Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases

 ,
,  ,
,
Abstract
1. Introduction
2. TNF-α Signaling in Neuroinflammation
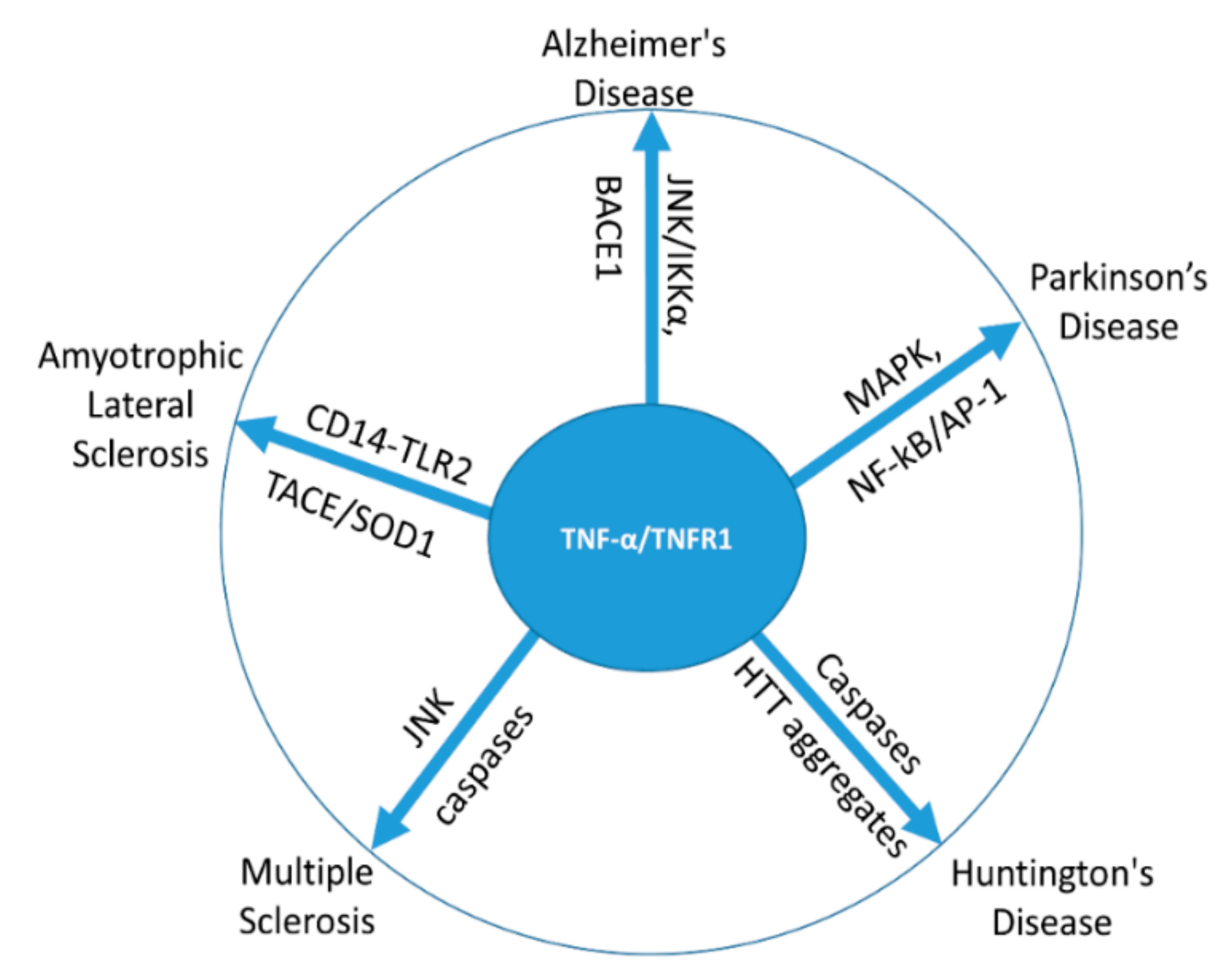
3. TNF-α in Neurodegenerative Disorders
4. Commercially Available TNF-α Inhibitors and Their Side Effects
5. Phytochemicals Inhibiting TNF-α for Lowering the Neuroinflammatory and Neurodegenerative Disorders
6. Plant Extract or Phytochemical for Neurological Complications
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Frankola, K.A.; Greig, N.H.; Luo, W.; Tweedie, D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2011, 10, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Gaire, B.P.; Park, S.J.; Shin, D.Y.; Choi, J.W. Identifying lysophosphatidic acid receptor subtype 1 (LPA1) as a novel factor to modulate microglial activation and their TNF-alpha production by activating ERK1/2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Raghav, S.K.; Gupta, B.; Agrawal, C.; Chaturvedi, V.P.; Das, H.R. Expression of TNF-alpha and related signaling molecules in the peripheral blood mononuclear cells of rheumatoid arthritis patients. Mediators Inflamm. 2006, 2006, 12682. [Google Scholar] [CrossRef]
- Chen, Z.; Palmer, T.D. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav. Immun. 2013, 30, 45–53. [Google Scholar] [CrossRef]
- Guo, G.; Morrissey, J.; McCracken, R.; Tolley, T.; Klahr, S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am. J. Physiol. 1999, 277, F766–F772. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef]
- Maddahi, A.; Kruse, L.S.; Chen, Q.W.; Edvinsson, L. The role of tumor necrosis factor-alpha and TNF-alpha receptors in cerebral arteries following cerebral ischemia in rat. J. Neuroinflammation 2011, 8, 107. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Gurzeler, U.; Federzoni, E.; Kaufmann, T.; Simon, H.U. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood 2011, 117, 5953–5962. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, Y.; Ekman, N.; Wu, Z.; Wu, J.; Alitalo, K.; Min, W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J. Biol. Chem. 2003, 278, 51267–51276. [Google Scholar] [CrossRef] [PubMed]
- Basuroy, S.; Tcheranova, D.; Bhattacharya, S.; Leffler, C.W.; Parfenova, H. Nox4 NADPH oxidase-derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-alpha-induced apoptosis. Am. J. Physiol. Cell Physiol. 2011, 300, C256–265. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Bowers, W.J. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. Neuroimmune. Pharmacol. 2012, 7, 42–59. [Google Scholar] [CrossRef]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef]
- Cheng, X.; Shen, Y.; Li, R. Targeting TNF: A therapeutic strategy for Alzheimer’s disease. Drug Discov. Today 2014, 19, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-kappaB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Allchorne, A.; Safieh-Garabedian, B.; Poole, S. Cytokines, nerve growth factor and inflammatory hyperalgesia: The contribution of tumour necrosis factor alpha. Br. J. Pharmacol. 1997, 121, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [Google Scholar] [CrossRef]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef]
- Tsuge, M.; Yasui, K.; Ichiyawa, T.; Saito, Y.; Nagaoka, Y.; Yashiro, M.; Yamashita, N.; Morishima, T. Increase of tumor necrosis factor-alpha in the blood induces early activation of matrix metalloproteinase-9 in the brain. Microbiol. Immunol. 2010, 54, 417–424. [Google Scholar] [CrossRef]
- Yang, X.; Nath, A.; Opperman, M.J.; Chan, C. The double-stranded RNA-dependent protein kinase differentially regulates insulin receptor substrates 1 and 2 in HepG2 cells. Mol. Biol. Cell 2010, 21, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Park, Y.; Zhang, H.; Xu, X.; Laine, G.A.; Dellsperger, K.C.; Zhang, C. Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, P.; Chang, Q.; Wang, J.; Liu, J.; Lv, Y.; Wang, T.T.Y.; Gao, B.; Zhang, Y.; Yu, L.L. Inhibitory Effect of Piceatannol on TNF-alpha-Mediated Inflammation and Insulin Resistance in 3T3-L1 Adipocytes. J. Agric. Food Chem. 2017, 65, 4634–4641. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, Y.; Liu, Y.; Akiyama, K.; Chen, C.; Qu, C.; Jin, Y.; Shi, S. IFN-gamma and TNF-alpha synergistically induce mesenchymal stem cell impairment and tumorigenesis via NFkappaB signaling. Stem Cells 2013, 31, 1383–1395. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef]
- Hsiao, H.Y.; Chiu, F.L.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Chen, Y.C.; Kuo, H.C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef]
- De Felice, F.G.; Lourenco, M.V. Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 94. [Google Scholar] [CrossRef]
- Chang, R.; Yee, K.L.; Sumbria, R.K. Tumor necrosis factor alpha Inhibition for Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517709278. [Google Scholar] [CrossRef]
- Blaylock, R.L. Parkinson’s disease: Microglial/macrophage-induced immunoexcitotoxicity as a central mechanism of neurodegeneration. Surg. Neurol. Int. 2017, 8, 65. [Google Scholar] [CrossRef]
- Spuler, S.; Yousry, T.; Scheller, A.; Voltz, R.; Holler, E.; Hartmann, M.; Wick, M.; Hohlfeld, R. Multiple sclerosis: Prospective analysis of TNF-alpha and 55 kDa TNF receptor in CSF and serum in correlation with clinical and MRI activity. J. Neuroimmunol. 1996, 66, 57–64. [Google Scholar]
- Tortarolo, M.; Lo Coco, D.; Veglianese, P.; Vallarola, A.; Giordana, M.T.; Marcon, G.; Beghi, E.; Poloni, M.; Strong, M.J.; Iyer, A.M.; et al. Amyotrophic Lateral Sclerosis, a Multisystem Pathology: Insights into the Role of TNFalpha. Mediators Inflamm. 2017, 2017, 2985051. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef]
- Ugolini, G.; Raoul, C.; Ferri, A.; Haenggeli, C.; Yamamoto, Y.; Salaun, D.; Henderson, C.E.; Kato, A.C.; Pettmann, B.; Hueber, A.O. Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J. Neurosci. 2003, 23, 8526–8531. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, J.H.; Gwag, B.J.; Choi, E.J. Iron accumulation promotes TACE-mediated TNF-alpha secretion and neurodegeneration in a mouse model of ALS. Neurobiol. Dis. 2015, 80, 63–69. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Armuzzi, A.; Lionetti, P.; Blandizzi, C.; Caporali, R.; Chimenti, S.; Cimino, L.; Gionchetti, P.; Girolomoni, G.; Lapadula, G.; Marchesoni, A.; et al. anti-TNF agents as therapeutic choice in immune-mediated inflammatory diseases: Focus on adalimumab. Int J. Immunopathol. Pharmacol. 2014, 27, 11–32. [Google Scholar] [CrossRef]
- Lindhaus, C.; Tittelbach, J.; Elsner, P. Cutaneous side effects of TNF-alpha inhibitors. J. Dtsch. Dermatol. Ges. 2017, 15, 281–288. [Google Scholar] [CrossRef]
- Parakkal, D.; Sifuentes, H.; Semer, R.; Ehrenpreis, E.D. Hepatosplenic T-cell lymphoma in patients receiving TNF-alpha inhibitor therapy: Expanding the groups at risk. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1150–1156. [Google Scholar] [CrossRef]
- Thabet, M.M.; Huizinga, T.W. Drug evaluation: Apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr. Opin. Investig. Drugs 2006, 7, 1014–1019. [Google Scholar] [PubMed]
- Dolapcioglu, C.; Dolapcioglu, H. Structural brain lesions in inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2015, 6, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Kumar, A. Meningococcal meningoencephalitis after certolizumab pegol treatment in a patient with Crohn’s disease. J. Crohns Colitis 2013, 7, e19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lis, K.; Kuzawinska, O.; Balkowiec-Iskra, E. Tumor necrosis factor inhibitors - state of knowledge. Arch. Med. Sci. 2014, 10, 1175–1185. [Google Scholar] [CrossRef]
- Andus, T.; Stange, E.F.; Hoffler, D.; Keller-Stanislawski, B. [Suspected cases of severe side effects after infliximab (Remicade) in Germany]. Med. Klin. (Munich) 2003, 98, 429–436. [Google Scholar] [CrossRef]
- Fanning, W.L.; Gump, D.W.; Sofferman, R.A. Side effects of minocycline: A double-blind study. Antimicrob. Agents Chemother. 1977, 11, 712–717. [Google Scholar] [CrossRef][Green Version]
- Svetlana Balkanov, K.; Sotirova, T.; Genadieva, S.S.; Cevreska, L.; Stojanovik, A.; Balkanov, T. Adverse effects of thalidomide administration, in patients with myeloma multiplex? Mater. Sociomed. 2014, 26, 134–136. [Google Scholar] [CrossRef]
- Stewart, F.A.; Gavino, A.C.; Elewski, B.E. New side effect of TNF-alpha inhibitors: Morphea. Skinmed 2013, 11, 59–60. [Google Scholar]
- Genovese, M.C.; Cohen, S.; Moreland, L.; Lium, D.; Robbins, S.; Newmark, R.; Bekker, P.; Study, G. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004, 50, 1412–1419. [Google Scholar] [CrossRef]
- Gupta, R.; Levin, E.; Wu, J.J.; Koo, J.; Liao, W. An update on drug-drug interactions with biologics for the treatment of moderate-to-severe psoriasis. J. Dermatolog. Treat. 2014, 25, 87–89. [Google Scholar] [CrossRef]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-alpha Blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez de la Camara, C.; Hernandez-Pinto, A.M.; Olivares-Gonzalez, L.; Cuevas-Martin, C.; Sanchez-Arago, M.; Hervas, D.; Salom, D.; Cuezva, J.M.; de la Rosa, E.J.; Millan, J.M.; et al. Adalimumab Reduces Photoreceptor Cell Death in A Mouse Model of Retinal Degeneration. Sci. Rep. 2015, 5, 11764. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, N. Adalimumab: A review of side effects. Expert Opin. Drug Saf. 2005, 4, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Sklair-Tavron, L.; Nudelman, R. Drug insight: Tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2008, 4, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Utari, A.; Chonchaiya, W.; Rivera, S.M.; Schneider, A.; Hagerman, R.J.; Faradz, S.M.; Ethell, I.M.; Nguyen, D.V. Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am. J. Intellect Dev. Disabil. 2010, 115, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. C Embryo Today 2015, 105, 140–156. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Rajkumar, S.V. Management of thalidomide toxicity. J. Support. Oncol. 2003, 1, 194–205. [Google Scholar]
- Ignatowski, T.A.; Spengler, R.N.; Dhandapani, K.M.; Folkersma, H.; Butterworth, R.F.; Tobinick, E. Perispinal etanercept for post-stroke neurological and cognitive dysfunction: Scientific rationale and current evidence. CNS Drugs 2014, 28, 679–697. [Google Scholar] [CrossRef]
- Tobinick, E. Rapid improvement of chronic stroke deficits after perispinal etanercept: Three consecutive cases. CNS Drugs 2011, 25, 145–155. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Sung, B. Curcumin: An orally bioavailable blocker of TNF and other pro-inflammatory biomarkers. Br. J. Pharmacol. 2013, 169, 1672–1692. [Google Scholar] [CrossRef]
- Isa, Y.; Miyakawa, Y.; Yanagisawa, M.; Goto, T.; Kang, M.S.; Kawada, T.; Morimitsu, Y.; Kubota, K.; Tsuda, T. 6-Shogaol and 6-gingerol, the pungent of ginger, inhibit TNF-alpha mediated downregulation of adiponectin expression via different mechanisms in 3T3-L1 adipocytes. Biochem. Biophys Res. Commun. 2008, 373, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Gou, Z.; Jiang, S.; Zheng, C.; Tian, Z.; Lin, X. Equol Inhibits LPS-Induced Oxidative Stress and Enhances the Immune Response in Chicken HD11 Macrophages. Cell Physiol. Biochem. 2015, 36, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Kwon, O.W.; Park, S.H.; Chun, K.H.; Kim, S.Y.; Shin, D.Y.; Choi, J.W. Neuroprotective effect of 6-paradol in focal cerebral ischemia involves the attenuation of neuroinflammatory responses in activated microglia. PLoS ONE 2015, 10, e0120203. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Weng, C.J.; Jhang, J.J.; Cheng, Y.T.; Huang, S.M.; Yen, G.C. Diallyl sulfide as a potential dietary agent to reduce TNF-alpha- and histamine-induced proinflammatory responses in A7r5 cells. Mol. Nutr. Food Res. 2014, 58, 1069–1078. [Google Scholar] [CrossRef]
- Chan, M.M. Inhibition of tumor necrosis factor by curcumin, a phytochemical. Biochem. Pharmacol. 1995, 49, 1551–1556. [Google Scholar] [CrossRef]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: The golden pigment from golden spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef]
- Subedi, L.; Venkatesan, R.; Kim, S.Y. Neuroprotective and Anti-Inflammatory Activities of Allyl Isothiocyanate through Attenuation of JNK/NF-kappaB/TNF-alpha Signaling. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Park, G.; Kim, H.G.; Ju, M.S.; Ha, S.K.; Park, Y.; Kim, S.Y.; Oh, M.S. 6-Shogaol, an active compound of ginger, protects dopaminergic neurons in Parkinson’s disease models via anti-neuroinflammation. Acta Pharmacol. Sin. 2013, 34, 1131–1139. [Google Scholar] [CrossRef]
- Han, Q.; Yuan, Q.; Meng, X.; Huo, J.; Bao, Y.; Xie, G. 6-Shogaol attenuates LPS-induced inflammation in BV2 microglia cells by activating PPAR-gamma. Oncotarget 2017, 8, 42001–42006. [Google Scholar] [CrossRef]
- Kim, D.H.; Jung, W.S.; Kim, M.E.; Lee, H.W.; Youn, H.Y.; Seon, J.K.; Lee, H.N.; Lee, J.S. Genistein inhibits proinflammatory cytokines in human mast cell activation through the inhibition of the ERK pathway. Int. J. Mol. Med. 2014, 34, 1669–1674. [Google Scholar] [CrossRef]
- Messina, S.; Bitto, A.; Aguennouz, M.; Vita, G.L.; Polito, F.; Irrera, N.; Altavilla, D.; Marini, H.; Migliorato, A.; Squadrito, F.; et al. The soy isoflavone genistein blunts nuclear factor kappa-B, MAPKs and TNF-alpha activation and ameliorates muscle function and morphology in mdx mice. Neuromuscul. Disord. 2011, 21, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Lai, C.S.; Ho, C.T. Anti-inflammatory activity of natural dietary flavonoids. Food Funct. 2010, 1, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Lopez, N.; Gutierrez-Grijalva, E.P.; Ambriz-Perez, D.L.; Heredia, J.B. Flavonoids as Cytokine Modulators: A Possible Therapy for Inflammation-Related Diseases. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Su, J.; Guo, B.; Wang, K.; Li, X.; Liang, G. Apigenin protects blood-brain barrier and ameliorates early brain injury by inhibiting TLR4-mediated inflammatory pathway in subarachnoid hemorrhage rats. Int. Immunopharmacol. 2015, 28, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Vafeiadou, K.; Vauzour, D.; Lee, H.Y.; Rodriguez-Mateos, A.; Williams, R.J.; Spencer, J.P. The citrus flavanone naringenin inhibits inflammatory signalling in glial cells and protects against neuroinflammatory injury. Arch. Biochem. Biophys. 2009, 484, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef]
- Lee, D.S.; Jeong, G.S. Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br. J. Pharmacol. 2016, 173, 2894–2909. [Google Scholar] [CrossRef]
- Lee, S.H.; Seo, G.S.; Jin, X.Y.; Ko, G.; Sohn, D.H. Butein blocks tumor necrosis factor alpha-induced interleukin 8 and matrix metalloproteinase 7 production by inhibiting p38 kinase and osteopontin mediated signaling events in HT-29 cells. Life Sci. 2007, 81, 1535–1543. [Google Scholar] [CrossRef]
- Yoshida, H.; Takamura, N.; Shuto, T.; Ogata, K.; Tokunaga, J.; Kawai, K.; Kai, H. The citrus flavonoids hesperetin and naringenin block the lipolytic actions of TNF-alpha in mouse adipocytes. Biochem. Biophys. Res. Commun. 2010, 394, 728–732. [Google Scholar] [CrossRef]
- Xagorari, A.; Papapetropoulos, A.; Mauromatis, A.; Economou, M.; Fotsis, T.; Roussos, C. Luteolin inhibits an endotoxin-stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J. Pharmacol. Exp. Ther. 2001, 296, 181–187. [Google Scholar] [PubMed]
- Lee, H.; Kim, Y.O.; Kim, H.; Kim, S.Y.; Noh, H.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Suk, K. Flavonoid wogonin from medicinal herb is neuroprotective by inhibiting inflammatory activation of microglia. FASEB J. 2003, 17, 1943–1944. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Aleisa, A.M.; Al-Rejaie, S.S.; Abuohashish, H.M.; Parmar, M.Y.; Alhomida, A.S.; Ahmed, M.M. Flavonoid, morin inhibits oxidative stress, inflammation and enhances neurotrophic support in the brain of streptozotocin-induced diabetic rats. Neurol. Sci. 2014, 35, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Jung, K.H.; Hong, S.W.; Park, I.S.; Lee, C.; Han, H.K.; Lee, D.H.; Hong, S.S. Morin protects acute liver damage by carbon tetrachloride (CCl(4)) in rat. Arch. Pharm. Res. 2008, 31, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.K.; Moon, E.; Kim, S.Y. Chrysin suppresses LPS-stimulated proinflammatory responses by blocking NF-kappaB and JNK activations in microglia cells. Neurosci. Lett. 2010, 485, 143–147. [Google Scholar] [CrossRef]
- Cho, J.Y.; Yoo, E.S.; Baik, K.U.; Park, M.H. Eudesmin inhibits tumor necrosis factor-alpha production and T cell proliferation. Arch. Pharm. Res. 1999, 22, 348–353. [Google Scholar] [CrossRef]
- Lee, C.J.; Lee, M.H.; Yoo, S.M.; Choi, K.I.; Song, J.H.; Jang, J.H.; Oh, S.R.; Ryu, H.W.; Lee, H.S.; Surh, Y.J.; et al. Magnolin inhibits cell migration and invasion by targeting the ERKs/RSK2 signaling pathway. BMC Cancer 2015, 15, 576. [Google Scholar] [CrossRef]
- Chae, S.H.; Kim, P.S.; Cho, J.Y.; Park, J.S.; Lee, J.H.; Yoo, E.S.; Baik, K.U.; Lee, J.S.; Park, M.H. Isolation and identification of inhibitory compounds on TNF-alpha production from Magnolia fargesii. Arch. Pharm. Res. 1998, 21, 67–69. [Google Scholar] [CrossRef]
- Ramachandran, C.; Wilk, B.; Melnick, S.J.; Eliaz, I. Synergistic Antioxidant and Anti-Inflammatory Effects between Modified Citrus Pectin and Honokiol. Evid. Based Complement. Alternat. Med. 2017, 2017, 8379843. [Google Scholar] [CrossRef]
- Kaushik, D.K.; Mukhopadhyay, R.; Kumawat, K.L.; Gupta, M.; Basu, A. Therapeutic targeting of Kruppel-like factor 4 abrogates microglial activation. J. Neuroinflammation 2012, 9, 57. [Google Scholar] [CrossRef]
- Zhu, J.; Mu, X.; Zeng, J.; Xu, C.; Liu, J.; Zhang, M.; Li, C.; Chen, J.; Li, T.; Wang, Y. Ginsenoside Rg1 prevents cognitive impairment and hippocampus senescence in a rat model of D-galactose-induced aging. PLoS ONE 2014, 9, e101291. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, S.H. Therapeutic Effects of Korean Red Ginseng Extract in a Murine Model of Atopic Dermatitis: Anti-pruritic and Anti-inflammatory Mechanism. J. Korean Med. Sci. 2017, 32, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Piao, W.H.; Campagnolo, D.; Dayao, C.; Lukas, R.J.; Wu, J.; Shi, F.D. Nicotine and inflammatory neurological disorders. Acta Pharmacol. Sin. 2009, 30, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zuo, X.; Li, Y.; Wang, Y.; Zhao, H.; Xiao, X. Nicotine inhibits tumor necrosis factor-alpha induced IL-6 and IL-8 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Rheumatol. Int. 2012, 32, 97–104. [Google Scholar] [CrossRef]
- Li, T.; Wu, S.; Zhang, H.; Wang, Y.; Luo, H.; Zuo, X.; Xiao, X. Activation of Nicotinic Receptors Inhibits TNF-alpha-Induced Production of Pro-inflammatory Mediators Through the JAK2/STAT3 Signaling Pathway in Fibroblast-Like Synoviocytes. Inflammation 2015, 38, 1424–1433. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, X.D.; Kolosov, V.P.; Perelman, J.M. Nicotine reduces TNF-alpha expression through a alpha7 nAChR/MyD88/NF-kB pathway in HBE16 airway epithelial cells. Cell Physiol. Biochem. 2011, 27, 605–612. [Google Scholar] [CrossRef]
- Chen, C.C.; Hung, T.H.; Lee, C.Y.; Wang, L.F.; Wu, C.H.; Ke, C.H.; Chen, S.F. Berberine protects against neuronal damage via suppression of glia-mediated inflammation in traumatic brain injury. PLoS ONE 2014, 9, e115694. [Google Scholar] [CrossRef]
- Zhu, F.; Wu, F.; Ma, Y.; Liu, G.; Li, Z.; Sun, Y.; Pei, Z. Decrease in the production of beta-amyloid by berberine inhibition of the expression of beta-secretase in HEK293 cells. BMC Neurosci. 2011, 12, 125. [Google Scholar] [CrossRef]
- Chen, F.L.; Yang, Z.H.; Liu, Y.; Li, L.X.; Liang, W.C.; Wang, X.C.; Zhou, W.B.; Yang, Y.H.; Hu, R.M. Berberine inhibits the expression of TNFalpha, MCP-1, and IL-6 in AcLDL-stimulated macrophages through PPARgamma pathway. Endocrine 2008, 33, 331–337. [Google Scholar] [CrossRef]
- Chung, Y.C.; Baek, J.Y.; Kim, S.R.; Ko, H.W.; Bok, E.; Shin, W.H.; Won, S.Y.; Jin, B.K. Capsaicin prevents degeneration of dopamine neurons by inhibiting glial activation and oxidative stress in the MPTP model of Parkinson’s disease. Exp. Mol. Med. 2017, 49, e298. [Google Scholar] [CrossRef]
- Park, J.Y.; Kawada, T.; Han, I.S.; Kim, B.S.; Goto, T.; Takahashi, N.; Fushiki, T.; Kurata, T.; Yu, R. Capsaicin inhibits the production of tumor necrosis factor alpha by LPS-stimulated murine macrophages, RAW 264.7: A PPARgamma ligand-like action as a novel mechanism. FEBS Lett. 2004, 572, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Natarajan, K.; Aggarwal, B.B. Capsaicin (8-methyl-N-vanillyl-6-nonenamide) is a potent inhibitor of nuclear transcription factor-kappa B activation by diverse agents. J. Immunol. 1996, 157, 4412–4420. [Google Scholar] [PubMed]
- Tzeng, Y.M.; Lee, M.J. Neuroprotective properties of kavalactones. Neural Regen. Res. 2015, 10, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, M.P.; Whitty, A.; Merrill, J.C.; Tang, X.; Ashton, T.D.; Amar, S. Identification and characterization of kava-derived compounds mediating TNF-alpha suppression. Chem. Biol. Drug Des. 2009, 74, 121–128. [Google Scholar] [CrossRef]
- Folmer, F.; Blasius, R.; Morceau, F.; Tabudravu, J.; Dicato, M.; Jaspars, M.; Diederich, M. Inhibition of TNFalpha-induced activation of nuclear factor kappaB by kava (Piper methysticum) derivatives. Biochem. Pharmacol. 2006, 71, 1206–1218. [Google Scholar] [CrossRef]
- Bi, X.L.; Yang, J.Y.; Dong, Y.X.; Wang, J.M.; Cui, Y.H.; Ikeshima, T.; Zhao, Y.Q.; Wu, C.F. Resveratrol inhibits nitric oxide and TNF-alpha production by lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2005, 5, 185–193. [Google Scholar] [CrossRef]
- de Sa Coutinho, D.; Pacheco, M.T.; Frozza, R.L.; Bernardi, A. Anti-Inflammatory Effects of Resveratrol: Mechanistic Insights. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Subedi, L.; Baek, S.H.; Kim, S.Y. Genetically Engineered Resveratrol-Enriched Rice Inhibits Neuroinflammation in Lipopolysaccharide-Activated BV2 Microglia Via Downregulating Mitogen-Activated Protein Kinase-Nuclear Factor Kappa B Signaling Pathway. Oxid. Med. Cell Longev. 2018, 2018, 8092713. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, T.H.; Wahedi, H.M.; Baek, S.H.; Kim, S.Y. Resveratrol-Enriched Rice Attenuates UVB-ROS-Induced Skin Aging via Downregulation of Inflammatory Cascades. Oxid. Med. Cell Longev. 2017, 2017, 8379539. [Google Scholar] [CrossRef]
- Subedi, L.; Gaire, B.P.; Do, M.H.; Lee, T.H.; Kim, S.Y. Anti-neuroinflammatory and neuroprotective effects of the Lindera neesiana fruit in vitro. Phytomedicine 2016, 23, 872–881. [Google Scholar] [CrossRef]
- Ju, H.K.; Baek, S.H.; An, R.B.; Bae, K.; Son, K.H.; Kim, H.P.; Kang, S.S.; Lee, S.H.; Son, J.K.; Chang, H.W. Inhibitory effects of nardostachin on nitric oxide, prostaglandin E2, and tumor necrosis factor-alpha production in lipopolysaccharide activated macrophages. Biol. Pharm. Bull. 2003, 26, 1375–1378. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ryu, J.H.; Son, H.J.; Lee, S.H.; Sohn, D.H. Two neolignans from Perilla frutescens and their inhibition of nitric oxide synthase and tumor necrosis factor-alpha expression in murine macrophage cell line RAW 264.7. Bioorg. Med. Chem. Lett. 2002, 12, 649–651. [Google Scholar] [CrossRef]
- Garcia-Pastor, P.; Randazzo, A.; Gomez-Paloma, L.; Alcaraz, M.J.; Paya, M. Effects of petrosaspongiolide M, a novel phospholipase A2 inhibitor, on acute and chronic inflammation. J. Pharmacol. Exp. Ther. 1999, 289, 166–172. [Google Scholar] [PubMed]
- Ma, Y.Q.; Chen, Y.R.; Leng, Y.F.; Wu, Z.W. Tanshinone IIA Downregulates HMGB1 and TLR4 Expression in a Spinal Nerve Ligation Model of Neuropathic Pain. Evid. Based Complement. Alternat. Med. 2014, 2014, 639563. [Google Scholar] [CrossRef]
- Huang, N.; Li, Y.; Zhou, Y.; Zhou, Y.; Feng, F.; Shi, S.; Ba, Z.; Luo, Y. Neuroprotective effect of tanshinone IIA-incubated mesenchymal stem cells on Abeta25-35-induced neuroinflammation. Behav. Brain Res. 2019, 365, 48–55. [Google Scholar] [CrossRef]
- Dheen, S.T.; Jun, Y.; Yan, Z.; Tay, S.S.; Ling, E.A. Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia 2005, 50, 21–31. [Google Scholar] [CrossRef]
- Ji, H.F.; Li, X.J.; Zhang, H.Y. Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009, 10, 194–200. [Google Scholar] [CrossRef]
- Gaire, B.P. Herbal Medicine in Ischemic Stroke: Challenges and Prospective. Chin. J. Integr. Med. 2018, 24, 243–246. [Google Scholar] [CrossRef]
- Roberts, G.K.; Gardner, D.; Foster, P.M.; Howard, P.C.; Lui, E.; Walker, L.; van Breemen, R.B.; Auerbach, S.S.; Rider, C. Finding the bad actor: Challenges in identifying toxic constituents in botanical dietary supplements. Food Chem. Toxicol. 2018, 124, 431–438. [Google Scholar] [CrossRef]
- Tang, Y.; Su, G.; Li, N.; Li, W.; Chen, G.; Chen, R.; Zhou, D.; Hou, Y. Preventive agents for neurodegenerative diseases from resin of Dracaena cochinchinensis attenuate LPS-induced microglia over-activation. J. Nat. Med. 2019, 73, 318–330. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Gawande, S.S.; Bodade, R.G.; Gawande, N.M.; Khobragade, C.N. Synthesis and biological evaluation of a novel series of pyrazole chalcones as anti-inflammatory, antioxidant and antimicrobial agents. Bioorg. Med. Chem. 2009, 17, 8168–8173. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Palanivelu, K. The effect of curcumin (turmeric) on Alzheimer’s disease: An overview. Ann. Indian Acad. Neurol. 2008, 11, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Du, Z.Y.; Zheng, X.; Li, D.L.; Zhou, R.P.; Zhang, K. Use of curcumin in diagnosis, prevention, and treatment of Alzheimer’s disease. Neural Regen. Res. 2018, 13, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Browne, A.; Child, D.; Tanzi, R.E. Curcumin decreases amyloid-beta peptide levels by attenuating the maturation of amyloid-beta precursor protein. J. Biol. Chem. 2010, 285, 28472–28480. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Z.; Gao, Z.; Xie, K.; Zhang, Q.; Jiang, H.; Pang, Q. Effects of curcumin on learning and memory deficits, BDNF, and ERK protein expression in rats exposed to chronic unpredictable stress. Behav. Brain Res. 2014, 271, 116–121. [Google Scholar] [CrossRef]
- Rege, S.D.; Geetha, T.; Griffin, G.D.; Broderick, T.L.; Babu, J.R. Neuroprotective effects of resveratrol in Alzheimer disease pathology. Front. Aging Neurosci. 2014, 6, 218. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys Acta. 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Zeng, G.F.; Zong, S.H.; Zhang, Z.Y.; Fu, S.W.; Li, K.K.; Fang, Y.; Lu, L.; Xiao, D.Q. The Role of 6-Gingerol on Inhibiting Amyloid beta Protein-Induced Apoptosis in PC12 Cells. Rejuvenation Res. 2015, 18, 413–421. [Google Scholar] [CrossRef]
- Shi, C.; Liu, J.; Wu, F.; Yew, D.T. Ginkgo biloba extract in Alzheimer’s disease: From action mechanisms to medical practice. Int. J. Mol. Sci. 2010, 11, 107–123. [Google Scholar] [CrossRef]
- Sabogal-Guaqueta, A.M.; Munoz-Manco, J.I.; Ramirez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gomez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.S.; Tiwari, N.R.; Tiwari, R.R.; Sharma, R.S. Neurocognitive Effect of Nootropic Drug Brahmi (Bacopa monnieri) in Alzheimer’s Disease. Ann. Neurosci. 2017, 24, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Iwata, N.; Yoshikawa, A.; Aizaki, Y.; Ishiura, S.; Saido, T.C.; Maruyama, K. Berberine alters the processing of Alzheimer’s amyloid precursor protein to decrease Abeta secretion. Biochem. Biophys Res. Commun. 2007, 352, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, J.L.; Liu, R.; Li, X.X.; Li, J.F.; Zhang, L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules 2013, 18, 9949–9965. [Google Scholar] [CrossRef]
- Choi, J.G.; Kim, S.Y.; Jeong, M.; Oh, M.S. Pharmacotherapeutic potential of ginger and its compounds in age-related neurological disorders. Pharmacol. Ther. 2018, 182, 56–69. [Google Scholar] [CrossRef]
- Kwon, G.; Lee, H.E.; Lee, D.H.; Woo, H.; Park, S.J.; Gao, Q.; Ahn, Y.J.; Son, K.H.; Ryu, J.H. Spicatoside A enhances memory consolidation through the brain-derived neurotrophic factor in mice. Neurosci. Lett. 2014, 572, 58–62. [Google Scholar] [CrossRef]
- Jin, X.; Liu, M.Y.; Zhang, D.F.; Zhong, X.; Du, K.; Qian, P.; Yao, W.F.; Gao, H.; Wei, M.J. Baicalin mitigates cognitive impairment and protects neurons from microglia-mediated neuroinflammation via suppressing NLRP3 inflammasomes and TLR4/NF-kappaB signaling pathway. CNS Neurosci. Ther. 2019, 25, 575–590. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, Y.; Lee, H.E.; Park, S.J.; Jeon, S.J.; Jeon, S.J.; Cheong, J.H.; Shin, C.Y.; Son, K.H.; Ryu, J.H. Oroxylin A enhances memory consolidation through the brain-derived neurotrophic factor in mice. Brain Res. Bull. 2014, 108, 67–73. [Google Scholar] [CrossRef]
- Subedi, L.; Cho, K.; Park, Y.U.; Choi, H.J.; Kim, S.Y. Sulforaphane-Enriched Broccoli Sprouts Pretreated by Pulsed Electric Fields Reduces Neuroinflammation and Ameliorates Scopolamine-Induced Amnesia in Mouse Brain through Its Antioxidant Ability via Nrf2-HO-1 Activation. Oxid. Med. Cell Longev. 2019, 2019, 3549274. [Google Scholar] [CrossRef]
- Kim, C.S.; Suh, W.S.; Subedi, L.; Kim, S.Y.; Choi, S.U.; Lee, K.R. Neuroprotective Fatty Acids from the Stem Bark of Sorbus commixta. Lipids 2016, 51, 989–995. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| TNF-α Inhibitor | Disease(s) | Toxicity/Side Effect | Mechanism of Action | Reference(s) |
|---|---|---|---|---|
| Adalimumab | CD, retinitis pigmentosa, psoriatic arthritis | Increased risk of infections, lymphoma | Blocks the effects of TNF-α, reduces oxidative stress | [62,63] |
| Apratastat | RA | Less effective, dropped from clinical trial | TACE and MMP inhibitor | [64] |
| Certolizumab | RA, CD | Meningococcal meningoencephalitis, palmoplantar pustulosis | Inhibits soluble TNF-α binding | [52,53] |
| Golimumab | RA, ankylosing spondylitis, psoriatic arthritis | Bacterial and viral infection, fungal infection, tuberculosis | Prevents TNF-α binding with TNFR1 and TNFR2 | [54] |
| Infliximab | CD, psoriasis, cognitive improvements, AD | Parkinsonism | Binds with high affinity to soluble and transmembrane forms of TNF-α, progressive MS | [54,55] |
| Minocycline | ALS, MS, AD, stroke, TBI, spinal cord injury | Dizziness, vertigo, lightheadedness | TNF-α synthesis inhibition | [56,65] |
| Thalidomide | Multiple myeloma, CD, Behcet’s disease, HIV, lupus, leprosy | Congenital abnormalities, birth defects, sensorimotor peripheral neuropathy, somnolence, rash, fatigue, constipation | Increases TNF-α degradation | [66,67] |
| GW3333 | RA, other inflammation | NA | TACE and MMP inhibitor | [64] |
| BMS-561392 | RA, other inflammation | NA | Specific TACE inhibitor | [64] |
| Etanercept | Acute and chronic stroke, post-stroke cognitive impairment, chronic brain injury | NA | Inhibits natural TNF-α, TNF blockade | [68,69] |
| Medicinal Plant | Active Compound | Compound Full Name | Mechanism of Action | Disease/ Pathogenesis | Experiment Model | Reference(s) |
|---|---|---|---|---|---|---|
| Wasabia japonica | AITC (isothiocyanate) | 3-isothiocyanatoprop-1-ene | Inhibits JNK/NF-κB signaling and inhibit TNF-α secretion | Neuroinflammation | In vitro | [77] |
| Zingiber officinale | 6-Shogaol (phenols) | (E)-1-(4-hydroxy-3-methoxyphenyl)dec-4-en-3-one | Attenuates LPS-induced TNF-alpha secretion, protects dopaminergic neurons | Neuroinflammation, PD | In vitro, In vivo | [78,79] |
| Glycine max | Genistein (isoflavone) | 5,7-dihydroxy-3-(4-hydroxyphenyl)chromen-4-one | Inhibits ERK activation and NF-κB regulation by blocking the cleavage of IκB-α | Inflammation, muscular dystrophy | In vitro | [80,81,82] |
| Brassica oleracea | Quercetin (aglycon) | 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one | Inhibits nuclear translocation of NF-κB and phosphorylated Akt | MPTP-induced neurotoxicity | In vitro, in vivo, human subject | [83,84] |
| Allium fistulosum | Kaempferol (flavonoid) | 3,5,7-trihydroxy-2-(4-hydroxyphenyl)chromen-4-one | Inhibits TLR4 and corresponding downstream activation of NF-κB, JNK, p38 MAPK, and Akt | Neuroinflammation | In vitro | [83] |
| Psidium guajava | Apigenin (flavone) | 5,7-dihydroxy-2-(4-hydroxyphenyl)chromen-4-one | Attenuates the upregulation of NF-κB gene | PD | In vitro, In vivo | [85] |
| Citrus paradisi | Naringenin (flavanone) | (2S)-5,7-dihydroxy-2-(4-hydroxyphenyl)-2,3-dihydrochromen-4-one | Inhibits iNOS/NO, decreases α-synuclein expression and neuroinflammation in PD | Neuroinflammatory injury | In vitro | [86] |
| Brassica oleracea | Myricetin (flavonoid) | 3,5,7-trihydroxy-2-(3,4,5-trihydroxyphenyl)chromen-4-one | Attenuates the activation of the MAPK and NF-κB signaling pathways | AD, PD | In vitro | [87] |
| Rhus verniciflua | Butein (polyphenol) | (E)-1-(2,4-dihydroxyphenyl)-3-(3,4-dihydroxyphenyl)prop-2-en-1-one | Inhibits the production of IL-1β, IL-6, and TNF-α | Neuroinflammation, neurotoxicity | In vitro | [88,89] |
| Citrus sinensis | Hesperetin (flavanone) | (2S)-5,7-dihydroxy-2-(3-hydroxy-4-methoxyphenyl)-2,3-dihydrochromen-4-one | Inhibits iNOS expression and TNF-α production | Neuroinflammatory injury | In vitro | [90,91] |
| Scutellaria baicalensis | Wogoni n(flavone) | 5,7-dihydroxy-8-methoxy-2-phenylchromen-4-one | Alteration of JAK/STAT pathways | AD, PD | In vitro | [92] |
| Maclura pomifera | Morin (flavonol) | 2-(2,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one | Inhibits NF-κB- and AP-1-mediated transcription and phosphorylation of MAPKs and Akt | Neuroinflammation, AD | In vivo | [93,94] |
| Honey, propolis | Chrysin (hydroxyflavone) | 5,7-dihydroxy-2-phenylchromen-4-one | Inhibits iNOS, COX2, NO | Neuroinflammation | In vitro | [95] |
| Zanthoxylum armatum | Eudesmin (lignan) | 3,6-bis(3,4-dimethoxyphenyl)-1,3,3a,4,6,6a-hexahydrofuro[3,4-c]furan | Suppression of NF-κB | Inflammation | In vitro | [96] |
| Magnolia fargesii | Magnolin (lignan) | (3S,3aR,6S,6aR)-3-(3,4-dimethoxyphenyl)-6-(3,4,5-trimethoxyphenyl)-1,3,3a,4,6,6a-hexahydrofuro[3,4-c]furan | Suppression of NF-κB, NO, PGE2 | Inflammation | In vitro | [97,98] |
| Magnolia officinalis | Honokiol (lignan) | 2-(4-hydroxy-3-prop-2-enylphenyl)-4-prop-2-enylphenol | Inhibits the phosphorylation of PI3K/Akt/MAP kinases, NF-κB, and CB2 receptor | Neurodegenerative diseases (e. g. AD) | In vitro | [99,100] |
| Panax ginseng | Ginsenoside Rg1 (triterpene glycosides) | (2R,3R,4S,5S,6R)-2-[[(3S,5R,6S,8R,9R,10R,12R,13R,14R,17S)-3,12-dihydroxy-4,4,8,10,14-pentamethyl-17-[(2S)-6-methyl-2-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyhept-5-en-2-yl]-2,3,5,6,7,9,11,12,13,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-6-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol | Reduces the levels of IL-1β, IL-6, and TNF-α | D-galactose-induced aging (related to AD) | In vito, in vivo | [101] |
| Panax ginseng | Ginsenoside Rb2 (triterpene glycoside) | (2S,3R,4S,5S,6R)-2-[(2R,3R,4S,5S,6R)-4,5-dihydroxy-6-(hydroxymethyl)-2-[[(3S,5R,8R,9R,10R,12R,13R,14R,17S)-12-hydroxy-4,4,8,10,14-pentamethyl-17-[(2S)-6-methyl-2-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2S,3R,4S,5S)-3,4,5-trihydroxyoxan-2-yl]oxymethyl]oxan-2-yl]oxyhept-5-en-2-yl]-2,3,5,6,7,9,11,12,13,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxy]oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol | Suppresses TNF-α production via NF-κB inhibition | AD, PD, MS | In vivo | [102] |
| Nicotiana tabacum | Nicotine (alkaloid) | 3-[(2S)-1-methylpyrrolidin-2-yl]pyridine | Immune modulation, alteration of MYD88/NF-κB downstream pathway | AD, PD, MS | In vitro, In vivo and in patients | [103,104,105,106] |
| Berberis vulgaris | Berberine (alkaloid) | 16,17-dimethoxy-5,7-dioxa-13-azoniapentacyclo[11.8.0.02,10.04,8.015,20]henicosa-1(13),2,4(8),9,14,16,18,20-octaene | Downregulates acetylcholinesterase and inhibits the activation of the NF-κB signaling pathway | AD | In vitro and In vivo | [107,108,109] |
| Capsicum annuum | Capsaicin (alkaloid) | (E)-N-[(4-hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide | Inhibits glial activation-mediated oxidative stress and neuroinflammation | PD | In vitro and In vivo | [110,111,112] |
| Piper methysticum | Kavalactones (polyketide) | Reduces intracellular oxidative stress | AD, stroke | In vitro and In vivo | [113,114,115] | |
| Vitis vinifera | Resveratrol (Polyphenol) | 5-[(E)-2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol | Upregulates the expression of the suppressor of SOCS-1 | PD | In vitro and In vivo | [116,117,118,119] |
| Lindera neesiana | Koaburaside (ether) | (2S,3R,4S,5S,6R)-2-(4-hydroxy-3,5-dimethoxyphenoxy)-6-(hydroxymethyl)oxane-3,4,5-triol | Inhibits inflammatory mediators, pro-inflammatory cytokines in LPS-activated microglia, prevents neuronal death | AD, PD, MS lesions | In vitro | [120] |
| Patrinia saniculaefolia | Nardostachin (iridoid) | [(1S,4aS,6S,7R,7aS)-6-hydroxy-7-methyl-1-(3-methylbutanoyloxy)-1,4a,5,6,7,7a-hexahydrocyclopenta[c]pyran-4-yl]methyl 3-methylbutanoate | Reduces cytokines, COX-2, and PGE2 | Inflammatory disorders such as neuroinflammation | In vitro | [121] |
| Perilla frutescens | Magnosalin, Andamanicin (neolignans) | 1-[(1R,2R,3R,4R)-2,3-dimethyl-4-(2,4,5-trimethoxyphenyl)cyclobutyl]-2,4,5-trimethoxybenzene | Inhibits neuroinflammation and cell death | Degenerative disease | In vitro | [122] |
| Petrosaspongianigra | Petrosaspongiolid M | [(1S,3R,4aR,4bS,6aS,10aS,10bS,12aS)-3-(2-hydroxy-5-oxo-2H-furan-3-yl)-4b,7,7,10a-tetramethyl-1,3,4,4a,5,6,6a,8,9,10,10b,11,12,12a-tetradecahydronaphtho[2,1-f]isochromen-1-yl] acetate | Reduces PGE2, NO, and TNF-α levels | Acute and Chronic Inflammation | In vivo | [123] |
| Salvia miltiorrhiza | Tanshinone (diterpene) | 1,6-dimethylnaphtho[1,2-g][1]benzofuran-10,11-dione | Selectively suppresses pro-inflammatory gene expression and partially decreased anti-inflammatory genes expression | Neuropathic pain, Neuroinflammation | In vitro and in vivo | [124,125] |
| Vitamin A | Retinoic acid (terpenes) | (2E,4E,6E,8E)-3,7-dimethyl-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraenoic acid | Inhibits TNF-α and iNOS in (Aβ) or LPS-induced microglia-mediated neuroinflammation | AD, activated microglia-mediated brain disorders | In vitro | [126] |
| Allium sativum | Diallyl sulfide (sulfide) | 3-prop-2-enylsulfanylprop-1-ene | Suppress pro-inflammatory cytokines by decreased ROS production through-induced PI3K/Akt and reduced NF-κB and AP-1 | Inflammation, Allergy | In vitro | [74] |
| Curcuma longa | Curcumin (polyphenol) | (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione | Reduction of NF-κB mediated transcription | Inflammation, | In vitro, in vivo and in human | [70,75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, L.; Lee, S.E.; Madiha, S.; Gaire, B.P.; Jin, M.; Yumnam, S.; Kim, S.Y. Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 764. https://doi.org/10.3390/ijms21030764
Subedi L, Lee SE, Madiha S, Gaire BP, Jin M, Yumnam S, Kim SY. Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases. International Journal of Molecular Sciences. 2020; 21(3):764. https://doi.org/10.3390/ijms21030764
Chicago/Turabian StyleSubedi, Lalita, Si Eun Lee, Syeda Madiha, Bhakta Prasad Gaire, Mirim Jin, Silvia Yumnam, and Sun Yeou Kim. 2020. "Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases" International Journal of Molecular Sciences 21, no. 3: 764. https://doi.org/10.3390/ijms21030764
APA StyleSubedi, L., Lee, S. E., Madiha, S., Gaire, B. P., Jin, M., Yumnam, S., & Kim, S. Y. (2020). Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases. International Journal of Molecular Sciences, 21(3), 764. https://doi.org/10.3390/ijms21030764