Sirtuin-1 and Its Relevance in Vascular Calcification

 , ,
, , 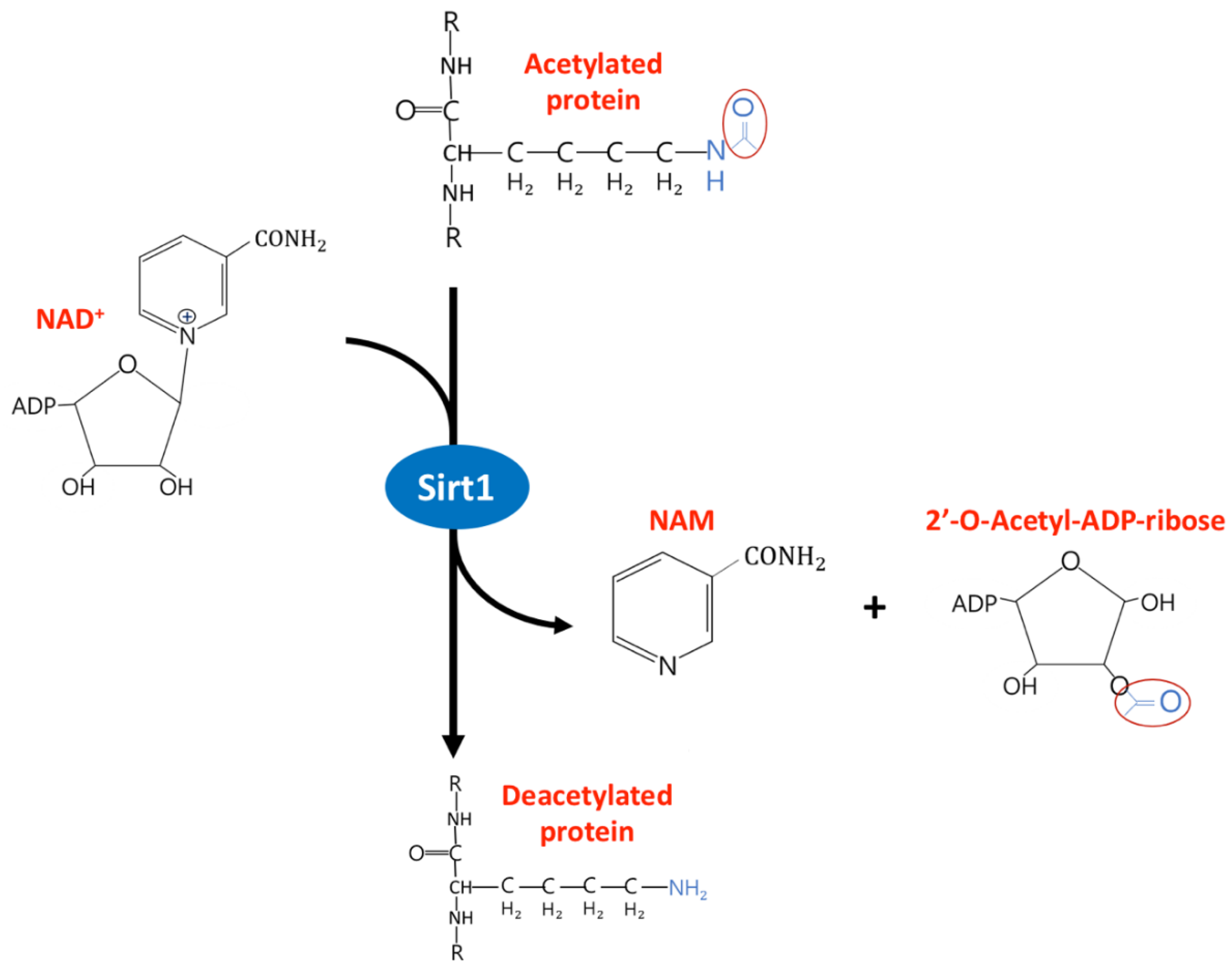{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Diverse Function of Sirtuin1 in Physiology and Clinical Disease
3. The Protective Role of Sirt1 Against Vascular Calcification
3.1. Sirt1 Regulate Nitric Oxide and eNOS Expression in Endothelium
3.2. Sirt1 Affect Endothelial Cells and VSMCs Function against Vascular Calcification
3.2.1. Sirt1 Abolish Endothelial Cells and VSMCs Senescence
3.2.2. Sirt1 Attenuates the Osteoblastic Phenotypic Transition of VSMCs
3.3. Sirt1 Restore Perivascular Adipose Tissue Dysregulation
4. The Interplay between Sirt1 and the Wnt/β-Catenin Pathway in Vascular Calcification
5. Role of Sirt1 and Sirt1 Modulator in Clinical Setting
5.1. Sirt1 Retard Hyperphosphatemia-Induced Medial Calcification in CKD
5.2. Vitamin D Supplement Is Beneficial in ECs and Adipose Tissue by Upregulation of Sirt1
5.3. Sirt1 Activator Modulates Vascular Disease in Preclinical and Clinical Settings
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | Adenosine monophosphate-activated protein kinase |
| BMP-2 | Bone morphogenetic protein-2 |
| CR | Calorie restriction |
| CKD | Chronic kidney disease |
| DM | Diabetes mellitus |
| ECM | Extracellular matrix |
| FoxO | Forkhead box O |
| GSK-3β | Glycogen Synthase Kinase-3 beta |
| MGP | Matrix Gla protein |
| NAM | Nicotinamide |
| NO | Nitric oxide |
| NOS | NO synthase |
| PPAR | Peroxisome proliferator-activated receptor |
| PVAT | Perivascular adipose tissue |
| ROS | Reactive oxygen species |
| RUNX2 | Runt-related transcription factor-2 |
| SMαA | Smooth muscle α-actin |
| SM-22α | Smooth muscle-22α |
| SIPS | Stress-induced premature senescence |
| Sirt1 | Sirtuin-1 |
| VC | Vascular calcification |
| VSMCs | Vascular smooth muscle cells |
References
- Ohtake, T.; Kobayashi, S. Impact of vascular calcification on cardiovascular mortality in hemodialysis patients: Clinical significance, mechanisms and possible strategies for treatment. Ren. Replace. Ther. 2017, 3, 55. [Google Scholar] [CrossRef][Green Version]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular Calcification. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Cano-Megías, M.; Vasco, P.G.; Bouarich, H.; Fuente, G.D.A.-D.L.; De Sequera-Ortiz, P.; Álvarez-Sanz, C.; Rodríguez-Puyol, D. Coronary calcification as a predictor of cardiovascular mortality in advanced chronic kidney disease: A prospective long-term follow-up study. BMC Nephrol. 2019, 20, 188. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; Hilaire, C.S.; Shanahan, C.M. Medial vascular calcification revisited: Review and perspectives. Eur. Hear J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Mönckeberg, J.G. Über die reine Mediaverkalkung der Extremitätenarterien und ihr Verhalten zur Arteriosklerose. Virchows Archiv 1903, 171, 141–167. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, K.; Chen, J.; Wang, M.-H.; Wang, J.; Liu, P.; Huang, H.-Q. Roles of aldosterone in vascular calcification: An update. Eur. J. Pharmacol. 2016, 786, 186–193. [Google Scholar] [CrossRef]
- Hung, S.; Kuo, K.; Wu, C.-C.; Tarng, D.-C. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Lu, C.-L.; Zheng, C.-M.; Chen, R.-M.; Lin, Y.-F.; Liu, W.-C.; Yen, T.-H.; Chen, R.; Lu, K.-C. Emerging Role of Vitamins D and K in Modulating Uremic Vascular Calcification: The Aspect of Passive Calcification. Nutrients 2019, 11, 152. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.B.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Viegas, C.; Santos, L.; Macedo, A.; De Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification. Atheroscler. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Richards, C. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013, 2013, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Albasanz-Puig, A.; Murray, J.; Preusch, M.; Coan, D.; Namekata, M.; Patel, Y.; Dong, Z.M.; Rosenfeld, M.E.; Wijelath, E. Oncostatin M is expressed in atherosclerotic lesions: A role for Oncostatin M in the pathogenesis of atherosclerosis. Atherosclerosis 2011, 216, 292–298. [Google Scholar] [CrossRef]
- Sorokin, V.; Woo, C.C. Role of Serpina3 in vascular biology. Int. J. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Park, J.E.; Kumar, S.; Hao, P.; Gallart-Palau, X.; Serra, A.; Ren, Y.; Sorokin, V.; Lee, C.N.; Ho, H.H.; et al. Monocyte adhesion to atherosclerotic matrix proteins is enhanced by Asn-Gly-Arg deamidation. Sci. Rep. 2017, 7, 5765. [Google Scholar] [CrossRef] [PubMed]
- Kupis, W.; Pałyga, J.; Tomal, E.; Niewiadomska, E. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Lu, C.-L.; Yuan, T.-H.; Liao, M.-T.; Chao, C.-T.; Lu, K.-C. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. Int. J. Mol. Sci. 2020, 21, 980. [Google Scholar] [CrossRef]
- Zhang, T.; Kraus, W.L. SIRT1-dependent regulation of chromatin and transcription: Linking NAD+ metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1804, 1666–1675. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Yu, J.; Auwerx, J. The role of sirtuins in the control of metabolic homeostasis. Ann. N. Y. Acad. Sci. 2009, 1173 (Suppl. 1), E10–E19. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, A.; Favero, G.; Rezzani, R. Resveratrol and SIRT1 Activators for the Treatment of Aging and Age-Related Diseases. Available online: https://www.intechopen.com/books/resveratrol-adding-life-to-years-not-adding-years-to-life/resveratrol-and-sirt1-activators-for-the-treatment-of-aging-and-age-related-diseases (accessed on 27 January 2020).
- Imai, S.-I.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. npj Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. HepatoBiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [PubMed]
- Ding, R.-B.; Bao, J.; Deng, C. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab. 2009, 20, 325–331. [Google Scholar] [CrossRef]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPAR to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2010, 90, 276–284. [Google Scholar] [CrossRef]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef]
- Rodgers, J.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Liu, Y.; Dentin, R.; Chen, D.; Hedrick, S.; Ravnskjaer, K.; Schenk, S.; Milne, J.; Meyers, D.J.; Cole, P.; Yates, J.; et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 2008, 456, 269–273. [Google Scholar] [CrossRef]
- Ponugoti, B.; Kim, N.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D.; Kemper, J.K. SIRT1 Deacetylates and Inhibits SREBP-1C Activity in Regulation of Hepatic Lipid Metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 Deacetylates and Positively Regulates the Nuclear Receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of Glycolytic Enzyme Phosphoglycerate Mutase-1 by Sirt1 Protein-mediated Deacetylation. J. Biol. Chem. 2011, 287, 3850–3858. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L.; Franklin, H. Epstein Lecture: Sirtuins, aging, and medicine. N. Engl. J. Med. 2011, 364, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Blatter, L.; Wier, W. Nitric oxide decreases [Ca2+]i in vascular smooth muscle by inhibition of the calcium current. Cell Calcium 1994, 15, 122–131. [Google Scholar] [CrossRef]
- Van Hove, C.; Van Der Donckt, C.; Herman, A.; Bult, H.; Fransen, P. Vasodilator efficacy of nitric oxide depends on mechanisms of intracellular calcium mobilization in mouse aortic smooth muscle cells. Br. J. Pharmacol. 2009, 158, 920–930. [Google Scholar] [CrossRef]
- Lee, R.; Channon, K.M.; Antoniades, C. Therapeutic strategies targeting endothelial function in humans: Clinical implications. Curr. Vasc. Pharmacol. 2012, 10, 77–93. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.-F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef]
- Haynes, W.G.; Noon, J.P.; Walker, B.R.; Webb, D.J. Inhibition of nitric oxide synthesis increases blood pressure in healthy humans. J. Hypertens. 1993, 11, 1142. [Google Scholar] [CrossRef]
- Lepori, M.; Sartori, C.; Trueb, L.; Owlya, R.; Nicod, P.; Scherrer, U. Haemodynamic and sympathetic effects of inhibition of nitric oxide synthase by systemic infusion of N(G)-monomethyl-L-arginine into humans are dose dependent. J. Hypertens. 1998, 16, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Collier, J.; Moncada, S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989, 334, 997–1000. [Google Scholar] [CrossRef]
- Pucci, M.L.; Lin, L.; Nasjletti, A. Pressor and renal vasoconstrictor effects of NG-nitro-L-arginine as affected by blockade of pressor mechanisms mediated by the sympathetic nervous system, angiotensin, prostanoids and vasopressin. J. Pharmacol. Exp. Ther. 1992, 261, 240–245. [Google Scholar]
- Deng, A.; Engels, K.; Baylis, C. Impact of nitric oxide deficiency on blood pressure and glomerular hemodynamic adaptations to pregnancy in the rat. Kidney Int. 1996, 50, 1132–1138. [Google Scholar] [CrossRef]
- Rapoport, R.M. Acute nitric oxide synthase inhibition and endothelin-1-dependent arterial pressure elevation. Front. Pharmacol. 2014, 5, 57. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis 2018, 271, 237–244. [Google Scholar] [CrossRef]
- Woo, C.C.; Liu, W.; Lin, X.Y.; Dorajoo, R.; Lee, K.W.; Richards, A.M.; Lee, C.N.; Wongsurawat, T.; Nookaew, I.; Sorokin, V.; et al. The Interaction between 30b-5p miRNA and MBNL1 mRNA is Involved in Vascular Smooth Muscle Cell Differentiation in Patients with Coronary Atherosclerosis. Int. J. Mol. Sci. 2019, 21, 11. [Google Scholar] [CrossRef]
- Derda, A.A.; Woo, C.C.; Wongsurawat, T.; Richards, M.; Lee, C.N.; Kofidis, T.; Kuznetsov, V.A.; Sorokin, V. Gene expression profile analysis of aortic vascular smooth muscle cells reveals upregulation of cadherin genes in myocardial infarction patients. Physiol. Genom. 2018, 50, 648–657. [Google Scholar] [CrossRef]
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef]
- Wallerath, T.; Deckert, G.; Ternes, T.; Anderson, H.; Li, H.; Witte, K.; Förstermann, U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation 2002, 106, 1652–1658. [Google Scholar] [CrossRef]
- Ota, H.; Eto, M.; Kano, M.R.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Cilostazol Inhibits Oxidative Stress–Induced Premature Senescence Via Upregulation of Sirt1 in Human Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1634–1639. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Tedesco, L.; Falcone, S.; Carruba, M.; Bracale, R.; Valerio, A.; Cantoni, O.; et al. Calorie Restriction Promotes Mitochondrial Biogenesis by Inducing the Expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, M.; Sakamoto, H.; Kawasuji, M.; Furuyama, T.; Ogawa, M. Different roles of Foxo1 and Foxo3 in the control of endothelial cell morphology. Genes Cells 2009, 14, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; You, H.-J.; Won, J.-Y.; Youn, S.-W.; Cho, H.-J.; Park, K.-W.; Park, W.-Y.; Seo, J.-S.; Park, Y.-B.; Walsh, K.; et al. Forkhead Factor, FOXO3a, Induces Apoptosis of Endothelial Cells Through Activation of Matrix Metalloproteinases. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ko, Y.S.; Park, J.; Choi, Y.; Park, J.-W.; Kim, Y.; Pyo, J.-S.; Yoo, Y.B.; Lee, J.-S.; Lee, B.L. Forkhead Transcription Factor FOXO1 Inhibits Angiogenesis in Gastric Cancer in Relation to SIRT1. Cancer Res. Treat. 2016, 48, 345–354. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Chung, J.-W.; Youn, S.-W.; Kim, J.-Y.; Park, K.-W.; Koo, B.-K.; Oh, B.-H.; Park, Y.-B.; Chaqour, B.; Walsh, K.; et al. Forkhead Transcription Factor FOXO3a Is a Negative Regulator of Angiogenic Immediate Early Gene CYR61, Leading to Inhibition of Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia. Circ. Res. 2007, 100, 372–380. [Google Scholar] [CrossRef]
- Potente, M.; Urbich, C.; Sasaki, K.-I.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; Depinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005, 115, 2382–2392. [Google Scholar] [CrossRef]
- Van Der Heide, L.P.; Hoekman, M.F.M.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Yun, J.-M.; Chien, A.; Jialal, I.; Devaraj, S. Resveratrol up-regulates SIRT1 and inhibits cellular oxidative stress in the diabetic milieu: Mechanistic insights. J. Nutr. Biochem. 2011, 23, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Cao, Q.; Wang, F.; Huang, L.-Y.; Sang, T.-T.; Liu, F.; Chen, S.-Y. SIRT1 Protects Against Oxidative Stress-Induced Endothelial Progenitor Cells Apoptosis by Inhibiting FOXO3a via FOXO3a Ubiquitination and Degradation. J. Cell. Physiol. 2015, 230, 2098–2107. [Google Scholar] [PubMed]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S.; et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology 2013, 15, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Chang, S. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab. Investig. 2007, 87, 1071–1076. [Google Scholar] [CrossRef]
- Tamrakar, S. Role of pRB dephosphorylation in cell cycle regulation. Front. Biosci. 2000, 5, d121. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Hansen, G.; Murray, D. Role of p16INK4A in Replicative Senescence and DNA Damage-Induced Premature Senescence in p53-Deficient Human Cells. Biochem. Res. Int. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K. Role of NADH/NADPH Oxidase–Derived H2O2in Angiotensin II–Induced Vascular Hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef]
- Herbert, K.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2007, 102, 201–208. [Google Scholar] [CrossRef]
- Griendling, K.; Sorescu, D.; Ushio-Fukai, M.; Lasseègue, B. Modulation of Protein Kinase Activity and Gene Expression by Reactive Oxygen Species and Their Role in Vascular Physiology and Pathophysiology. Atheroscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef]
- Thompson, A.M.; Wagner, R.; Rzucidlo, E.M. Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am. J. Physiol. Circ. Physiol. 2014, 307, H533–H541. [Google Scholar] [CrossRef]
- Durham, A.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Alves, R.D.A.M.; Eijken, M.; Van De Peppel, J.; Van Leeuwen, J. Calcifying vascular smooth muscle cells and osteoblasts: Independent cell types exhibiting extracellular matrix and biomineralization-related mimicries. BMC Genom. 2014, 15, 965. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Wiemann, S.; Buer, J.; Lauber, J.; Dittmar, K.; Wüstefeld, T.; Blasco, M.A.; Manns, M.; Rudolph, K.L. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003, 22, 4003–4013. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; Von Zglinicki, T.; Passos, J. Quantitative assessment of markers for cell senescence. Exp. Gerontol. 2010, 45, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Nakano-Kurimoto, R.; Ikeda, K.; Uraoka, M.; Nakagawa, Y.; Yutaka, K.; Koide, M.; Takahashi, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am. J. Physiol. Circ. Physiol. 2009, 297, H1673–H1684. [Google Scholar] [CrossRef] [PubMed]
- Drüeke, T.B.; Massy, Z.A. Role of vitamin D in vascular calcification: Bad guy or good guy? Nephrol. Dial. Transplant. 2012, 27, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Schiro, A.; Inglott, F.S.; Alexander, M.Y.; Weston, R. Suppression of SIRT1 in Diabetic Conditions Induces Osteogenic Differentiation of Human Vascular Smooth Muscle Cells via RUNX2 Signalling. Sci. Rep. 2019, 9, 878. [Google Scholar] [CrossRef]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; et al. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Atheroscler. Thromb. Vasc. Biol. 2018, 38, 2079–2090. [Google Scholar] [CrossRef]
- Sharma, A.; Gautam, V.; Costantini, S.; Paladino, A.; Colonnaa, G. Interactomic and Pharmacological Insights on Human Sirt-1. Front. Pharmacol. 2012, 3, 3. [Google Scholar] [CrossRef]
- Costa, R.M.; Neves, K.B.; Tostes, R.C.; Lobato, N.S. Perivascular Adipose Tissue as a Relevant Fat Depot for Cardiovascular Risk in Obesity. Front. Physiol. 2018, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Meziat, C.; Boulghobra, D.; Strock, E.; Battault, S.; Bornard, I.; Walther, G.; Reboul, C. Exercise training restores eNOS activation in the perivascular adipose tissue of obese rats: Impact on vascular function. Nitric Oxide 2019, 86, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Villacorta, L.; Chang, L. The role of perivascular adipose tissue in vasoconstriction, arterial stiffness, and aneurysm. Horm. Mol. Biol. Clin. Investig. 2015, 21, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, C.T.; Jiang, Y.; Yun, C.-H.; Debanne, S.; Funderburg, N.; Lederman, M.M.; Storer, N.; Labbato, D.E.; Bezerra, H.G.; Mccomsey, G.A. Perivascular fat, inflammation, and cardiovascular risk in HIV-infected patients on antiretroviral therapy. Int. J. Cardiol. 2013, 168, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Shields, K.J.; Barinas-Mitchell, E.; Gingo, M.R.; Tepper, P.; Goodpaster, B.H.; Kao, A.H.; Manzi, S.; Sutton-Tyrrell, K. Perivascular adipose tissue of the descending thoracic aorta is associated with systemic lupus erythematosus and vascular calcification in women. Atherosclerosis 2013, 231, 129–135. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xiao, N.; Wang, M.; Kou, J.; Qi, L.; Huang, F.; Liu, B.; Liu, K. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol. Res. 2014, 89, 19–28. [Google Scholar] [CrossRef]
- Fleenor, B.S.; Eng, J.S.; Sindler, A.L.; Pham, B.T.; Kloor, J.D.; Seals, U.R. Superoxide signaling in perivascular adipose tissue promotes age-related artery stiffness. Aging Cell 2014, 13, 576–578. [Google Scholar] [CrossRef]
- Qiao, L.; Shao, J. SIRT1 Regulates Adiponectin Gene Expression through Foxo1-C/Enhancer-binding Protein Transcriptional Complex. J. Biol. Chem. 2006, 281, 39915–39924. [Google Scholar] [CrossRef]
- Yoo, J.-K.; Hwang, M.-H.; Luttrell, M.J.; Kim, H.-K.; Meade, T.H.; English, M.; Segal, M.S.; Christou, D.D. Higher levels of adiponectin in vascular endothelial cells are associated with greater brachial artery flow-mediated dilation in older adults. Exp. Gerontol. 2015, 63, 1–7. [Google Scholar] [CrossRef]
- Man, A.W.C.; Li, H.; Xia, N. The Role of Sirtuin1 in Regulating Endothelial Function, Arterial Remodeling and Vascular Aging. Front. Physiol. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed]
- Gay, A.; Towler, D. Wnt signaling in cardiovascular disease. Curr. Opin. Lipidol. 2017, 28, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef] [PubMed]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.N.; Komm, B.S.; Javed, A.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT Signaling Promotes Osteogenesis by Directly StimulatingRunx2Gene Expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Jia, H.; Meyers, C.; Shrestha, S.; Asatrian, G.; Ding, C.; Tsuei, R.; Zhang, X.; Peault, B.; et al. Effects of WNT3A and WNT16 on the Osteogenic and Adipogenic Differentiation of Perivascular Stem/Stromal Cells. Tissue Eng. Part A 2018, 24, 68–80. [Google Scholar] [CrossRef]
- Baschant, U.; Rauner, M.; Balaian, E.; Weidner, H.; Roetto, A.; Platzbecker, U.; Hofbauer, L.C. Wnt5a is a key target for the pro-osteogenic effects of iron chelation on osteoblast progenitors. Haematologica 2016, 101, 1499–1507. [Google Scholar] [CrossRef]
- Mill, C.; George, S.J. Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovasc. Res. 2012, 95, 233–240. [Google Scholar] [CrossRef]
- Hecht, A.; Vleminckx, K.; Stemmler, M.P.; Van Roy, F.; Kemler, R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000, 19, 1839–1850. [Google Scholar] [CrossRef]
- Bouras, T.; Fu, M.; Sauve, A.A.; Wang, F.; Quong, A.A.; Perkins, N.D.; Hay, R.T.; Gu, W.; Pestell, R.G. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 2005, 280, 10264–10276. [Google Scholar] [CrossRef]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Langford-Smith, A.W.W.; Alexander, M.Y.; Weston, R. The Interplay of SIRT1 and Wnt Signaling in Vascular Calcification. Front. Cardiovasc. Med. 2018, 5, 183. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, F.; Herencia, C.; Almadén, Y.; Martínez-Moreno, J.M.; de Oca, A.M.; Rodriguez-Ortiz, M.E.; Diaz-Tocados, J.M.; Canalejo, A.; Florio, M.; López, I.; et al. TGF-beta prevents phosphate-induced osteogenesis through inhibition of BMP and Wnt/beta-catenin pathways. PLoS ONE 2014, 9, e89179. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, M.M.; Xavier, S.; Vasko, R.; Kaur, K.; Goligorksy, M.S.; Ratliff, B.B. High-mobility group box 1 is a novel deacetylation target of Sirtuin1. Kidney Int. 2014, 87, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci. Rep. 2015, 5, 15971. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, e10–e17. [Google Scholar] [CrossRef]
- Mathew, S.; Tustison, K.S.; Sugatani, T.; Chaudhary, L.R.; Rifas, L.; Hruska, K.A. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J. Am. Soc. Nephrol. 2008, 19, 1092–1105. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Speer, M.Y.; Li, X.; Hiremath, P.G.; Giachelli, C.M. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell. Biochem. 2010, 110, 935–947. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, B.; Zhou, P.; Zhang, R.; He, M.; Yang, Z.; Wen, J. Vascular calcification is coupled with phenotypic conversion of vascular smooth muscle cells through Klf5-mediated transactivation of the Runx2 promoter. Biosci. Rep. 2014, 34, 663–672. [Google Scholar] [CrossRef]
- Zhang, D.; Bi, X.; Liu, Y.; Huang, Y.; Xiong, J.; Xu, X.; Xiao, T.; Yu, Y.; Jiang, W.; Huang, Y.; et al. High Phosphate-Induced Calcification of Vascular Smooth Muscle Cells is Associated with the TLR4/NF-kappab Signaling Pathway. Kidney Blood Press Res. 2017, 42, 1205–1215. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.; Lu, J.; Tintut, Y.; Demer, L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2010, 79, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Melaragno, M.G.; Cavet, M.E.; Yan, C.; Tai, L.-K.; Jin, Z.-G.; Haendeler, J.; Berk, B.C. Gas6 inhibits apoptosis in vascular smooth muscle: Role of Axl kinase and Akt. J. Mol. Cell. Cardiol. 2004, 37, 881–887. [Google Scholar] [CrossRef]
- Collett, G.; Wood, A.; Alexander, Y.; Varnum, B.C.; Boot-Handford, R.P.; Ohanian, V.; Ohanian, J.; Fridell, Y.-W.; Canfield, A. Receptor Tyrosine Kinase Axl Modulates the Osteogenic Differentiation of Pericytes. Circ. Res. 2003, 92, 1123–1129. [Google Scholar] [CrossRef]
- Kaesler, N.; Immendorf, S.; Ouyang, C.; Herfs, M.; Drummen, N.; Carmeliet, P.; Vermeer, C.; Floege, J.; Krüger, T.; Schlieper, G. Gas6 protein: Its role in cardiovascular calcification. BMC Nephrol. 2016, 17, 52. [Google Scholar] [CrossRef]
- Son, B.-K.; Kozaki, K.; Iijima, K.; Eto, M.; Kojima, T.; Ota, H.; Senda, Y.; Maemura, K.; Nakano, T.; Akishita, M.; et al. Statins Protect Human Aortic Smooth Muscle Cells From Inorganic Phosphate-Induced Calcification by Restoring Gas6-Axl Survival Pathway. Circ. Res. 2006, 98, 1024–1031. [Google Scholar] [CrossRef]
- Ewence, A.E.; Bootman, M.; Roderick, H.L.; Skepper, J.N.; McCarthy, G.; Epple, M.; Neumann, M.; Shanahan, C.M.; Proudfoot, D. Calcium Phosphate Crystals Induce Cell Death in Human Vascular Smooth Muscle Cells. Circ. Res. 2008, 103, e28–e34. [Google Scholar] [CrossRef]
- Takemura, A.; Iijima, K.; Ota, H.; Son, B.-K.; Ito, Y.; Ogawa, S.; Eto, M.; Akishita, M.; Ouchi, Y. Sirtuin 1 Retards Hyperphosphatemia-Induced Calcification of Vascular Smooth Muscle Cells. Atheroscler. Thromb. Vasc. Biol. 2011, 31, 2054–2062. [Google Scholar] [CrossRef]
- Ganesh, S.K.; Stack, A.G.; Levin, N.W.; Hulbert-Shearon, T.; Port, F.K. Association of elevated serum PO(4), Ca x PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J. Am. Soc. Nephrol. 2001, 12, 2131–2138. [Google Scholar]
- Young, E.W.; Albert, J.; Satayathum, S.; Goodkin, D.A.; Pisoni, R.L.; Akiba, T.; Akizawa, T.; Kurokawa, K.; Bommer, J.; Piera, L.; et al. Predictors and consequences of altered mineral metabolism: The Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2005, 67, 1179–1187. [Google Scholar] [CrossRef]
- Sehgal, A.R.; Sullivan, C.; Leon, J.B.; Bialostosky, K. Public health approach to addressing hyperphosphatemia among dialysis patients. J. Ren. Nutr. 2008, 18, 256–261. [Google Scholar] [CrossRef][Green Version]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G. Relation Between Serum Phosphate Level and Cardiovascular Event Rate in People With Coronary Disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef]
- Dhingra, R.; Sullivan, L.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Relations of Serum Phosphorus and Calcium Levels to the Incidence of Cardiovascular Disease in the Community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Tatsumoto, N.; Tokumoto, M.; Noguchi, H.; Ooboshi, H.; Kitazono, T.; Tsuruya, K. Phosphate Binders Prevent Phosphate-Induced Cellular Senescence of Vascular Smooth Muscle Cells and Vascular Calcification in a Modified, Adenine-Based Uremic Rat Model. Calcif. Tissue Int. 2014, 96, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Kim, J.; Ireland, E.; Chazot, C.; Drüeke, T.B.; De Francisco, A.; Kronenberg, F.; Marcelli, D.; Passlick-Deetjen, J.; Schernthaner, G.; et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol. Dial. Transplant. 2010, 26, 1948–1955. [Google Scholar] [CrossRef]
- Hoe, E.; Nathanielsz, J.; Toh, Z.Q.; Spry, L.; Marimla, R.; Balloch, A.; Mulholland, E.K.; Licciardi, P. Anti-Inflammatory Effects of Vitamin D on Human Immune Cells in the Context of Bacterial Infection. Nutrients 2016, 8, 806. [Google Scholar] [CrossRef] [PubMed]
- Saputo, S.; Faustoferri, R.C.; Quivey, R.G. Vitamin D Compounds Are Bactericidal against Streptococcus mutans and Target the Bacitracin-Associated Efflux System. Antimicrob. Agents Chemother. 2017, 62, e01675-17. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, K.; Moore, C.G.; Khalid, A.T.; Vallejo, A.N.; A Virji, M.; Holick, M.F.; Greenspan, S.L.; Arslanian, S.; E Reis, S. Effect of vitamin D3 supplementation on vascular and metabolic health of vitamin D–deficient overweight and obese children: A randomized clinical trial. Am. J. Clin. Nutr. 2020. [Google Scholar] [CrossRef]
- Kalmarzi, R.N.; Ahmadi, S.; Rahehagh, R.; Fathallahpour, A.; Khalafi, B.; Kashefi, H.; Roshani, D.; Zaryan, R.N.; Mohamadi, S.; Kooti, W. The Effect of Vitamin D Supplementation on Clinical Outcomes of Asthmatic Children with Vitamin D Insufficiency. Endocrine Metab. Immune Disord. Drug Targets 2020, 20, 149–155. [Google Scholar] [CrossRef]
- Bhatt, S.P.; Misra, A.; Pandey, R.M.; Upadhyay, A.D.; Gulati, S.; Singh, N. Vitamin D Supplementation in Overweight/obese Asian Indian Women with Prediabetes Reduces Glycemic Measures and Truncal Subcutaneous Fat: A 78 Weeks Randomized Placebo-Controlled Trial (PREVENT-WIN Trial). Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Mony, A.; Chandrashekar, L.; Rajappa, M.; Munisamy, M.; Sahoo, J.P.; Selvarajan, S. Effect of vitamin D supplementation on clinical outcome and biochemical profile in South Indian population with vitamin D-deficient chronic urticaria- a randomized double-blind placebo controlled trial. Clin. Chim. Acta Int. J. Clin. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- LeBoff, M.S.; Chou, S.H.; Murata, E.M.; Donlon, C.M.; Cook, N.R.; Mora, S.; Lee, I.M.; Kotler, G.; Bubes, V.; Buring, J.E.; et al. Effects of Supplemental Vitamin D on Bone Health Outcomes in Women and Men in the VITamin D and OmegA-3 TriaL (VITAL). J. Bone Miner. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Brito, R.B.; Rebello, J.F.; Grabulosa, C.C.; Pinto, W.; Morales, A.; Elias, R.M.; Moyses, R.M.A.; Dalboni, M.A. 25-vitamin D reduces inflammation in uremic environment. Sci. Rep. 2020, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-Q.; Hou, Y.-C.; Zheng, C.-M.; Lu, C.-L.; Liu, W.-C.; Wu, C.-C.; Huang, M.-T.; Lin, Y.-F.; Lu, K.-C. Cholecalciferol Additively Reduces Serum Parathyroid Hormone and Increases Vitamin D and Cathelicidin Levels in Paricalcitol-Treated Secondary Hyperparathyroid Hemodialysis Patients. Nutrients 2016, 8, 708. [Google Scholar] [CrossRef]
- Zheng, C.-M.; Wu, C.-C.; Hung, C.-F.; Liao, M.-T.; Shyu, J.-F.; Hsu, Y.-H.; Lu, C.-L.; Wang, Y.-H.; Zheng, J.-Q.; Chang, T.-J.; et al. Cholecalciferol Additively Reduces Serum Parathyroid Hormone Levels in Severe Secondary Hyperparathyroidism Treated with Calcitriol and Cinacalcet among Hemodialysis Patients. Nutrients 2018, 10, 196. [Google Scholar] [CrossRef]
- Polidoro, L.; Properzi, G.; Marampon, F.; Gravina, G.L.; Festuccia, C.; Di Cesare, E.; Scarsella, L.; Ciccarelli, C.; Zani, B.M.; Ferri, C. Vitamin D Protects Human Endothelial Cells from H2O2 Oxidant Injury Through the Mek/Erk-Sirt1 Axis Activation. J. Cardiovasc. Transl. Res. 2012, 6, 221–231. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Festuccia, C.; Popov, V.M.; Colapietro, E.A.; Sanitá, P.; Musio, D.; De Felice, F.; Lenzi, A.; Jannini, E.A.; et al. Vitamin D protects endothelial cells from irradiation-induced senescence and apoptosis by modulating MAPK/SirT1 axis. J. Endocrinol. Investig. 2015, 39, 411–422. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats. Nutrients 2017, 9, 338. [Google Scholar] [CrossRef]
- Gunasekar, P.; Swier, V.J.; Fleegel, J.P.; Boosani, C.S.; Radwan, M.M.; Agrawal, D.K. Vitamin D and macrophage polarization in epicardial adipose tissue of atherosclerotic swine. PLoS ONE 2018, 13, e0199411. [Google Scholar] [CrossRef]
- Manna, P.; Achari, A.E.; Jain, S.K. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch. Biochem. Biophys. 2017, 615, 22–34. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Ameliorates Fat Accumulation with AMPK/SIRT1 Activity in C2C12 Skeletal Muscle Cells. Nutrients 2019, 11, 2806. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Donato, A.J.; Henson, G.D.; Hearon Jr, C.M.; Hamza, M.; Seals, D.R. Treatment with the SIRT1 activator SRT1720 reduces large elastic artery stiffness, superoxide production and inflammation in old mice. FASEB J. 2011, 25 (1_supplement), lb485-lb485. [Google Scholar]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M.; Sindler, A.L.; Seals, U.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef] [PubMed]
- Lamichane, S.; Baek, S.H.; Kim, Y.-J.; Park, J.H.; Lamichane, B.D.; Jang, W.; Ji, S.; Lee, N.K.; Dehua, L.; Kim, D.Y.; et al. MHY2233 Attenuates Replicative Cellular Senescence in Human Endothelial Progenitor Cells via SIRT1 Signaling. Oxid. Med. Cell. Longev. 2019, 2019, 6492029. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Minor, R.K.; Baur, J.A.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.-K.; Cantó, C.; Scheibye-Knudsen, M.; et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1, 70. [Google Scholar] [CrossRef] [PubMed]
- Baksi, A.; Kraydashenko, O.; Zalevkaya, A.; Stets, R.; Elliott, P.; Haddad, J.; Hoffmann, E.; Vlasuk, G.P.; Jacobson, E.W. A phase II, randomized, placebo-controlled, double-blind, multi-dose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br. J. Clin. Pharmacol. 2014, 78, 69–77. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-L.; Liao, M.-T.; Hou, Y.-C.; Fang, Y.-W.; Zheng, C.-M.; Liu, W.-C.; Chao, C.-T.; Lu, K.-C.; Ng, Y.-Y. Sirtuin-1 and Its Relevance in Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 1593. https://doi.org/10.3390/ijms21051593
Lu C-L, Liao M-T, Hou Y-C, Fang Y-W, Zheng C-M, Liu W-C, Chao C-T, Lu K-C, Ng Y-Y. Sirtuin-1 and Its Relevance in Vascular Calcification. International Journal of Molecular Sciences. 2020; 21(5):1593. https://doi.org/10.3390/ijms21051593
Chicago/Turabian StyleLu, Chien-Lin, Min-Tser Liao, Yi-Chou Hou, Yu-Wei Fang, Cai-Mei Zheng, Wen-Chih Liu, Chia-Ter Chao, Kuo-Cheng Lu, and Yee-Yung Ng. 2020. "Sirtuin-1 and Its Relevance in Vascular Calcification" International Journal of Molecular Sciences 21, no. 5: 1593. https://doi.org/10.3390/ijms21051593
APA StyleLu, C.-L., Liao, M.-T., Hou, Y.-C., Fang, Y.-W., Zheng, C.-M., Liu, W.-C., Chao, C.-T., Lu, K.-C., & Ng, Y.-Y. (2020). Sirtuin-1 and Its Relevance in Vascular Calcification. International Journal of Molecular Sciences, 21(5), 1593. https://doi.org/10.3390/ijms21051593