. Reduced Circulating miR-15b Is Correlated with Phosphate Metabolism in Patients with End-Stage Renal Disease on Maintenance Hemodialysis. Ren. Fail. 2012, 34, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Mangino, M.; Cecelja, M.; Menni, C.; Tsai, P.C.; Yuan, W.; Small, K.; Bell, J.; Mitchell, G.F.; Chowienczyk, P.; Spector, T.D. Integrated multiomics approach identifies calcium and integrin-binding protein-2 as a novel gene for pulse wave velocity. J. Hypertens. 2016, 34, 79. [Google Scholar] [CrossRef] [PubMed]
- Ulbing, M.; Kirsch, A.H.; Leber, B.; Lemesch, S.; Münzker, J.; Schweighofer, N.; Hofer, D.; Trummer, O.; Rosenkranz, A.R.; Muller, H.; et al. MicroRNAs 223-3p and 93-5p in patients with chronic kidney disease before and after renal transplantation. Bone 2017, 95, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cai, B.; Zhang, Z.; Zhang, Y.; Wang, L.; Liu, K.; Zhang, H.; Sun, L.; Cai, H.; Lu, G.; et al. CDKN2B Methylation and Aortic Arch Calcification in Patients with Ischemic Stroke. J. Atheroscler. Thromb. 2017, 24, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Nishikawa, Y.; Yamada, H.; Yamada, K.; Mase, M. Differential Expression of microRNAs in Severely Calcified Carotid Plaques. J. Stroke Cerebrovasc. Dis. 2018, 27, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Lee, Y.T.; Tain, Y.L.; Ng, H.Y.; Kuo, W.H. Circulating microRNAs and vascular calcification in hemodialysis patients. J. Int. Med. Res. 2019, 47, 2929–2939. [Google Scholar] [CrossRef]
- Chao, C.T.; Yuan, T.H.; Yeh, H.Y.; Chen, H.Y.; Huang, J.W.; Chen, H.W. Risk Factors Associated With Altered Circulating Micro RNA -125b and Their Influences on Uremic Vascular Calcification Among Patients With End-Stage Renal Disease. J. Am. Heart Assoc. 2019, 8, e010805. [Google Scholar] [CrossRef] [PubMed]
- Dudunk, E.; Florijn, B.; Weijs, B.; Duijs, J.; Luermans, J.; Peeters, F.; Schurgers, L.; Wildberger, J.; Schotten, U.; Bijkerk, R.; et al. Vascular Calcification and not Arrhythmia in Idiopathic Atrial Fibrillation Associates with Sex Differences in Diabetic Microvascular Injury miRNA Profiles. MicroRNA 2019, 8, 127–134. [Google Scholar] [CrossRef]
- Pickering, M.E.; Millet, M.; Rousseau, J.C.; Croset, M.; Szulc, P.; Borel, O.; Sornay Rendu, E.; Chapurlat, R. Selected serum microRNA, abdominal aortic calcification and risk of osteoporotic fracture. PLoS ONE 2019, 14, e0216947. [Google Scholar] [CrossRef]
- Mao, L.; Liu, S.; Hu, L.; Jia, L.; Wang, H.; Guo, M.; Chen, C.; Liu, Y.; Xu, L. miR-30 Family: A Promising Regulator in Development and Disease. Biomed. Res. Int. 2018, 2018, 9623412. [Google Scholar] [CrossRef]
- Wu, T.; Zhou, H.; Hong, Y.; Li, J.; Jiang, X.; Huang, H. miR-30 family members negatively regulate osteoblast differentiation. J. Biol. Chem. 2012, 287, 7503–7511. [Google Scholar] [CrossRef]
- Bridge, G.; Monteiro, R.; Henderson, S.; Emuss, V.; Lagos, D.; Georgopoulou, D.; Patient, R.; Boshoff, C. The microRNA-30 family targets DLL4 to modulate endothelial cell behavior during angiogenesis. Blood 2012, 120, 5063–5072. [Google Scholar] [CrossRef]
- Yu, H.; Lu, Y.; Li, Z.; Wang, Q. microRNA-133: Expression, Function and Therapeutic Potential in Muscle Diseases and Cancer. Curr. Drug Targets 2014, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhou, H.; Tang, Q. miR-133: A Suppressor of Cardiac Remodeling? Front. Pharmacol. 2018, 9, 903. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Yeh, H.Y.; Yuan, T.H.; Chiang, C.K.; Chen, H.W. MicroRNA-125b in vascular diseases: An updated systematic review of pathogenetic implications and clinical applications. J. Cell. Mol. Med. 2019, 23, 5884–5894. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Pan, H.; Li, R. The dual regulatory role of miR-204 in cancer. Tumour Biol. 2016, 37, 11667–11677. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, L.; Xing, L.; Chen, D. MicroRNA-204 regulates Runx2 protein expression and mesenchymal progenitor cell differentiation. Stem Cells 2010, 28, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, R.S.; Rajagopalan, S.; Natarajan, R.; Deiuliis, J.A. Noncoding RNAs in Cardiovascular Disease: Pathological Relevance and Emerging Role as Biomarkers and Therapeutics. Am. J. Hypertens. 2018, 31, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Sárközy, M.; Kahán, Z.; Csont, T. A myriad of roles of miR-25 in health and disease. Oncotarget 2018, 9, 21580–21612. [Google Scholar] [CrossRef]
- Li, X.; Pan, X.; Fu, X.; Yang, Y.; Chen, J.; Lin, W. MicroRNA-26a: An Emerging Regulator of Renal Biology and Disease. Kidney Blood Press. Res. 2019, 44, 287–297. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, Y.P.; Wu, J.; Jie, L.G.; Deng, J.X.; Zhao, D.B.; Yu, Q.H. Downregulated microRNA-135a ameliorates rheumatoid arthritis by inactivation of the phosphatidylinositol 3-kinase/AKT signaling pathway via phosphatidylinositol 3-kinase regulatory subunit 2. J. Cell. Physiol. 2019, 234, 17663–17676. [Google Scholar] [CrossRef]
- Shrestha, A.; Mukhametshina, R.T.; Taghizadeh, S.; Vásquez-Pacheco, E.; Cabrera-Fuentes, H.; Rizvanov, A.; Mari, B.; Carraro, G.; Bellusci, S. MicroRNA-142 is a multifaceted regulator in organogenesis, homeostasis, and disease. Dev. Dyn. 2017, 246, 285–290. [Google Scholar] [CrossRef]
- Vacante, F.; Denby, L.; Sluimer, J.C.; Baker, A.H. The function of miR-143, miR-145 and the MiR-143 host gene in cardiovascular development and disease. Vasc. Pharmacol. 2019, 112, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Park, S.J.; Jung, S.H.; Kim, E.J.; Jogeswar, G.; Ajita, J.; Rhee, Y.; Kim, C.H.; Lim, S.K. miR-182 is a negative regulator of osteoblast proliferation, differentiation, and skeletogenesis through targeting FoxO1. J. Bone Miner. Res. 2012, 27, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, J. MiR-211-5p contributes to chondrocyte differentiation by suppressing Fibulin-4 expression to play a role in osteoarthritis. J. Biochem. 2019, 166, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yang, J.; Feng, X.; Wang, H.; Ye, S.; Yang, P.; Tan, W.; Wei, G.; Zhou, Y. MicroRNA-32 (miR-32) regulates phosphatase and tensin homologue (PTEN) expression and promotes growth, migration, and invasion in colorectal carcinoma cells. Mol. Cancer 2013, 12, 30. [Google Scholar] [CrossRef]
- Cai, J.; Fang, L.; Huang, Y.; Li, R.; Xu, X.; Hu, Z.; Zhang, L.; Yang, Y.; Zhu, X.; Zhang, H.; et al. Simultaneous overactivation of Wnt/β-catenin and TGFβ signalling by miR-128-3p confers chemoresistance-associated metastasis in NSCLC. Nat. Commun. 2017, 8, 15870. [Google Scholar] [CrossRef]
- Icli, B.; Wu, W.; Ozdemir, D.; Li, H.; Haemmig, S.; Liu, X.; Giatsidis, G.; Cheng, H.S.; Avci, S.N.; Kurt, M.; et al. MicroRNA-135a-3p regulates angiogenesis and tissue repair by targeting p38 signaling in endothelial cells. FASEB J. 2019, 33, 5599–5614. [Google Scholar] [CrossRef]
- Gulei, D.; Raduly, L.; Broseghini, E.; Ferracin, M.; Berindan-Neagoe, I. The extensive role of miR-155 in malignant and non-malignant diseases. Mol. Asp. Med. 2019, 70, 33–56. [Google Scholar] [CrossRef]
- Alizadeh, M.; Safarzadeh, A.; Beyranvand, F.; Ahmadpour, F.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Baradaran, B. The potential role of miR-29 in health and cancer diagnosis, prognosis, and therapy. J. Cell. Physiol. 2019, 234, 19280–19297. [Google Scholar] [CrossRef]
- Agostini, M.; Knight, R.A. miR-34: From bench to bedside. Oncotarget 2014, 5, 872–881. [Google Scholar] [CrossRef]
- Lefort, K.; Brooks, Y.; Ostano, P.; Cario-André, M.; Calpini, V.; Guinea-Viniegra, J.; Albinger-Hegyi, A.; Hoetzenecker, W.; Kolfschoten, I.; Wagner, E.F.; et al. A miR-34a-SIRT6 axis in the squamous cell differentiation network. EMBO J. 2013, 32, 2248–2263. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Kim, N.G.; Lee, I.; Choi, H.S.; Li, X.Y.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Na, J.M.; et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal 2011, 4, ra71. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.J.; Masters, S.L. miR-223: Infection, inflammation and cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, K.; Chen, X. Inflammation-regulatory microRNAs: Valuable targets for intracranial atherosclerosis. J. Neurosci. Res. 2019, 97, 1242–1252. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA Species | VC Models | VC Agonistic or Antagonistic | Molecular Influences | Reference |
|---|---|---|---|---|
| miR-21 | Human ASMC | Antagonistic | Down-regulate OPN | [40] |
| miR-25 | Primary mouse ASMC | Antagonistic (potentially) | Down-regulate MOAP1 | [51] |
| miR-26a | Human VSMC | Antagonistic | Down-regulate CTGF and RANKL | [35] |
| miR-29a/29b/29c, miR-29b-3p | Rat VSMC, uremic rat arteries, uremic patient arteries | Antagonistic | Down-regulate ADAMTS-7 (direct target, miR-29a/b) | [14] |
| Human VSMC | Agonistic | Down-regulate elastin | [26] | |
| Primary rat ASMC, Nephrectomized rat with VC | Agonistic | Down-regulate HDAC4, CTNNBIP1, and ACVR2A | [32] | |
| Rat VSMC, rat calcified arteries | Antagonistic | Down-regulate MMP2 (direct target, 29b-3p) | [34] | |
| Human ASMC | Antagonistic | Down-regulate Wnt7b/ß-catenin | [44] | |
| miR-30b | Human coronary SMC, human calcified coronary arteries | Antagonistic | Down-regulate RUNX2 (direct target) | [16] |
| Human ASMC | Antagonistic (potentially) | [28] | ||
| Rat VSMC, nephrectomized rat with VC | Antagonistic | Down-regulate SOX9 (direct target), up-regulate MMP, autophagy and mTOR | [47] | |
| miR-30c/30e | Human coronary SMC | Antagonistic | Down-regulate RUNX2 (direct target, 30c) | [16] |
| Mouse ASMC, ApoE KO mouse aorta | Antagonistic | Down-regulate IGF-2 (direct target, 30e) | [25] | |
| miR-32 | Mouse ASMC, OPG KO mouse, plasma from human with CAC | Agonistic | Up-regulate RUNX2, BMP2, OPN, and ALP | [37] |
| miR-34a/34b/34c | Aldosterone-treated rat VSMC | Antagonistic | Down-regulate SATB2 (direct target, 34b/34c) | [29] |
| miR-34a KO mice | Agonistic | Down-regulate SIRT1 and Axl (direct target, 34a) | [39] | |
| Rat VSMC, Nephrectomized rat with VC, uremic calcified renal arteries | Antagonistic | Down-regulate Notch1 (direct target, 34b) | [64] | |
| miR-125b | Human coronary SMC, ApoE KO mouse calcified aorta | Antagonistic | Down-regulate osterix (direct target) | [13] |
| Primary rat ASMC | Antagonistic | Down-regulate Ets1 (direct target) | [22] | |
| Rat ASMC, adenine-feeding CKD rat with VC, sera from uremic patients | Antagonistic | Down-regulate RUNX2 and osteocalcin | [33] | |
| miR-128-3p | Type 2 diabetic rats | Agonistic | Down-regulate ISL1 (direct target), up-regulate Wnt-1/ß-catenin and GSK-3ß | [48] |
| miR-133a/133b | Primary mouse VSMC | Antagonistic | Down-regulate RUNX2 (direct target, 133a), osteocalcin, and ALP | [19] |
| Human ASMC | Antagonistic (potentially) | [28] | ||
| Primary rat ASMC, Nephrectomized rat with VC | Antagonistic | Down-regulate RUNX2 | [32] | |
| Rat ASMC, adenine-feeding rat with VC | Agonistic (potentially) | [68] | ||
| miR-135a, miR-135a-3p | Mouse ASMC, Klotho KO mouse aorta | Agonistic | Down-regulate NCX1 (135a-3p) | [17] |
| Primary rat ASMC | Antagonistic | Down-regulate KLF4 (direct target) (135a) | [30] | |
| miR-142-3p | DAB/2 mouse aorta, sera from uremic patients | Antagonistic (potentially) | [45] | |
| miR-143 | Human ASMC | Antagonistic (potentially) | [28] | |
| miR-155 | Rat ASMC, adenine-feeding rat with VC | Agonistic (potentially) | [68] | |
| miR-182 | Rat ASMC, Calcified arteries from VitD-treated rat | Antagonistic | Down-regulate SORT1 (direct target) | [42] |
| miR-204 | Mouse ASMC, VitD-treated mouse aorta | Antagonistic | Down-regulating RUNX2 (direct target) | [15] |
| Mouse VSMC, Nephrectomized mouse with VC, calcified renal arteries from uremic patients | Antagonistic | Down-regulating DNMT3a (direct target) | [41] | |
| Rat VSMC, adenine-feeding rat with VC, renal arteries from uremic patients | Antagonistic | Down-regulating RUNX2 (direct target) | [50] | |
| miR-205 | Human ASMC | Antagonistic | Down-regulating RUNX2 and Smad1 (direct target) | [21] |
| miR-211 | Primary rat ASMC, Nephrectomized rat with VC | Antagonistic | Down-regulate RUNX2 | [32] |
| miR-221/222 | Primary mouse VSMC | Agonistic | Up-regulate ENPP1, PiT-1 | [66] |
| miR-223 | Human primary VSMC, ApoE KO mouse aorta | Agonistic (potentially) | Down-regulate Mef2c, RhoB | [18] |
| RAW264.7 cells | Antagonistic | Up-regulate osteoclastogenesis-related genes | [23] | |
| RAW264.7 cells | Antagonistic | Down-regulate NF1A, RhoB | [36] | |
| Human VSMC, plasma from uremic patients | Agonistic | - | [49] | |
| miR-297a | VitD-treated rat with VC | Antagonistic | Down-regulate FGF-23 | [60] |
| miR-302b | Nephrectomized rat with VC | Antagonistic | Down-regulate BMP-2, RUNX2, Osterix | [38] |
| miR-712-3p | Mouse ASMC, Klotho KO mouse aorta | Agonistic | Down-regulate NCKX4 | [17] |
| miR-714 | Mouse ASMC, Klotho KO mouse aorta | Agonistic | Down-regulate PMCA1 | [17] |
| miR-762 | Mouse ASMC, Klotho KO mouse aorta | Agonistic | Down-regulate NCX1 | [17] |
| miR-2861 | Mouse primary ASMC | Agonistic | Down-regulate HDAC5 | [24] |
| miR-3960 | Mouse primary ASMC | Agonistic | Down-regulate HOXA2 | [24] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.-C.; Lu, C.-L.; Yuan, T.-H.; Liao, M.-T.; Chao, C.-T.; Lu, K.-C. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. Int. J. Mol. Sci. 2020, 21, 980. https://doi.org/10.3390/ijms21030980
Hou Y-C, Lu C-L, Yuan T-H, Liao M-T, Chao C-T, Lu K-C. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. International Journal of Molecular Sciences. 2020; 21(3):980. https://doi.org/10.3390/ijms21030980
Chicago/Turabian StyleHou, Yi-Chou, Chien-Lin Lu, Tzu-Hang Yuan, Min-Tser Liao, Chia-Ter Chao, and Kuo-Cheng Lu. 2020. "The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective" International Journal of Molecular Sciences 21, no. 3: 980. https://doi.org/10.3390/ijms21030980
APA StyleHou, Y.-C., Lu, C.-L., Yuan, T.-H., Liao, M.-T., Chao, C.-T., & Lu, K.-C. (2020). The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. International Journal of Molecular Sciences, 21(3), 980. https://doi.org/10.3390/ijms21030980