Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease

 , , and
, , and
Abstract
1. Introduction
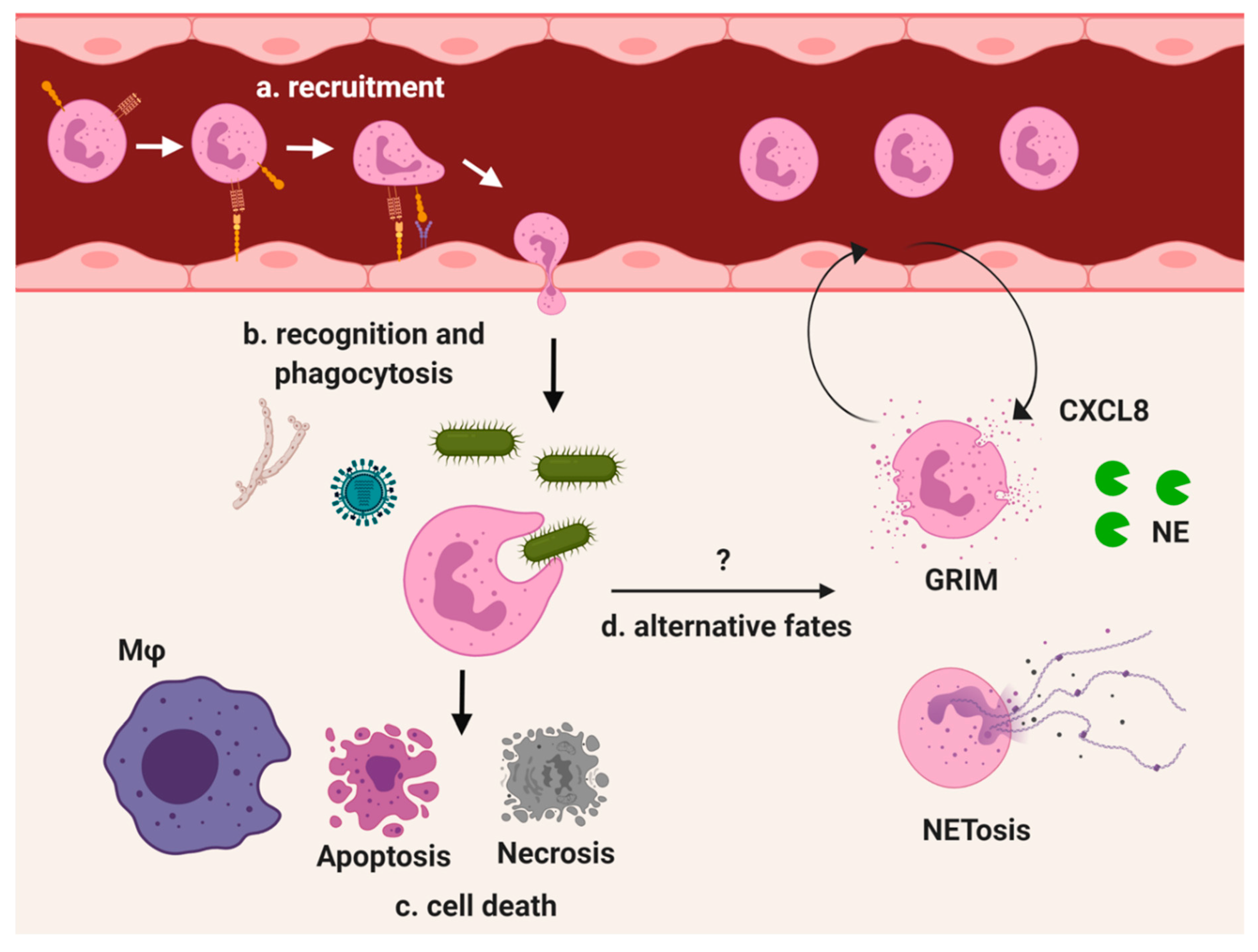
2. Homeostasis
3. Stress Response
3.1. Bacterial Infections
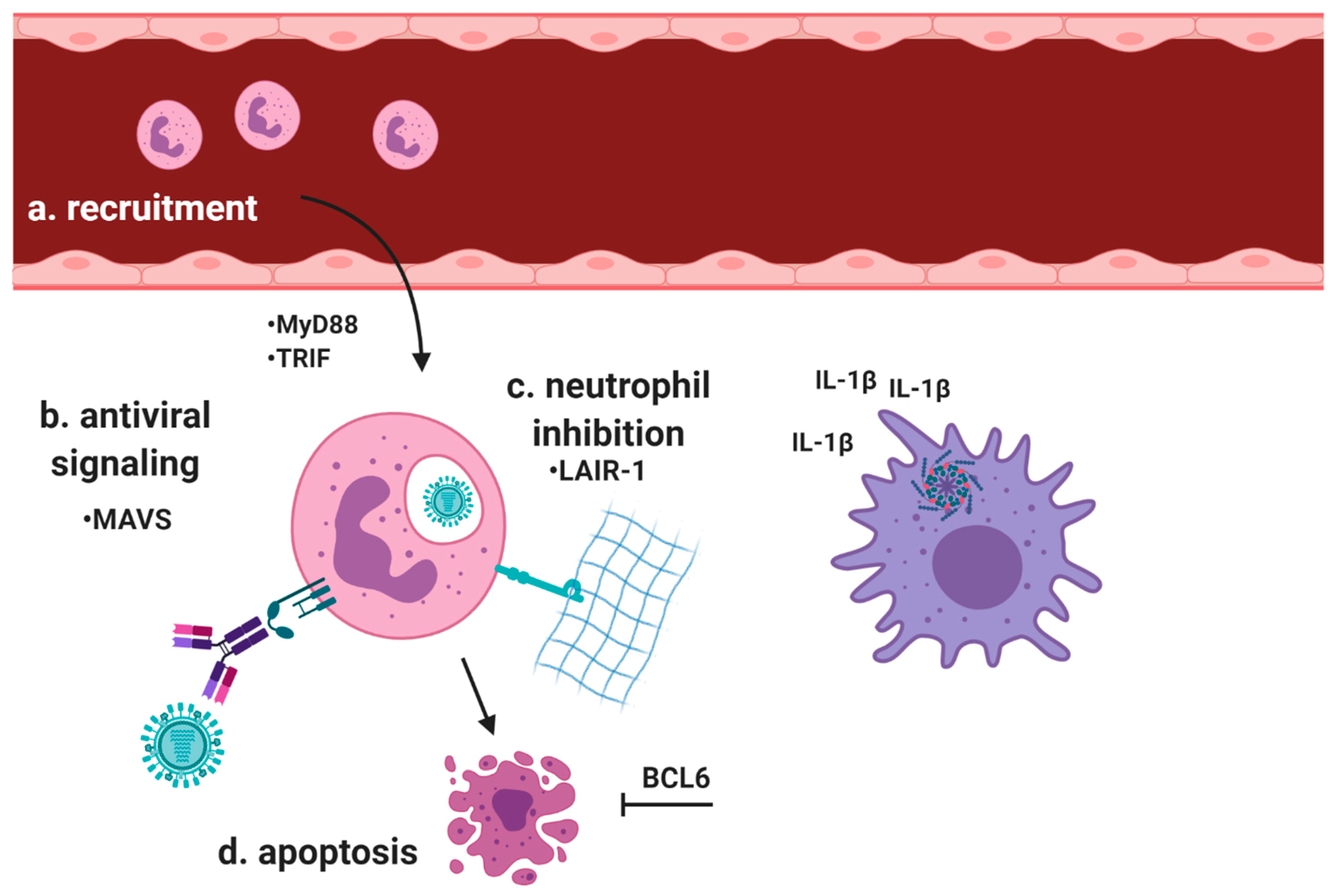
3.2. Viral Infections
3.3. Fungal Infections
4. Neutrophils in Chronic Respiratory Pathologies
4.1. Cystic Fibrosis (CF)
4.2. Asthma
4.3. Chronic Obstructive Pulmonary Disease (COPD)
4.4. Transfusion-Related Acute Lung Injury (TRALI)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARDS | acute respiratory distress syndrome |
| Arg1 | arginase-1 |
| CF | cystic fibrosis |
| COPD | chronic obstructive pulmonary disease |
| GRIM | granule releasing, immunomodulatory, and metabolically active |
| IAV | influenza A virus |
| iNOS | inducible nitric oxide synthase |
| LAIR-1 | leukocyte-associated Ig-like receptor 1 |
| MAVS | mitochondrial antiviral-signaling protein |
| MMP-9 | matrix metalloproteinase-9 |
| MPO | myeloperoxidase |
| mTOR | mechanistic target of rapamycin |
| MyD88 | myeloid differentiation primary response 88 |
| NE | neutrophil elastase |
| NETs | neutrophil extracellular traps; |
| NO | nitric oxide |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| oxCAMKII | oxidized calmodulin-dependent protein kinase II |
| PAD4 | peptidyl arginine deiminase 4 |
| ROS | reactive oxygen species |
| RSV | respiratory syncytial virus |
| TLR | Toll-like receptor |
| TRALI | transfusion-related acute lung injury |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
References
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef]
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Ingersoll, S.A.; Laval, J.; Forrest, O.A.; Preininger, M.; Brown, M.R.; Arafat, D.; Gibson, G.; Tangpricha, V.; Tirouvanziam, R. Mature cystic fibrosis airway neutrophils suppress T cell function: Evidence for a role of arginase 1 but not programmed death-ligand 1. J. Immunol. 2015, 194, 5520–5528. [Google Scholar] [CrossRef] [PubMed]
- Sippel, T.R.; White, J.; Nag, K.; Tsvankin, V.; Klaassen, M.; Kleinschmidt-DeMasters, B.K.; Waziri, A. Neutrophil degranulation and immunosuppression in patients with GBM: Restoration of cellular immune function by targeting arginase I. Clin. Cancer Res. 2011, 17, 6992–7002. [Google Scholar] [CrossRef]
- Domon, H.; Nagai, K.; Maekawa, T.; Oda, M.; Yonezawa, D.; Takeda, W.; Hiyoshi, T.; Tamura, H.; Yamaguchi, M.; Kawabata, S.; et al. Neutrophil elastase subverts the immune response by cleaving Toll-like receptors and cytokines in pneumococcal pneumonia. Front. Immunol. 2018, 9, 732. [Google Scholar] [CrossRef]
- Doring, G.; Frank, F.; Boudier, C.; Herbert, S.; Fleischer, B.; Bellon, G. Cleavage of lymphocyte surface antigens CD2, CD4, and CD8 by polymorphonuclear leukocyte elastase and cathepsin G in patients with cystic fibrosis. J. Immunol. 1995, 154, 4842–4850. [Google Scholar]
- Folds, J.D.; Prince, H.; Spitznagel, J.K. Limited cleavage of human immunoglobulins by elastase of human neutrophil polymorphonuclear granulocytes. Possible modulator of immune complex disease. Lab. Investig. 1978, 39, 313–321. [Google Scholar]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil diversity in health and disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Geng, S.; Zhang, Y.; Lee, C.; Li, L. Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci. Adv. 2019, 5, eaav2309. [Google Scholar] [CrossRef]
- Margaroli, C.; Tirouvanziam, R. Neutrophil plasticity enables the development of pathological microenvironments: Implications for cystic fibrosis airway disease. Mol. Cell Pediatr. 2016, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Ralhan, A.; Hartl, D. Neutrophils in cystic fibrosis. Biol. Chem. 2016, 397, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Doerschuk, C.M.; Wiggs, B.; Minshall, D. Neutrophil retention during a single transit through the pulmonary circulation. J. Appl. Physiol. 1992, 73, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
- Sibille, Y.; Reynolds, H.Y. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am. Rev. Respir. Dis. 1990, 141, 471–501. [Google Scholar] [CrossRef]
- GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir. Med. 2017, 5, 691–706. [Google Scholar] [CrossRef]
- Sapey, E.; Patel, J.M.; Greenwood, H.L.; Walton, G.M.; Hazeldine, J.; Sadhra, C.; Parekh, D.; Dancer, R.C.A.; Nightingale, P.; Lord, J.M.; et al. Pulmonary infections in the elderly lead to impaired neutrophil targeting, which is improved by simvastatin. Am. J. Respir. Crit. Care Med. 2017, 196, 1325–1336. [Google Scholar] [CrossRef]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394. [Google Scholar] [CrossRef]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, R.; Li, C.; Jiang, L.; Xiang, M.; Ye, Z.; Kita, H.; Melnick, A.M.; Dent, A.L.; Sun, J. BCL6 modulates tissue neutrophil survival and exacerbates pulmonary inflammation following influenza virus infection. Proc. Natl. Acad. Sci. USA 2019, 116, 11888–11893. [Google Scholar] [CrossRef]
- Casulli, J.; Fife, M.E.; Houston, S.A.; Rossi, S.; Dow, J.; Williamson, E.D.; Clark, G.C.; Hussell, T.; D’Elia, R.V.; Travis, M.A. CD200R deletion promotes a neutrophil niche for Francisella tularensis and increases infectious burden and mortality. Nat. Commun. 2019, 10, 2121. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, U.; Zemans, R.L.; Smith, C.A.; Wood, S.C.; Deng, J.C.; Goldstein, D.R. Excessive neutrophil levels in the lung underlie the age-associated increase in influenza mortality. Mucosal Immunol. 2019, 12, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Wang, Y.; Chew, W.K.; Lima, R.; A-González, N.; Mattar, C.N.; Chong, S.Z.; Schlitzer, A.; Bakocevic, N.; Chew, S.; et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J. Exp. Med. 2013, 210, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.S.; Thompson, A.; Stott, M.; Benny, A.; Lewis, N.A.; Taylor, P.R.; Forton, J.; Herrick, S.; Orr, S.J.; McGreal, E.P. Differential susceptibility of Dectin-1 isoforms to functional inactivation by neutrophil and fungal proteases. FASEB J. 2018, 32, 3385–3397. [Google Scholar] [CrossRef]
- Adams, L.; Franco, M.C.; Estevez, A.G. Reactive nitrogen species in cellular signaling. Exp. Biol. Med. 2015, 240, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, A.; de Ruiter, T.; Geest, C.; Coffer, P.J.; Meyaard, L. Differential expression of leukocyte-associated Ig-like receptor-1 during neutrophil differentiation and activation. J. Leukoc. Biol. 2006, 79, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, K.; Geerdink, R.J.; Hennus, M.P.; Roda, M.A.; van Ark, I.; Leusink-Muis, T.; Folkerts, G.; van Oort-Jansen, A.; Mazharian, A.; Watson, S.P.; et al. LAIR-1 limits neutrophilic airway inflammation. Front. Immunol. 2019, 10, 842. [Google Scholar] [CrossRef]
- Van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327, 487–492. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Patel, K.D. Matrix metalloproteinase-2 (MMP-2) and MMP-9 in pulmonary pathology. Exp. Lung Res. 2005, 31, 599–621. [Google Scholar] [CrossRef]
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080. [Google Scholar] [CrossRef]
- Andrewes, F.W. The Croonian lectures on the behaviour of the leucocytes in infection and immunity. Lancet 1910, 175, 1737–1743. [Google Scholar] [CrossRef]
- Cartwright, G.E.; Athens, J.W.; Wintrobe, M.M. The kinetics of granulopoiesis in normal man. Blood 1964, 24, 780–803. [Google Scholar] [CrossRef] [PubMed]
- Doerschuk, C.M.; Allard, M.F.; Martin, B.A.; MacKenzie, A.; Autor, A.P.; Hogg, J.C. Marginated pool of neutrophils in rabbit lungs. J. Appl. Physiol. (1985) 1987, 63, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.A.; Wright, J.L.; Thommasen, H.; Hogg, J.C. Effect of pulmonary blood flow on the exchange between the circulating and marginating pool of polymorphonuclear leukocytes in dog lungs. J. Clin. Investig. 1982, 69, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.J.; Salamand, A.; Merhi, Y.; Simard, A.; Dupuis, J. Kinetic analysis of pulmonary neutrophil retention in vivo using the multiple-indicator-dilution technique. J. Appl. Physiol. (1985) 2003, 95, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Lien, D.C.; Wagner, W.W., Jr.; Capen, R.L.; Haslett, C.; Hanson, W.L.; Hofmeister, S.E.; Henson, P.M.; Worthen, G.S. Physiological neutrophil sequestration in the lung: Visual evidence for localization in capillaries. J. Appl. Physiol. (1985) 1987, 62, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, B.R.; English, D.; Quinlan, W.M.; Doyle, N.A.; Hogg, J.C.; Doerschuk, C.M. Contributions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J. Appl. Physiol. (1985) 1994, 77, 463–470. [Google Scholar] [CrossRef]
- Downey, G.P.; Worthen, G.S. Neutrophil retention in model capillaries: Deformability, geometry, and hydrodynamic forces. J. Appl. Physiol. (1985) 1988, 65, 1861–1871. [Google Scholar] [CrossRef]
- Downey, G.P.; Doherty, D.E.; Schwab, B., 3rd; Elson, E.L.; Henson, P.M.; Worthen, G.S. Retention of leukocytes in capillaries: Role of cell size and deformability. J. Appl. Physiol. (1985) 1990, 69, 1767–1778. [Google Scholar] [CrossRef]
- Dimasi, D.; Sun, W.Y.; Bonder, C.S. Neutrophil interactions with the vascular endothelium. Int. Immunopharmacol. 2013, 17, 1167–1175. [Google Scholar] [CrossRef]
- Permutt, S.; Bromberger-Barnea, B.; Bane, H.N. Alveolar pressure, pulmonary venous pressure, and the vascular waterfall. Med. Thorac. 1962, 19, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Permutt, S.; Riley, R.L. Hemodynamics of collapsible vessels with tone: The vascular waterfall. J. Appl. Physiol. 1963, 18, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C.; Checkley, L.L.; Giger, U.; Hanson, W.L.; Kirk, K.R.; Capen, R.L.; Wagner, W.W., Jr. Pulmonary microcirculatory kinetics of neutrophils deficient in leukocyte adhesion-promoting glycoproteins. J. Appl. Physiol. (1985) 1990, 69, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Doyle, N.A.; Bhagwan, S.D.; Meek, B.B.; Kutkoski, G.J.; Steeber, D.A.; Tedder, T.F.; Doerschuk, C.M. Neutrophil margination, sequestration, and emigration in the lungs of L-selectin-deficient mice. J. Clin. Investig. 1997, 99, 526–533. [Google Scholar] [CrossRef]
- Kuebler, W.M.; Kuhnle, G.E.; Groh, J.; Goetz, A.E. Contribution of selectins to leucocyte sequestration in pulmonary microvessels by intravital microscopy in rabbits. J. Physiol. 1997, 50, 375–386. [Google Scholar] [CrossRef]
- Singh, N.R.; Johnson, A.; Peters, A.M.; Babar, J.; Chilvers, E.R.; Summers, C. Acute lung injury results from failure of neutrophil de-priming: A new hypothesis. Eur. J. Clin. Investig. 2012, 42, 1342–1349. [Google Scholar] [CrossRef]
- Ekpenyong, A.E.; Toepfner, N.; Fiddler, C.; Herbig, M.; Li, W.; Cojoc, G.; Summers, C.; Guck, J.; Chilvers, E.R. Mechanical deformation induces depolarization of neutrophils. Sci. Adv. 2017, 3, e1602536. [Google Scholar] [CrossRef]
- Summers, C.; Singh, N.R.; White, J.F.; Mackenzie, I.M.; Johnston, A.; Solanki, C.; Balan, K.K.; Peters, A.M.; Chilvers, E.R. Pulmonary retention of primed neutrophils: A novel protective host response, which is impaired in the acute respiratory distress syndrome. Thorax 2014, 69, 623–629. [Google Scholar] [CrossRef]
- Kim, J.H.; Podstawka, J.; Lou, Y.; Li, L.; Lee, E.K.S.; Divangahi, M.; Petri, B.; Jirik, F.R.; Kelly, M.M.; Yipp, B.G. Aged polymorphonuclear leukocytes cause fibrotic interstitial lung disease in the absence of regulation by B cells. Nat. Immunol. 2018, 19, 192–201. [Google Scholar] [CrossRef]
- Granton, E.; Kim, J.H.; Podstawka, J.; Yipp, B.G. The lung microvasculature is a functional immune niche. Trends Immunol. 2018, 39, 890–899. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Naumenko, V.; Turk, M.; Jenne, C.N.; Kim, S.J. Neutrophils in viral infection. Cell Tissue Res. 2018, 371, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Andreakos, E. Neutrophils in viral infections: Current concepts and caveats. J. Leukoc. Biol. 2015, 98, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Godinez, C.; Carrero, J.C. The state of art of neutrophil extracellular traps in protozoan and helminthic infections. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Lei, B.; Minor, D.; Feng, W.; Jerome, M.; Quinn, M.T.; Jutila, M.A.; Liu, M. Tissue tropism in Streptococcal infection: Wild-type M1T1 group A Streptococcus is efficiently cleared by neutrophils using an NADPH oxidase-dependent mechanism in the lung but not in the skin. Infect. Immun. 2019. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Hoetzenecker, W.; Echtenacher, B.; Guenova, E.; Hoetzenecker, K.; Woelbing, F.; Bruck, J.; Teske, A.; Valtcheva, N.; Fuchs, K.; Kneilling, M.; et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 2011, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Stambas, J.; Selemidis, S. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol. Sci. 2012, 33, 3–8. [Google Scholar] [CrossRef]
- Subramaniam, R.; Barnes, P.F.; Fletcher, K.; Boggaram, V.; Hillberry, Z.; Neuenschwander, P.; Shams, H. Protecting against post-influenza bacterial pneumonia by increasing phagocyte recruitment and ROS production. J. Infect. Dis. 2014, 209, 1827–1836. [Google Scholar] [CrossRef]
- Khan, Z.; Shen, X.Z.; Bernstein, E.A.; Giani, J.F.; Eriguchi, M.; Zhao, T.V.; Gonzalez-Villalobos, R.A.; Fuchs, S.; Liu, G.Y.; Bernstein, K.E. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 2017, 130, 328–339. [Google Scholar] [CrossRef]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.D.; Mulsch, A.; Busse, R.; Osswald, H. Generation of nitric oxide by human neutrophils. Biochem. Biophys. Res. Commun. 1989, 160, 813–819. [Google Scholar] [CrossRef]
- Ichinose, M.; Sugiura, H.; Yamagata, S.; Koarai, A.; Shirato, K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 2000, 162, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Smith, S.D.; Garcia-Cardena, G.; Nathan, C.F.; Weiss, R.M.; Sessa, W.C. Bacterial infection induces nitric oxide synthase in human neutrophils. J. Clin. Investig. 1997, 99, 110–116. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef]
- Chang, C.I.; Liao, J.C.; Kuo, L. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. 1998, 274, H342–H348. [Google Scholar] [CrossRef]
- Rotondo, R.; Barisione, G.; Mastracci, L.; Grossi, F.; Orengo, A.M.; Costa, R.; Truini, M.; Fabbi, M.; Ferrini, S.; Barbieri, O. IL-8 induces exocytosis of arginase 1 by neutrophil polymorphonuclears in nonsmall cell lung cancer. Int. J. Cancer 2009, 125, 887–893. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, E.; Srivastava, V.K.; Kaushik, S.; Saxena, J.; Goyal, L.K.; Mehta, S.; Jyoti, A. Nitrosative stress and cytokines are linked with the severity of sepsis and organ dysfunction. Br. J. Biomed. Sci. 2019, 76, 29–34. [Google Scholar] [CrossRef]
- Shelton, J.L.; Wang, L.; Cepinskas, G.; Sandig, M.; Scott, J.A.; North, M.L.; Inculet, R.; Mehta, S. Inducible NO synthase (iNOS) in human neutrophils but not pulmonary microvascular endothelial cells (PMVEC) mediates septic protein leak in vitro. Microvasc. Res. 2007, 74, 23–31. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, A.L.; Gao, S.; Ma, S.; Guo, S.B. Neutrophil dysfunction in sepsis. Chin. Med. J. (Engl.) 2016, 129, 2741–2744. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef]
- Claushuis, T.A.M.; van der Donk, L.E.H.; Luitse, A.L.; van Veen, H.A.; van der Wel, N.N.; van Vught, L.A.; Roelofs, J.; de Boer, O.J.; Lankelma, J.M.; Boon, L.; et al. Role of peptidylarginine deiminase 4 in neutrophil extracellular trap formation and host defense during Klebsiella pneumoniae-induced pneumonia-derived sepsis. J. Immunol. 2018, 201, 1241–1252. [Google Scholar] [CrossRef]
- Guiducci, E.; Lemberg, C.; Kung, N.; Schraner, E.; Theocharides, A.P.A.; LeibundGut-Landmann, S. Candida albicans-induced NETosis is independent of peptidylarginine deiminase 4. Front. Immunol. 2018, 9, 1573. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Douda, D.N.; Bruggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce Th17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- Desai, J.; Mulay, S.R.; Nakazawa, D.; Anders, H.J. Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell Mol. Life Sci. 2016, 73, 2211–2219. [Google Scholar] [CrossRef]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef]
- Mikacenic, C.; Moore, R.; Dmyterko, V.; West, T.E.; Altemeier, W.A.; Liles, W.C.; Lood, C. Neutrophil extracellular traps (NETs) are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit. Care 2018, 22, 358. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Lal, C.V.; Li, J.D.; Lou, X.Y.; Viera, L.; Abdallah, T.; King, R.W.; Sethi, J.; Kanagarajah, P.; Restrepo-Jaramillo, R.; et al. The neutrophil chemoattractant peptide proline-glycine-proline is associated with acute respiratory distress syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L653–L661. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.D.; Margaroli, C.; Horati, H.; Kilgore, M.B.; Veltman, M.; Liu, H.K.; Taurone, A.J.; Peng, L.; Guglani, L.; Uppal, K.; et al. Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed]
- Dickerhof, N.; Pearson, J.F.; Hoskin, T.S.; Berry, L.J.; Turner, R.; Sly, P.D.; Kettle, A.J.; Arest, C.F. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic. Biol. Med. 2017, 113, 236–243. [Google Scholar] [CrossRef]
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef]
- Pylaeva, E.; Bordbari, S.; Spyra, I.; Decker, A.S.; Haussler, S.; Vybornov, V.; Lang, S.; Jablonska, J. Detrimental effect of type I IFNs during acute lung infection with Pseudomonas aeruginosa is mediated through the stimulation of neutrophil NETosis. Front. Immunol. 2019, 10, 2190. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 2017, 46, 875–890 e6. [Google Scholar] [CrossRef]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef]
- Niu, J.; Wu, S.; Chen, M.; Xu, K.; Guo, Q.; Lu, A.; Zhao, L.; Sun, B.; Meng, G. Hyperactivation of the NLRP3 inflammasome protects mice against influenza A virus infection via IL-1beta mediated neutrophil recruitment. Cytokine 2019, 120, 115–124. [Google Scholar] [CrossRef]
- Keeler, S.P.; Agapov, E.V.; Hinojosa, M.E.; Letvin, A.N.; Wu, K.; Holtzman, M.J. Influenza A virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J. Immunol. 2018, 201, 2354–2368. [Google Scholar] [CrossRef]
- Kirsebom, F.C.M.; Kausar, F.; Nuriev, R.; Makris, S.; Johansson, C. Neutrophil recruitment and activation are differentially dependent on MyD88/TRIF and MAVS signaling during RSV infection. Mucosal Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.D.; Unger, S.A.; Walton, M.; Schwarze, J. The human immune response to respiratory syncytial virus infection. Clin. Microbiol. Rev. 2017, 30, 481–502. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Raftery, M.J. Neutrophil extracellular traps go viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Papayannopoulos, V. Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol. 2013, 35, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef]
- Toussaint, M.; Jackson, D.J.; Swieboda, D.; Guedan, A.; Tsourouktsoglou, T.D.; Ching, Y.M.; Radermecker, C.; Makrinioti, H.; Aniscenko, J.; Bartlett, N.W.; et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 2017, 23, 681–691. [Google Scholar] [CrossRef]
- Moran, G.; Uberti, B.; Ortloff, A.; Folch, H. Aspergillus fumigatus-sensitive IgE is associated with bronchial hypersensitivity in a murine model of neutrophilic airway inflammation. J. Mycol. Med. 2018, 28, 128–136. [Google Scholar] [CrossRef]
- Alflen, A.; Prufer, S.; Ebner, K.; Reuter, S.; Aranda Lopez, P.; Scharrer, I.; Banno, F.; Stassen, M.; Schild, H.; Jurk, K.; et al. ADAMTS-13 regulates neutrophil recruitment in a mouse model of invasive pulmonary aspergillosis. Sci. Rep. 2017, 7, 7184. [Google Scholar] [CrossRef]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; Hoogenboezem, M.; van den Berg, J.M.; Prins, J.M.; Vitkov, L.; van de Veerdonk, F.L.; van den Berg, T.K.; Roos, D.; et al. Human neutrophils use different mechanisms to kill Aspergillus fumigatus conidia and hyphae: Evidence from phagocyte defects. J. Immunol. 2016, 196, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Burdick, M.D.; Mehrad, B. Neutrophils mediate maturation and efflux of lung dendritic cells in response to Aspergillus fumigatus germ tubes. Infect. Immun. 2012, 80, 1759–1765. [Google Scholar] [CrossRef]
- Engel, T.G.P.; Slabbers, L.; de Jong, C.; Melchers, W.J.G.; Hagen, F.; Verweij, P.E.; Merkus, P.; Meis, J.F.; Dutch Cystic Fibrosis Fungal Collection Consortium. Prevalence and diversity of filamentous fungi in the airways of cystic fibrosis patients—A Dutch, multicentre study. J. Cyst. Fibros. 2019, 18, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Gernez, Y.; Conrad, C.K.; Moss, R.B.; Schrijver, I.; Dunn, C.E.; Davies, Z.A.; Herzenberg, L.A.; Herzenberg, L.A. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2008, 105, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Brown, G.D.; Reid, D.M.; Willment, J.A.; Martinez-Pomares, L.; Gordon, S.; Wong, S.Y. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J. Immunol. 2002, 169, 3876–3882. [Google Scholar] [CrossRef]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006, 6, 33–43. [Google Scholar] [CrossRef]
- Meyer, K.C.; Zimmerman, J. Neutrophil mediators, Pseudomonas, and pulmonary dysfunction in cystic fibrosis. J. Lab. Clin. Med. 1993, 121, 654–661. [Google Scholar]
- McKelvey, M.C.; Weldon, S.; McAuley, D.F.; Mall, M.A.; Taggart, C.C. Targeting proteases in cystic fibrosis lung disease: Paradigms, progress, and potential. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef]
- Cowley, A.C.; Thornton, D.J.; Denning, D.W.; Horsley, A. Aspergillosis and the role of mucins in cystic fibrosis. Pediatr. Pulmonol. 2017, 52, 548–555. [Google Scholar] [CrossRef]
- Grunwell, J.R.; Giacalone, V.D.; Stephenson, S.; Margaroli, C.; Dobosh, B.S.; Brown, M.R.; Fitzpatrick, A.M.; Tirouvanziam, R. Neutrophil dysfunction in the airways of children with acute respiratory failure due to lower respiratory tract viral and bacterial coinfections. Sci. Rep. 2019, 9, 2874. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.F.; Hwang, T.L. Neutrophil elastase inhibitors: A patent review and potential applications for inflammatory lung diseases (2010–2014). Expert. Opin Ther. Pat. 2015, 25, 1145–1158. [Google Scholar] [CrossRef]
- Hoenderdos, K.; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Takahashi, H.; Kobayashi, M.; Hanafusa, T.; Herndon, D.N.; Suzuki, F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 2004, 21, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Guglani, L. Changing the paradigm-treating the basic defect in cystic fibrosis. Indian J. Pediatr. 2015, 82, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Forrest, O.A.; Ingersoll, S.A.; Preininger, M.K.; Laval, J.; Limoli, D.H.; Brown, M.R.; Lee, F.E.; Bedi, B.; Sadikot, R.T.; Goldberg, J.B.; et al. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J. Leukoc. Biol. 2018, 104, 665–675. [Google Scholar] [CrossRef]
- Mitchell, T.C. A GRIM fate for human neutrophils in airway disease. J. Leukoc. Biol. 2018, 104, 657–659. [Google Scholar] [CrossRef]
- Margaroli, C.; Garratt, L.W.; Horati, H.; Dittrich, A.S.; Rosenow, T.; Montgomery, S.T.; Frey, D.L.; Brown, M.R.; Schultz, C.; Guglani, L.; et al. Elastase exocytosis by airway neutrophils is associated with early lung damage in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 873–881. [Google Scholar] [CrossRef]
- Gehrig, S.; Duerr, J.; Weitnauer, M.; Wagner, C.J.; Graeber, S.Y.; Schatterny, J.; Hirtz, S.; Belaaouaj, A.; Dalpke, A.H.; Schultz, C.; et al. Lack of neutrophil elastase reduces inflammation, mucus hypersecretion, and emphysema, but not mucus obstruction, in mice with cystic fibrosis-like lung disease. Am. J. Respir. Crit. Care Med. 2014, 189, 1082–1092. [Google Scholar] [CrossRef]
- Guerra, M.; Frey, D.; Hagner, M.; Dittrich, S.; Paulsen, M.; Mall, M.A.; Schultz, C. Cathepsin G activity as a new marker for detecting airway inflammation by microscopy and flow cytometry. ACS Cent. Sci. 2019, 5, 539–548. [Google Scholar] [CrossRef]
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Touhami, J.; Herzenberg, L.A.; Conrad, C.; Taylor, N.; Battini, J.L.; Sitbon, M.; Tirouvanziam, R. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J. Immunol. 2013, 190, 6043–6050. [Google Scholar] [CrossRef] [PubMed]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.L.; Brosius, F.C., 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar] [CrossRef]
- Baker, E.H.; Clark, N.; Brennan, A.L.; Fisher, D.A.; Gyi, K.M.; Hodson, M.E.; Philips, B.J.; Baines, D.L.; Wood, D.M. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J. Appl. Physiol. (1985) 2007, 102, 1969–1975. [Google Scholar] [CrossRef]
- Forrest, O.A.; Chopyk, D.M.; Gernez, Y.; Brown, M.R.; Conrad, C.K.; Moss, R.B.; Tangpricha, V.; Peng, L.; Tirouvanziam, R. Resistin is elevated in cystic fibrosis sputum and correlates negatively with lung function. J. Cyst. Fibros. 2019, 18, 64–70. [Google Scholar] [CrossRef]
- Park, H.K.; Kwak, M.K.; Kim, H.J.; Ahima, R.S. Linking resistin, inflammation, and cardiometabolic diseases. Korean J. Intern. Med. 2017, 32, 239–247. [Google Scholar] [CrossRef]
- Miller, L.; Singbartl, K.; Chroneos, Z.C.; Ruiz-Velasco, V.; Lang, C.H.; Bonavia, A. Resistin directly inhibits bacterial killing in neutrophils. Intensive Care Med. Exp. 2019, 7, 30. [Google Scholar] [CrossRef]
- Aleman, F.; Lim, H.F.; Nair, P. Eosinophilic endotype of asthma. Immunol. Allergy Clin. North. Am. 2016, 36, 559–568. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.W.; Icitovic, N.; Boushey, H.A.; Lazarus, S.C.; Sutherland, E.R.; Chinchilli, V.M.; Fahy, J.V.; Asthma Clinical Research Network of the National Heart, Lung, and Blood Institute. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am. J. Respir. Crit. Care Med. 2012, 185, 612–619. [Google Scholar] [CrossRef]
- Ray, A.; Kolls, J.K. Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. 2017, 38, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Vicencio, A.G.; Du, Z.; Tsirilakis, K.; Salva, P.S.; Webley, W.C. Infectious Chlamydia pneumoniae is associated with elevated interleukin-8 and airway neutrophilia in children with refractory asthma. Pediatr. Infect. Dis. J. 2010, 29, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Webley, W.C. Respiratory Chlamydia infection induce release of hepoxilin a3 and histamine production by airway neutrophils. Front. Immunol. 2018, 9, 2357. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.S.; Van Ly, D.; Spann, K.; Reading, P.C.; Burgess, J.K.; Hartl, D.; Baines, K.J.; Oliver, B.G. Differential neutrophil activation in viral infections: Enhanced TLR-7/8-mediated CXCL8 release in asthma. Respirology 2016, 21, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Merckx, J.; Ducharme, F.M.; Martineau, C.; Zemek, R.; Gravel, J.; Chalut, D.; Poonai, N.; Quach, C.; Pediatric Emergency Research Canada (PERC) DOORWAY Team. Respiratory viruses and treatment failure in children with asthma exacerbation. Pediatrics 2018, 142. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Powell, H.; Boyle, M.J.; Scott, R.J.; Gibson, P.G. Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am. J. Respir. Crit. Care Med. 2008, 177, 148–155. [Google Scholar] [CrossRef]
- Seys, S.F.; Lokwani, R.; Simpson, J.L.; Bullens, D.M.A. New insights in neutrophilic asthma. Curr. Opin. Pulm. Med. 2019, 25, 113–120. [Google Scholar] [CrossRef]
- Dragonieri, S.; Lacedonia, D.; Scioscia, G.; Palladino, G.P.; Quaranta, V.N.; Carratu, P.; Resta, O.; Foschino Barbaro, M.P.; Carpagnano, G.E. Assessment of induced sputum cellularity in COPD patients belonging to two different classes of air pollution exposure. Arch. Bronconeumol. 2019. [Google Scholar] [CrossRef]
- Chang, W.A.; Tsai, M.J.; Jian, S.F.; Sheu, C.C.; Kuo, P.L. Systematic analysis of transcriptomic profiles of COPD airway epithelium using next-generation sequencing and bioinformatics. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 2387–2398. [Google Scholar] [CrossRef]
- Russell, D.W.; Wells, J.M.; Blalock, J.E. Disease phenotyping in chronic obstructive pulmonary disease: The neutrophilic endotype. Curr. Opin. Pulm. Med. 2016, 22, 91–99. [Google Scholar] [CrossRef]
- Thulborn, S.J.; Mistry, V.; Brightling, C.E.; Moffitt, K.L.; Ribeiro, D.; Bafadhel, M. Neutrophil elastase as a biomarker for bacterial infection in COPD. Respir. Res. 2019, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Contoli, M.; Baraldo, S.; Conti, V.; Gnesini, G.; Marku, B.; Casolari, P.; Scrigner, P.; Morelli, P.; Saetta, M.; Spanevello, A.; et al. Airway inflammatory profile is correlated with symptoms in stable COPD: A longitudinal proof-of-concept cohort study. Respirology 2019. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Walton, G.M.; Sapey, E. Neutrophilic inflammation in the pathogenesis of chronic obstructive pulmonary disease. COPD 2018, 15, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Abdul Roda, M.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN exosomes: Pathogenic entities causing matrix destruction and disease in the lung. Cell 2019, 176, 113–126 e115. [Google Scholar] [CrossRef]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Buckley, A.G.; Kicic-Starcevich, E.; Lannigan, F.J.; et al. Alpha-1 antitrypsin mitigates the inhibition of airway epithelial cell repair by neutrophil elastase. Am. J. Respir Cell Mol. Biol. 2016, 54, 341–349. [Google Scholar] [CrossRef]
- DiCamillo, S.J.; Carreras, I.; Panchenko, M.V.; Stone, P.J.; Nugent, M.A.; Foster, J.A.; Panchenko, M.P. Elastase-released epidermal growth factor recruits epidermal growth factor receptor and extracellular signal-regulated kinases to down-regulate tropoelastin mRNA in lung fibroblasts. J. Biol. Chem. 2002, 277, 18938–18946. [Google Scholar] [CrossRef]
- Cosgrove, S.; Chotirmall, S.H.; Greene, C.M.; McElvaney, N.G. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/Toll-like receptor pathway. J. Biol. Chem. 2011, 286, 7692–7704. [Google Scholar] [CrossRef]
- Hwang, J.H.; Lyes, M.; Sladewski, K.; Enany, S.; McEachern, E.; Mathew, D.P.; Das, S.; Moshensky, A.; Bapat, S.; Pride, D.T.; et al. Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria. J. Mol. Med. (Berl.) 2016, 94, 667–679. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, S.; Prasad, G.L.; Li, L. Suppression of neutrophil antimicrobial functions by total particulate matter from cigarette smoke. Front. Immunol. 2018, 9, 2274. [Google Scholar] [CrossRef]
- Higham, A.; Rattray, N.J.; Dewhurst, J.A.; Trivedi, D.K.; Fowler, S.J.; Goodacre, R.; Singh, D. Electronic cigarette exposure triggers neutrophil inflammatory responses. Respir. Res. 2016, 17, 56. [Google Scholar] [CrossRef] [PubMed]
- Reidel, B.; Radicioni, G.; Clapp, P.W.; Ford, A.A.; Abdelwahab, S.; Rebuli, M.E.; Haridass, P.; Alexis, N.E.; Jaspers, I.; Kesimer, M. E-Cigarette use causes a unique innate immune response in the lung, involving increased neutrophilic activation and altered mucin secretion. Am. J. Respir. Crit. Care Med. 2018, 197, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.Y. Nobel prize and the history of blood transfusion. Transfus. Clin. Biol. 2019, 26, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Knopfelmacher, A.M. Transfusion-related acute lung injury (TRALI). In Oncologic Critical Care; Nates, J.L., Price, K.J., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1191–1196. [Google Scholar] [CrossRef]
- Popovsky, M.A.; Abel, M.D.; Moore, S.B. Transfusion-related acute lung injury associated with passive transfer of antileukocyte antibodies. Am. Rev. Respir. Dis. 1983, 128, 185–189. [Google Scholar] [CrossRef]
- Silliman, C.C.; Voelkel, N.F.; Allard, J.D.; Elzi, D.J.; Tuder, R.M.; Johnson, J.L.; Ambruso, D.R. Plasma and lipids from stored packed red blood cells cause acute lung injury in an animal model. J. Clin. Investig. 1998, 101, 1458–1467. [Google Scholar] [CrossRef]
- Toy, P.; Gajic, O.; Bacchetti, P.; Looney, M.R.; Gropper, M.A.; Hubmayr, R.; Lowell, C.A.; Norris, P.J.; Murphy, E.L.; Weiskopf, R.B.; et al. Transfusion-related acute lung injury: Incidence and risk factors. Blood 2012, 119, 1757–1767. [Google Scholar] [CrossRef]
- Popovsky, M.A. Transfusion-related acute lung injury: Three decades of progress but miles to go before we sleep. Transfusion 2015, 55, 930–934. [Google Scholar] [CrossRef]
- Rebetz, J.; Semple, J.W.; Kapur, R. The pathogenic involvement of neutrophils in acute respiratory distress syndrome and transfusion-related acute lung injury. Transfus. Med. Hemother. 2018, 45, 290–298. [Google Scholar] [CrossRef]
- Silliman, C.C.; Curtis, B.R.; Kopko, P.M.; Khan, S.Y.; Kelher, M.R.; Schuller, R.M.; Sannoh, B.; Ambruso, D.R. Donor antibodies to HNA-3a implicated in TRALI reactions prime neutrophils and cause PMN-mediated damage to human pulmonary microvascular endothelial cells in a two-event in vitro model. Blood 2007, 109, 1752–1755. [Google Scholar] [CrossRef]
- Ussov, W.Y.; Peters, A.M.; Savill, J.; Pusey, C.D.; Gaskin, G.; Hodgson, H.J.; Goldman, J.M.; Hughes, J.M. Relationship between granulocyte activation, pulmonary granulocyte kinetics and alveolar permeability in extrapulmonary inflammatory disease. Clin. Sci. (Lond.) 1996, 91, 329–335. [Google Scholar] [CrossRef]
- Kapur, R.; Kim, M.; Aslam, R.; McVey, M.J.; Tabuchi, A.; Luo, A.; Liu, J.; Li, Y.; Shanmugabhavananthan, S.; Speck, E.R.; et al. T regulatory cells and dendritic cells protect against transfusion-related acute lung injury via IL-10. Blood 2017, 129, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- Looney, M.R.; Nguyen, J.X.; Hu, Y.; Van Ziffle, J.A.; Lowell, C.A.; Matthay, M.A. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J. Clin. Investig. 2009, 119, 3450–3461. [Google Scholar] [CrossRef] [PubMed]
- Curtis, B.R.; McFarland, J.G. Mechanisms of transfusion-related acute lung injury (TRALI): Anti-leukocyte antibodies. Crit. Care Med. 2006, 34, S118–S123. [Google Scholar] [CrossRef]
- Sachs, U.J.; Wasel, W.; Bayat, B.; Bohle, R.M.; Hattar, K.; Berghofer, H.; Reil, A.; Bux, J.; Bein, G.; Santoso, S.; et al. Mechanism of transfusion-related acute lung injury induced by HLA class II antibodies. Blood 2011, 117, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Vlaar, A.P.; Hofstra, J.J.; Determann, R.M.; Veelo, D.P.; Paulus, F.; Kulik, W.; Korevaar, J.; de Mol, B.A.; Koopman, M.M.; Porcelijn, L.; et al. The incidence, risk factors, and outcome of transfusion-related acute lung injury in a cohort of cardiac surgery patients: A prospective nested case-control study. Blood 2011, 117, 4218–4225. [Google Scholar] [CrossRef]
- Peters, A.L.; Van Stein, D.; Vlaar, A.P. Antibody-mediated transfusion-related acute lung injury; from discovery to prevention. Br. J. Haematol. 2015, 170, 597–614. [Google Scholar] [CrossRef]
- Peters, A.L.; van Hezel, M.E.; Juffermans, N.P.; Vlaar, A.P. Pathogenesis of non-antibody mediated transfusion-related acute lung injury from bench to bedside. Blood Rev. 2015, 29, 51–61. [Google Scholar] [CrossRef]
- Alexander, P.E.; Barty, R.; Fei, Y.; Vandvik, P.O.; Pai, M.; Siemieniuk, R.A.; Heddle, N.M.; Blumberg, N.; McLeod, S.L.; Liu, J.; et al. Transfusion of fresher vs older red blood cells in hospitalized patients: A systematic review and meta-analysis. Blood 2016, 127, 400–410. [Google Scholar] [CrossRef]
- Peters, A.L.; van Hezel, M.E.; Cortjens, B.; Tuip-de Boer, A.M.; van Bruggen, R.; de Korte, D.; Jonkers, R.E.; Bonta, P.I.; Zeerleder, S.S.; Lutter, R.; et al. Transfusion of 35-day stored RBCs in the presence of endotoxemia does not result in lung injury in humans. Crit. Care Med. 2016, 44, e412–e419. [Google Scholar] [CrossRef]
- Maslanka, K.; Smolenska-Sym, G.; Michur, H.; Wrobel, A.; Lachert, E.; Brojer, E. Lysophosphatidylcholines: Bioactive lipids generated during storage of blood components. Arch. Immunol. Ther. Exp. (Warsz.) 2012, 60, 55–60. [Google Scholar] [CrossRef]
- Tzounakas, V.L.; Kriebardis, A.G.; Papassideri, I.S.; Antonelou, M.H. Donor-variation effect on red blood cell storage lesion: A close relationship emerges. Proteom. Clin. Appl. 2016, 10, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Chasse, M.; McIntyre, L.; English, S.W.; Tinmouth, A.; Knoll, G.; Wolfe, D.; Wilson, K.; Shehata, N.; Forster, A.; van Walraven, C.; et al. Effect of blood donor characteristics on transfusion outcomes: A systematic review and meta-analysis. Transfus. Med. Rev. 2016, 30, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Zimring, J.C.; Smith, N.; Stowell, S.R.; Johnsen, J.M.; Bell, L.N.; Francis, R.O.; Hod, E.A.; Hendrickson, J.E.; Roback, J.D.; Spitalnik, S.L. Strain-specific red blood cell storage, metabolism, and eicosanoid generation in a mouse model. Transfusion 2014, 54, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Jongerius, I.; Porcelijn, L.; van Beek, A.E.; Semple, J.W.; van der Schoot, C.E.; Vlaar, A.P.J.; Kapur, R. The role of complement in transfusion-related acute lung injury. Transfus. Med. Rev. 2019, 33, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Kim, M.; Rebetz, J.; Hallstrom, B.; Bjorkman, J.T.; Takabe-French, A.; Kim, N.; Liu, J.; Shanmugabhavananthan, S.; Milosevic, S.; et al. Gastrointestinal microbiota contributes to the development of murine transfusion-related acute lung injury. Blood Adv. 2018, 2, 1651–1663. [Google Scholar] [CrossRef]
- Kapur, R.; Kasetty, G.; Rebetz, J.; Egesten, A.; Semple, J.W. Osteopontin mediates murine transfusion-related acute lung injury via stimulation of pulmonary neutrophil accumulation. Blood 2019, 134, 74–84. [Google Scholar] [CrossRef]
- Finlayson, J.; Grey, D.; Kavanagh, L.; Witt, C. Transfusion-related acute lung injury in a neutropenic patient. Intern. Med. J. 2011, 41, 638–641. [Google Scholar] [CrossRef]
- Danielson, C.; Benjamin, R.J.; Mangano, M.M.; Mills, C.J.; Waxman, D.A. Pulmonary pathology of rapidly fatal transfusion-related acute lung injury reveals minimal evidence of diffuse alveolar damage or alveolar granulocyte infiltration. Transfusion 2008, 48, 2401–2408. [Google Scholar] [CrossRef]
- Bayat, B.; Tjahjono, Y.; Sydykov, A.; Werth, S.; Hippenstiel, S.; Weissmann, N.; Sachs, U.J.; Santoso, S. Anti-human neutrophil antigen-3a induced transfusion-related acute lung injury in mice by direct disturbance of lung endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2538–2548. [Google Scholar] [CrossRef]
- Sly, P.D.; Brennan, S.; Gangell, C.; de Klerk, N.; Murray, C.; Mott, L.; Stick, S.M.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 2009, 180, 146–152. [Google Scholar] [CrossRef]
- Balazs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S5–S12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Protein | Role | Function |
|---|---|---|
| Arg1 | pro/anti-inflammatory | suppresses T-cell proliferation [4] |
| BCL6 | pro/anti-inflammatory | suppresses neutrophil apoptosis [20] |
| CD200R | anti-inflammatory | attenuates oxidant production by neutrophils [21] |
| CXCR2 | pro-inflammatory | promotes chemotaxis as receptor for CXCL1 [22] |
| CXCR4 | homeostatic | promotes retention in bone marrow/lung as receptor to CXCL12, [23] |
| Dectin-1 | pro-inflammatory | promotes phagocytosis of fungi [24] |
| iNOS | pro-inflammatory | supports the generation of nitric oxide [25] |
| LAIR-1 | anti-inflammatory | suppresses neutrophil recruitment [26,27] |
| MPO | pro-inflammatory | supports generation of hypochlorous acid [28] |
| MMP-9 | pro-inflammatory | degrades the extracellular matrix [29] |
| NOX | pro-inflammatory | supports the generation of superoxide [1] |
| NE | pro-inflammatory | degrades phagocytosed microbes [1] and extracellular matrix [30] |
| oxCAMKII | pro-inflammatory | activates STAT1 and generation of inflammatory mediators [10] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacalone, V.D.; Margaroli, C.; Mall, M.A.; Tirouvanziam, R. Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease. Int. J. Mol. Sci. 2020, 21, 851. https://doi.org/10.3390/ijms21030851
Giacalone VD, Margaroli C, Mall MA, Tirouvanziam R. Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease. International Journal of Molecular Sciences. 2020; 21(3):851. https://doi.org/10.3390/ijms21030851
Chicago/Turabian StyleGiacalone, Vincent D., Camilla Margaroli, Marcus A. Mall, and Rabindra Tirouvanziam. 2020. "Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease" International Journal of Molecular Sciences 21, no. 3: 851. https://doi.org/10.3390/ijms21030851
APA StyleGiacalone, V. D., Margaroli, C., Mall, M. A., & Tirouvanziam, R. (2020). Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease. International Journal of Molecular Sciences, 21(3), 851. https://doi.org/10.3390/ijms21030851