1. Introduction
Vascular calcification (VC) is common in advanced chronic kidney disease (CKD), and is associated with poor cardiovascular (CV) outcomes [
1]. The pathogenesis of VC is multifactorial and incompletely understood. CKD patients are at risk of VC because of multiple risk factors. High phosphate (Pi) levels are associated with a high risk of VC and CV disease in the general population and, in particular, in CKD patients [
2]. It has been clearly demonstrated that high Pi induces vascular smooth muscle cells (VSMCs) osteoblastic differentiation [
3]. Beside high Pi, other factors potentially cause the phenotypical change of VSMCs into an osteoblast-like cell, such as high total body burden of calcium (Ca) and Pi, low levels of circulating and locally-produced inhibitors, impaired renal excretion, and CKD-MBD (Mineral Bone Disorder) treatments [
4].
Commonly, treatment of hyperphosphatemia requires a multimodal approach: Dialysis, dietary restriction of Pi intake (i.e., food with low Pi content, balancing sufficient protein intake and restricting Pi intake, counteracting “hidden” phosphate due to insufficient labelling of processed food), and the prescription of Pi binders [
5]. Considerations for the choice of Pi binder may include age, gender, diabetes, low bone turnover, vascular and/or valvular calcification, and inflammation [
6]. Recently, two new iron-based Pi binders have become available to dialysis patients to treat hyperphosphatemia [
7]. In addition, we demonstrated: (1) the effect of iron citrate in preventing calcification in an in vitro model of high Pi-induced VC, finding that iron reduces Ca deposition through the prevention of apoptosis and the enhancement of autophagy [
8]; (2) that iron is able to both prevent and partially revert extracellular matrix osteo-chondrogenic shifts induced by high Pi treatment acting on elastinolysis, fibrosis, mucins synthesis, and glycolytic VSMC metabolism [
9]. Although the effect of iron citrate on the progression of calcification in established calcified VSMCs has already been presented in a previous study of our research group [
8], the aim of the present set of data is to better elucidate the mechanisms of the direct effect of iron citrate on the progression of calcification in established high Pi-calcified VSMCs.
3. Discussion
VC is a multifactorial process due to the pathological deposition of minerals in the vascular system. Several causes have been identified in the pathogenesis of VC, such as an imbalance between pro- and anti- calcific factors; osteo-chondrogenic differentiation of VSMCs induced by inflammation, lipids, calcium and phosphate; apoptotic process; matrix degradation; and elastolysis [
10].
In CKD, together with the traditional factors inducing VC, some nontraditional factors exist, namely uremic milieu and hyperphosphatemia. It is accepted that among all of the pathophysiologic mechanisms in CKD, VC is strongly associated with CV mortality, and it appears that high serum Pi levels are the most likely culprit behind VC [
11].
Many efforts have been made in recent years to try to properly investigate and elucidate the mechanisms of high Pi-induced VC. The most in vitro-utilized approach is to study the effect of different molecules on the prevention of VC through different aspects such as calcium deposition, VSMCs osteoblastic transdifferentiation, apoptosis, elastolysis, matrix degradation, and authophagy. Applying the prophylactic approach in vitro, we published a study on the effect of iron on VC, demonstrating that ferric citrate prevents high Pi-induced calcium deposition by preventing apoptosis and by the potentiation of authophagy [
8]. Interestingly, we also found that iron acts on the extracellular matrix osteo-chondrogenic shift induced by high Pi treatment, both preventing and blocking/partially reverting it [
9]. The interest in the action of iron on calcification is also legitimated by the recent clinical use of this molecule as a Pi binder. In fact, two new Ca-free, iron-based P binders are now available to treat hyperphosphatemia in CKD, namely iron citrate and sucroferric oxyhydroxide [
7].
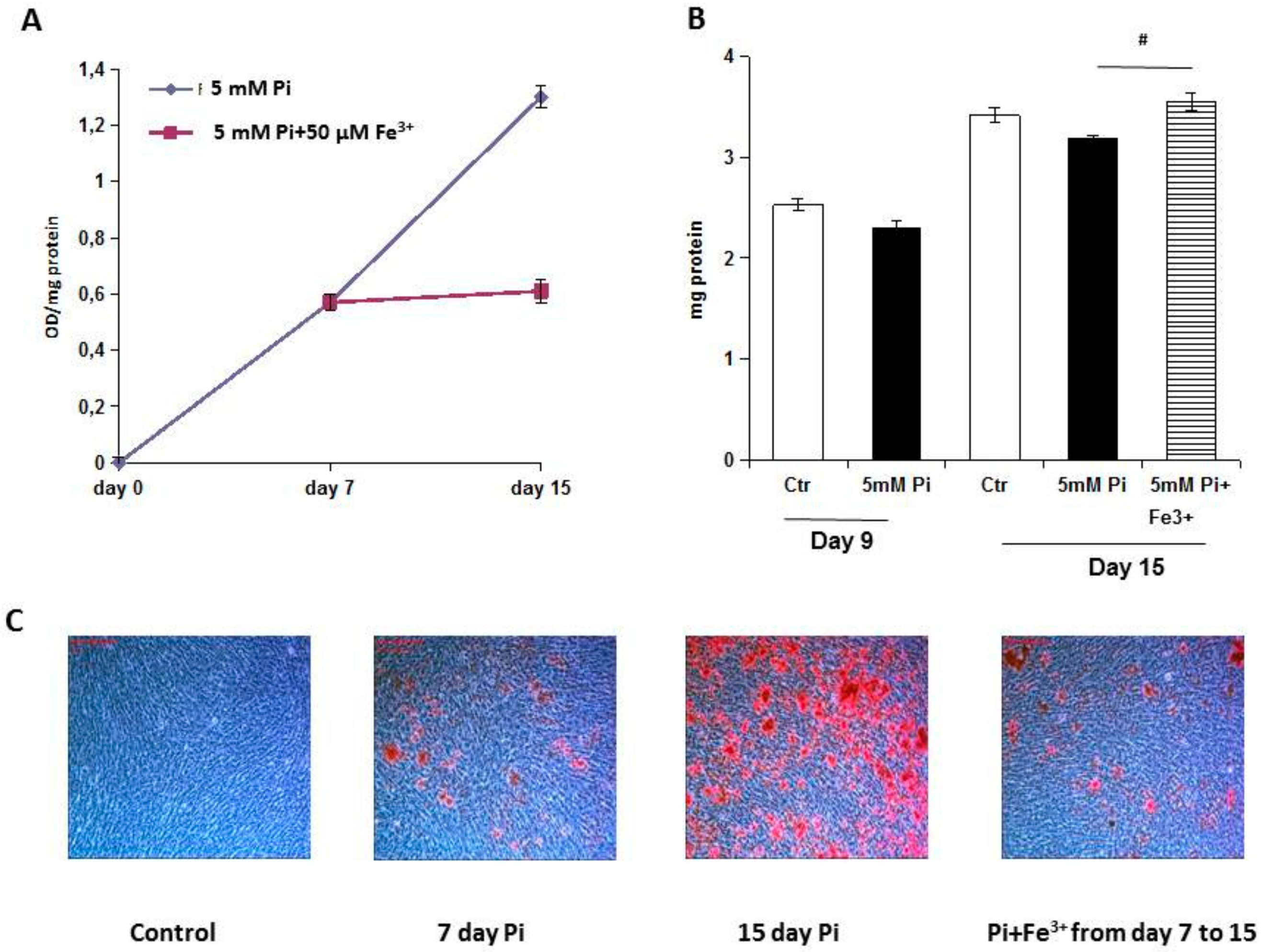
Trying to investigate the direct effect of iron on VC, in the present study, we evaluated the effect of ferric citrate on high Pi-induced calcification mechanisms in established calcified VSMCs, elucidating the data of the effects of iron on calcium deposition [
8]. We performed an in vitro therapeutic approach to evaluate how iron can modulate calcification in an established pathological model of VC. The addition of ferric citrate to 7-day calcified VSMCs completely blocked additional calcium deposition induced by high Pi in the following 8 days of treatment. This result is highly surprising, considering that after 7 days of high Pi challenge, the VSMCs that received iron were already transformed into simil-osteoblasts, and actively deposited calcium-phosphate crystals [
8]. In contrast, in the prophylactic approach, when VSMCs receive high Pi and iron, their phenotype is muscular, and iron may act on the prevention of high Pi-induced VC mechanisms. Thus, performing experiments with the prophylactic and therapeutic administration of iron, we demonstrate that iron can completely block calcium deposition regardless of the grade of high Pi-induced simil-osteoblastic transformation of VSMCs.
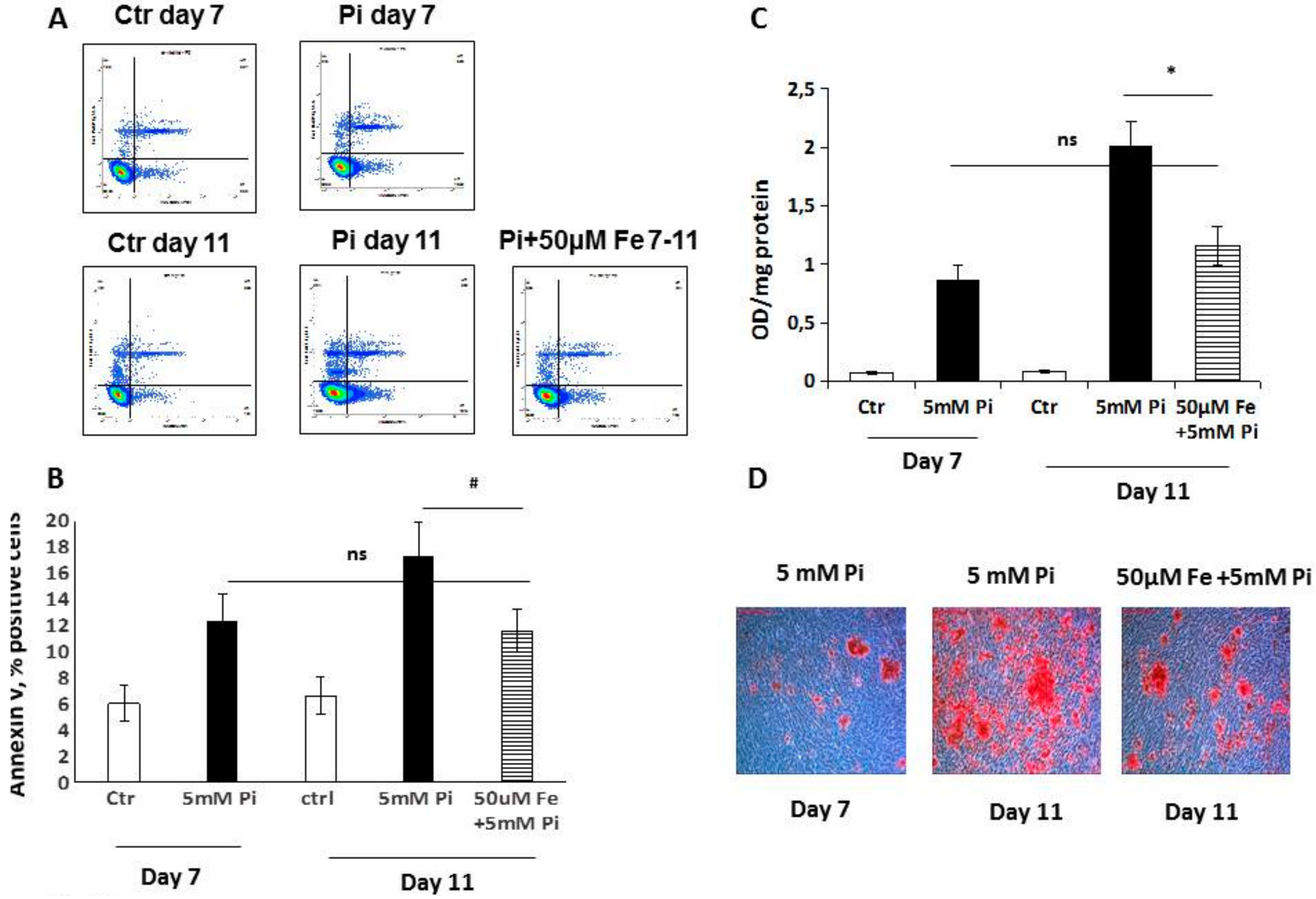
Trying to elucidate the mechanisms responsible for this therapeutic action, we studied apoptosis and autophagy, since we already demonstrated their involvement in the prevention of high Pi-induced calcification [
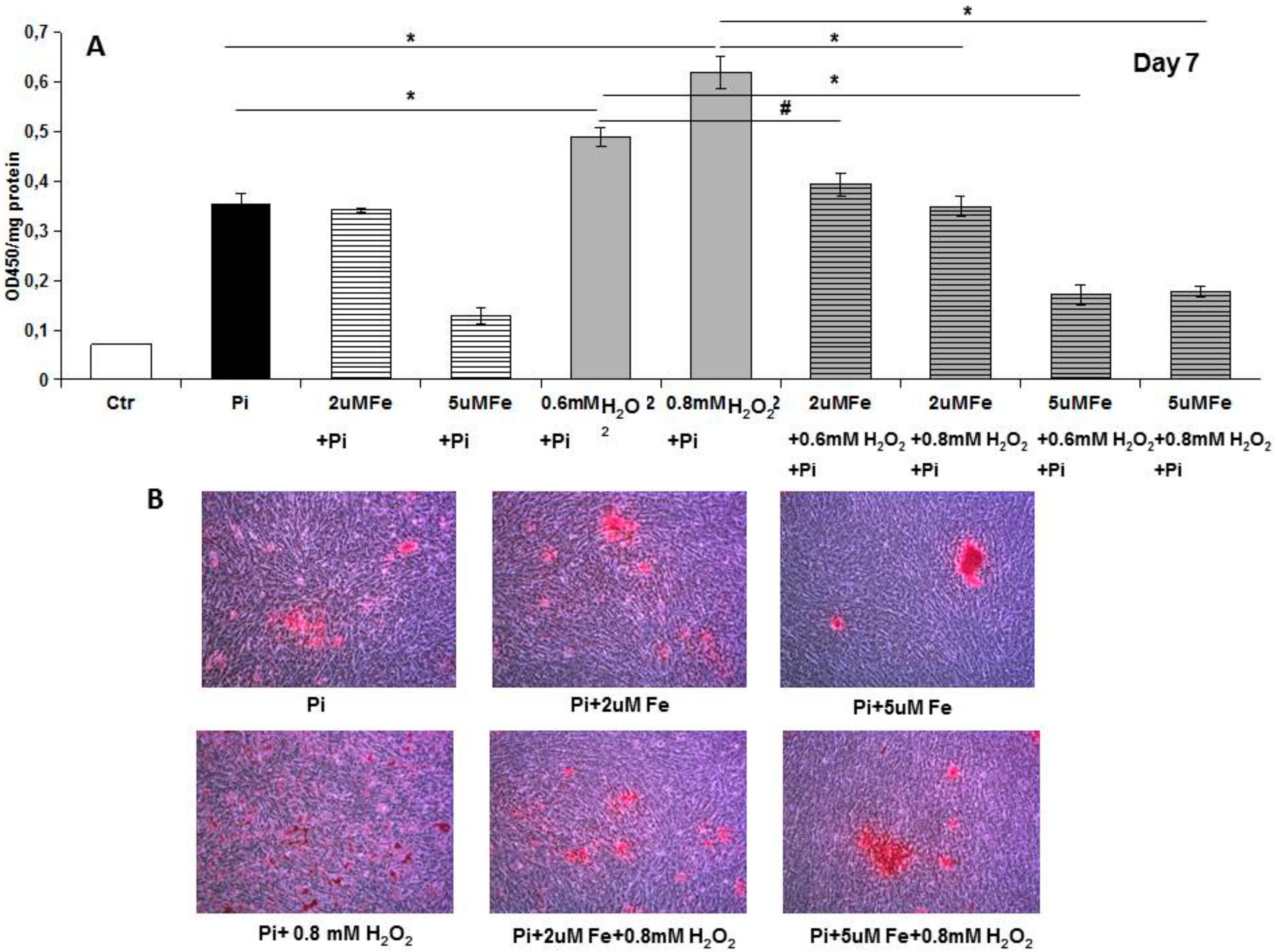
8]. Analyzing apoptosis in the early phase, we found that iron treatment for 4 days on high Pi-calcified VSMCs blocked the progression of apoptosis with a number of total apoptotic cells which was not statistically different from that at the beginning of the addition of iron. These data are surprising if we consider that iron is added together with high Pi, and that in 11-day, high Pi-treated VSMCs, apoptotic cell number increases. Thus, iron is able not only to prevent high Pi-induced apoptosis, but also to delay and block the progression of apoptosis, even if the process is already started and the apoptosis-inducing agent is present. Confirming the anti-apoptotic effect of iron are the data on H
2O
2, a recognized proapoptotic agent. After 7 days of high Pi treatment, H
2O
2 induced a worsening of calcification that was completely counteracted by iron. Interestingly, even a concentration of iron of 2 μM, i.e., one that is ineffective in preventing high Pi calcification, was able to completely prevent a H
2O
2-induced increase in calcium deposition. Moreover, iron is also effective at blocking H
2O
2-induced exacerbation of calcification on already calcified VSMCs, demonstrating an iron anti-apoptotic effect in our in vitro experimental model.
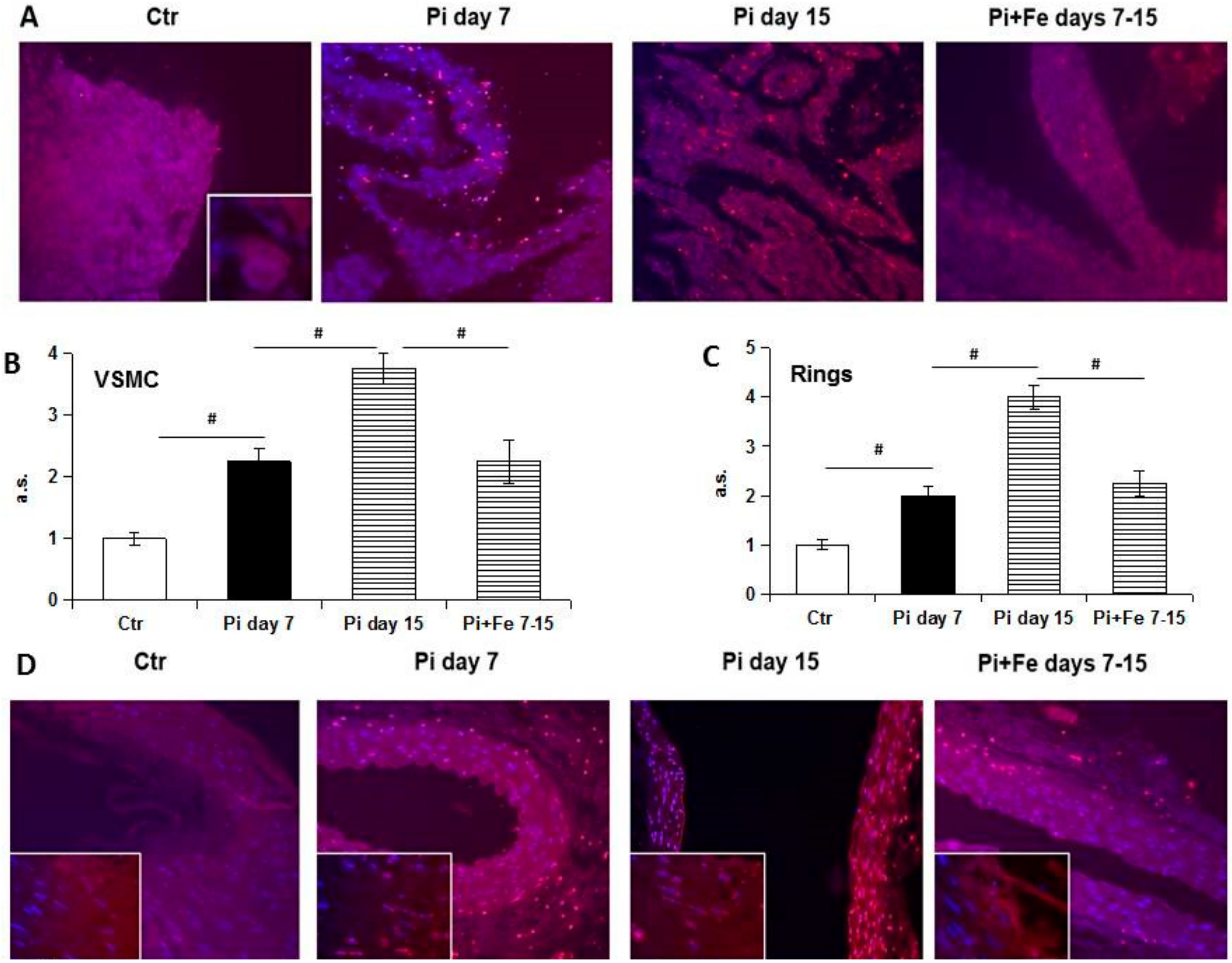
Study of the late stages of the apoptotic process by TUNEL confirms this concept. In fact, increased apoptotic nuclei were detected during high Pi treatment with a massive release of fragmented apoptotic bodies at late time points [
12]. Even if in this work only nuclear bodies were taken into account in evaluating apoptosis, fragmented red dust, at the cytoplasmic level and in the extracellular matrix, was detected. This kind of signal can be interpreted as fragmented apoptotic bodies excreted from dying cells in the extracellular space and/or possibly taken up by phagocytic cells [
13]. We decided to evaluate only the nuclear fragmentation of nuclei during apoptotic death, but interestingly, the specific increase of nuclear red signals consensually paralleled the increase of extra-nuclear apoptotic signals, thus confirming the pattern of apoptosis during high Pi and Fe treatment.
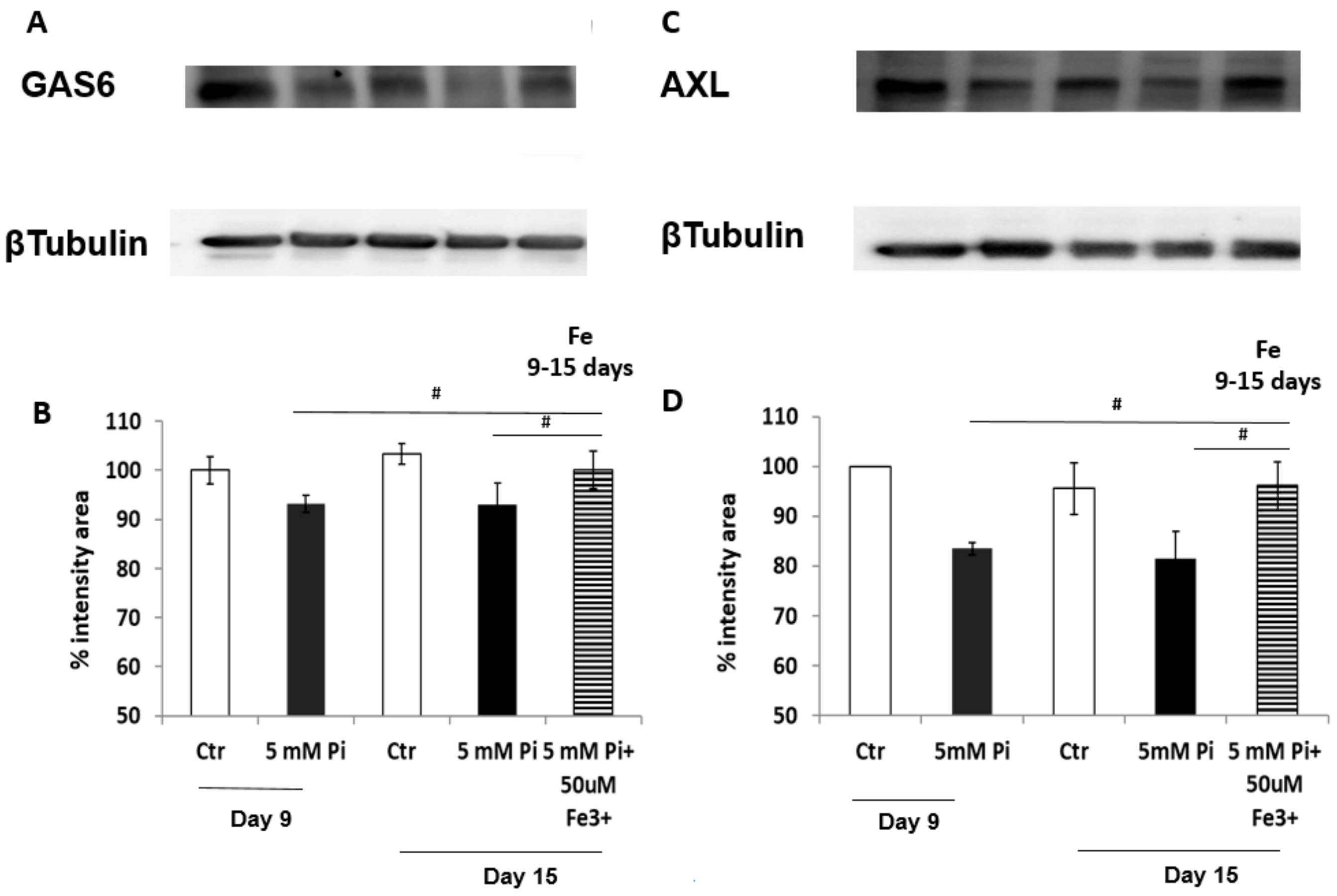
We next investigated the GAS6/AXL prosurvival pathway, which has been shown to be affected by high Pi treatment [
13]. Our data confirmed the involvement of these proteins in the iron citrate protective mechanism. In fact, we found an iron-induced positive modulation of GAS6/AXL synthesis. In our previous study on prophylactic treatment with iron, we demonstrated a preventative action exerted by iron on high Pi-induced GAS6/AXL down-regulation. Interestingly, therapeutic-added iron not only protects by further high Pi-induced down-regulation of GAS6/AXL proteins, but strongly stimulates their de novo synthesis, reactivating this prosurvival and anti-apoptotic pathway. Therefore, one of the mechanisms by which iron citrate exerts its anti-apoptotic action could be the restoration of the GAS6/AXL prosurvival axis. Apoptosis is a relevant process to VC. In fact, apoptotic cells and released apoptotic bodies form a nidus for calcification, and increase the propensity of VSMCs to calcify after calcium and phosphate treatment [
14]. Apoptosis, in calcifying VSMCs, is driven mainly by calcium rather than phosphate alone, as in our model. Nevertheless, a source of calcium is calcium phosphate crystals that undergo lysosomal degradation by VSMCs, leading to very high intracellular calcium levels and subsequent cell death, beginning a vicious cycle of progressive calcification [
15].
In this study, we show that one of the mechanisms of high Pi-induced calcification, apoptosis, can be reverted and counteracted by iron added therapeutically in vitro. These findings demonstrate that iron has an anti-apoptotic effect in high Pi-stimulated VSMCs, independent of the grade of calcification and simil-osteoblastic cell transformation.
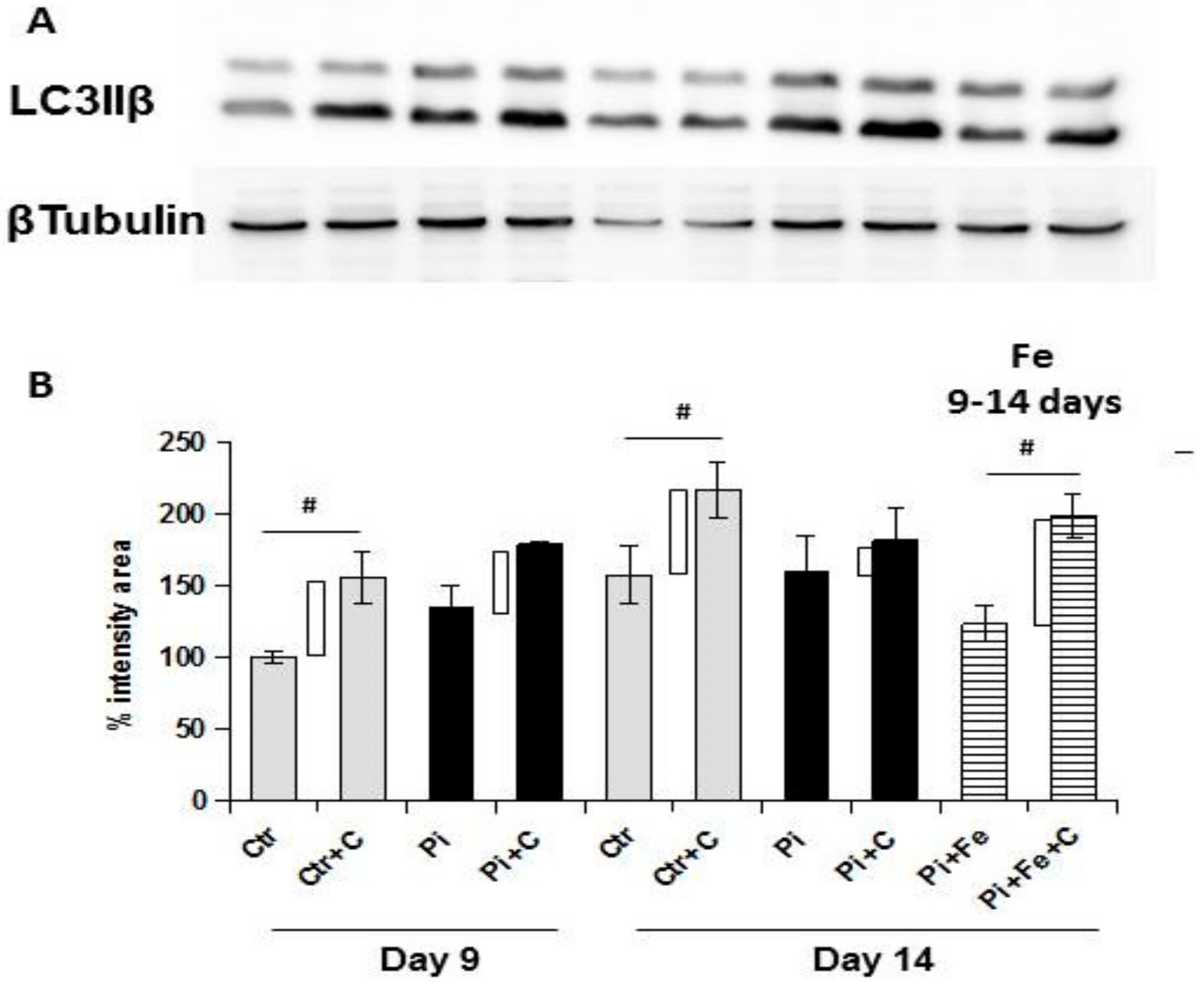
Apoptosis is strictly linked to autophagy, and can be considered as a dramatic consequence of the failure of autophagy to re-establish a positive balance for the cells evolving in survival. Since iron is prophylactically able to modulate autophagy in high Pi-treated VSMCs, we investigated the autophagic process. Autophagy is a highly-conserved cellular process which is responsible for the removal or recycling of long-lived proteins and organelles aimed to give an alternative source of nutrients to cells in conditions of either starvation or stress [
16]. To some extent, autophagy can prevent the activation of apoptotic pathways through the removal of damaged mitochondria [
17], although, in some systems, autophagy can enhance an apoptotic response [
18]. It has been demonstrated that autophagy is a protective phenomenon with respect of high Pi-induced calcification, playing a pivotal role in arterial calcification [
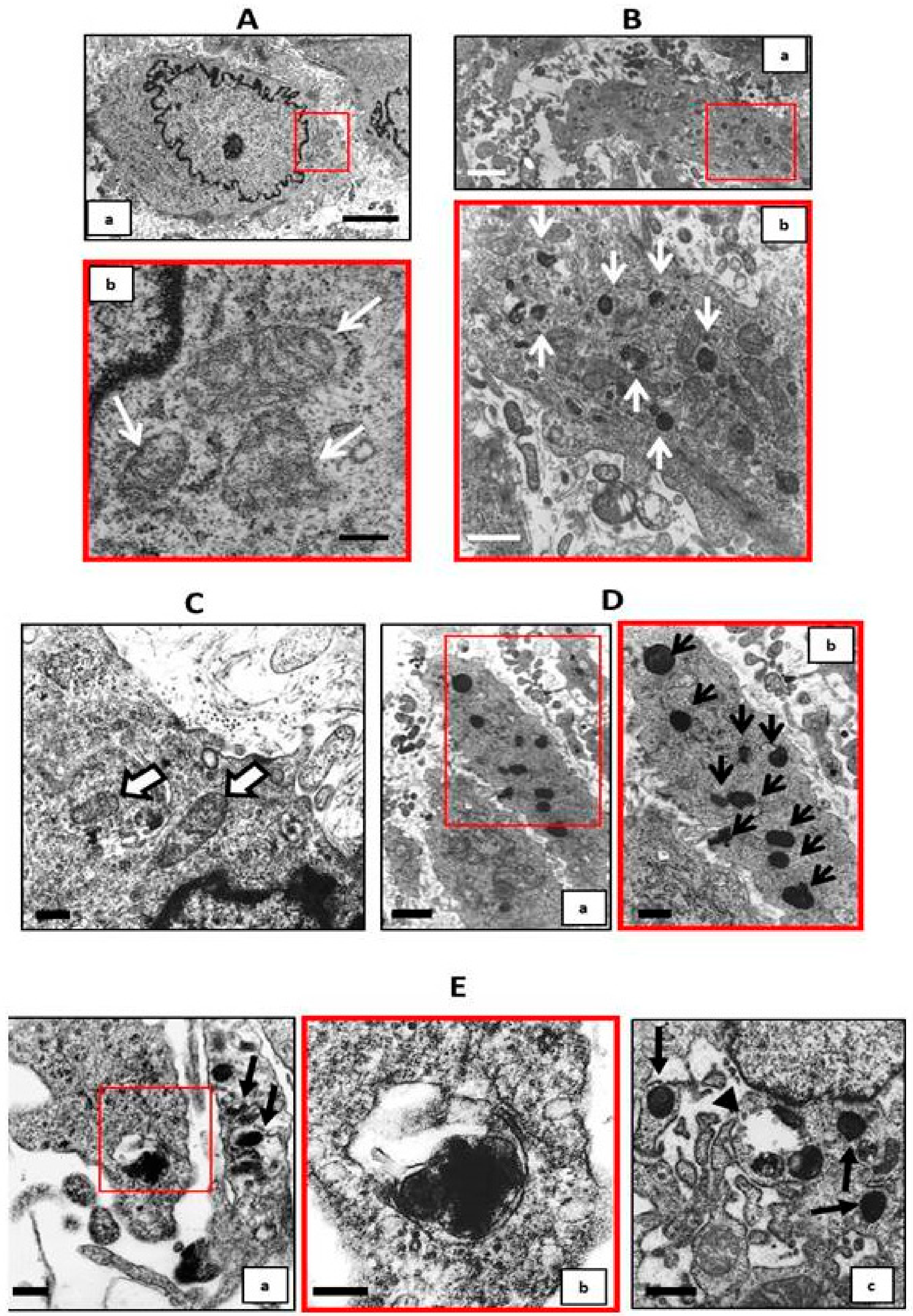
19]. We found that the therapeutic addition of iron was able to stimulate autophagy, even in the presence of high Pi challenge on already calcified VSMCs. In fact, from day 9 to day 14 of calcification, there was a significant reduction of autophagic flux, and treating cells in the same 6 days with iron, even in presence of high Pi, resulted not only in the prevention of further autophagic flux downregulation, but also in the re-induction of the autophagic process. These data were confirmed by electron microscopy. In fact, at 9 days of calcification, there were many calcified mitochondria and no or few autophagosomes, showing the degree of difficulty for the cells to remove these completely damaged organelles. After 6 days of therapeutic iron treatment, on day 14 of calcification, there were still many calcified mitochondria, but there were also autophagosomes filled with damaged organelles, indicating that iron reactivates the autophagic process, helping cells to eliminate damaged mitochondria. As demonstrated for apoptosis, also with respect to autophagy, the therapeutic addition of iron is able to revert the high Pi-induced decrease of the autophagic flux, reactivating the autophagic process.
Iron is an oxidative agent and is rapidly stored by cells because of its potential toxic effect. The addition of iron citrate may affect the red-ox state of high Pi-stimulated VSMCs, and a link between oxidative stress and autophagy in different cellular models has been demonstrated. In fact, autophagy is induced as a response to the toxic effect of reactive oxygen species (ROS), and contributes to clearing the cells of all irreversibly-oxidized biomolecules (proteins, DNA and lipids); this is why it can be included in antioxidant and DNA damage repair systems [
20]. The balance between ROS production and cellular antioxidant mechanisms in high Pi-stimulated VSMCs has been poorly investigated. Probably, a better understanding of oxidative stress/antioxidant defense processes in VC is needed to determine whether iron’s oxidant nature is linked to its mechanism of action and to its effect on autophagy and apoptosis. An important observation is that iron entry into cells induces ferritin synthesis for the storage of this ion to prevent the Fenton reaction, which is dangerous for cell viability due to the production of free radicals. Interestingly, there is evidence that the feroxidase activity of the ferritin heavy chain can protect from both calcification [
21,
22] and apoptosis [
23]. Thus, further studies are needed to deeply investigate whether ferritin heavy chain feroxidase activity can account for the block of calcium deposition and the anti-apoptotic and proautophagic effects of iron in our model of therapeutic in vitro progression of high Pi-induced calcification.
In summary, we studied the effect of iron treatment on established calcified VSMCs, and consequently, on the progression of calcification in vitro. We demonstrated that iron blocks the progression of calcium deposition by affecting apoptosis and autophagy. In fact, iron citrate can revert the high Pi-induced apoptotic process and re-induce autophagy; the combination of these two effects is probably responsible for blocking the calcification progression. These findings, together with the data on iron’s ability to partially revert the high Pi-induced osteo-chondrogenic shift of the VSMC extracellular matrix, help to better elucidate the complex effects of iron and add a piece to the mosaic of its action.
VSMCs are cells with some plasticity, and share their mesenchymal origin with osteoblasts. This may explain why continuous exposition at high Pi levels induces a VSMC simil-osteoblastic differentiation and their ability to deposit calcium phosphate crystals in the vasculature. Nevertheless, we have already demonstrated that suspending the high Pi challenge for brief periods is enough to significantly decrease calcium deposition, meaning that VSMCs can stop the calcium deposition activity, even if already transformed, if the pro calcifying stimulus is suspended, even temporally, at least in vitro [
24]. Here we demonstrate that even if VSMCs are already transformed and the procalcifying stimulus is present, reverting apoptosis and inducing autophagy probably contribute to stopping calcium deposition. Taken together, these data suggest that already transformed VSMCs are prone to stopping the calcium deposition process if calcification mechanisms are adequately targeted.
From these in vitro data, we can conclude that iron can block the progression of calcification and, in line with the iron effect on the extracellular matrix osteochondrogenic shift, we demonstrate that some high Pi-induced processes can be reverted, not only prevented, in vitro. The importance of this reversion concept is in the arrest of additional calcium deposition that is fundamental if we consider the detrimental role of VC in CV disease and its association with increased mortality in CKD patients.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







