The Endocannabinoid System: A Target for Cancer Treatment


 , , ,
, , , 
{kind=link}
Abstract
1. Introduction
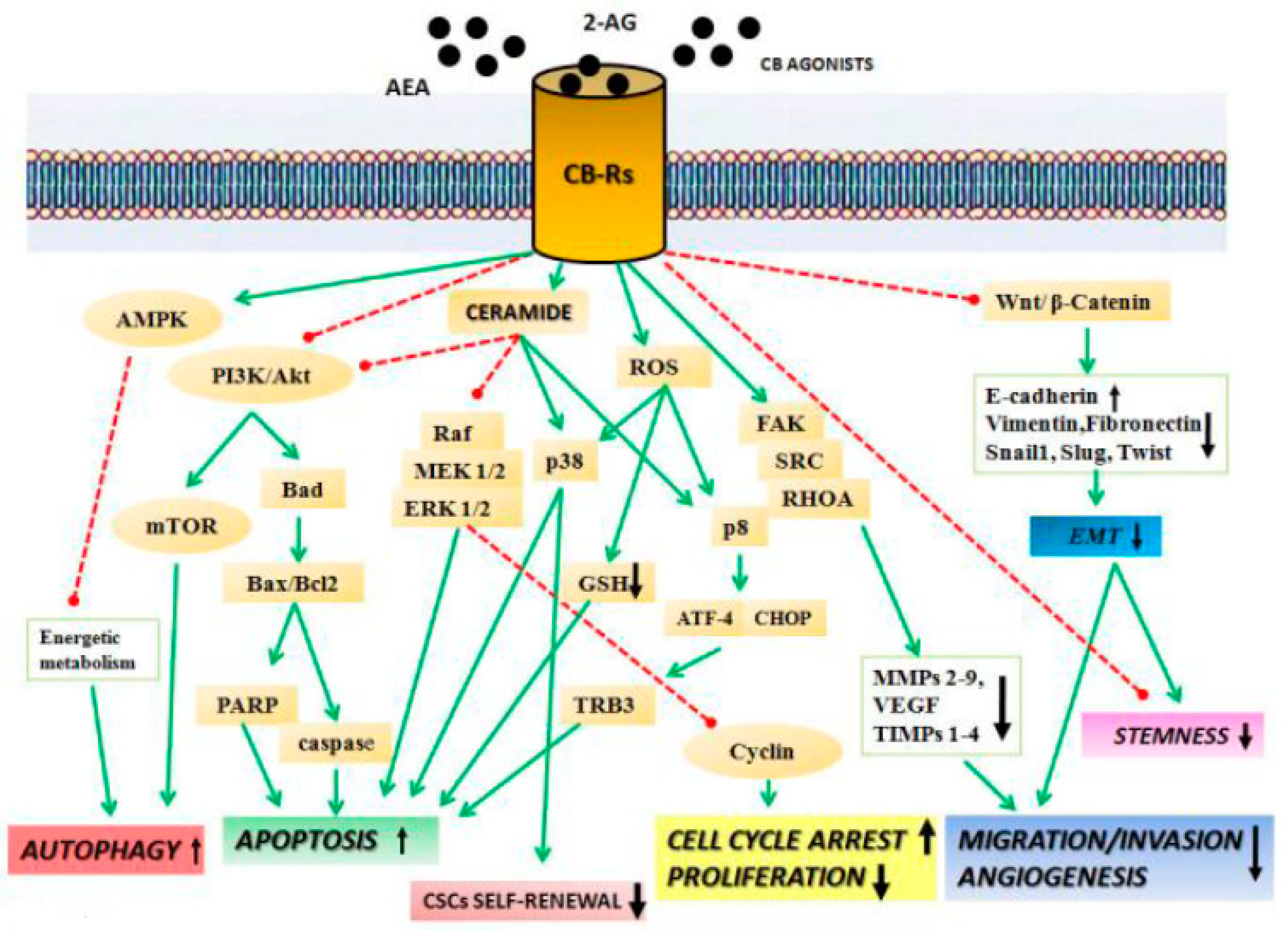
2. Anticancer Effects of Cannabinoids
2.1. Cannabinoids Inhibit Migration, Invasion, and Angiogenesis
2.2. Cannabinoids Affect non-CB1/CB2 Receptors
3. Gastrointestinal Cancers
4. Lung Cancer
5. Breast and Prostate Cancers
6. Pancreatic and Thyroid Cancers
7. Brain Cancer
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Andre, C.M.; Hausman, J.F.; Guerriero, G. Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 2016, 4, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. B. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Sobocinska, A.A.; Czarnecka, A.M.; Krol, M.; Botta, B.; Szczylik, C. The therapeutic aspects of the endocannabinoid system (ECS) for cancer and their development: From nature to laboratory. Curr. Pharmaceut. Des. 2016, 22, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Maurya, N.; Velmurugan, B.K. Therapeutic applications of cannabinoids. Chem. Biol. Interact. 2018, 293, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the Endocannabinoid System as a Potential Anticancer Strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Javid, F.A.; Phillips, R.M.; Afshinjavid, S.; Verde, R.; Ligresti, A. Cannabinoid pharmacology in cancer research: A new hope for cancer patients? Eur. J. Pharmacol. 2016, 775, 1–14. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Teixeira, N.A.; Correia-da-Silva, G. Cannabinoids as Modulators of Cell Death: Clinical Applications and Future Directions. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 63–88. [Google Scholar]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Casadó, V.; Canela, E. The Endocannabinoid System as a Target in Cancer Diseases: Are We There Yet? Front. Pharmacol. 2019, 10, 339. [Google Scholar] [CrossRef]
- Laezza, C.; D’Alessandro, A.; Paladino, S.; Malfitano, M.A.; Proto, M.C.; Gazzerro, P.; Pisanti, S.; Santoro, A.; Ciaglia, E.; Bifulco, M. Anandamide inhibits the Wnt/β-catenin signaling pathway in human breast cancer MDA MB 231 cells. Eur. J. Cancer. 2012, 16, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 2, S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.L.; Lau, D.T.; Allen, J.D.; Arnold, J.C. The multidrug transporter ABCG2 (BCRP) is inhibited by plant-derived cannabinoids. Br. J. Pharmacol. 2007, 152, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Vago, R.; Bettiga, A.; Salonia, A.; Ciuffreda, P.; Ottria, R. Development of new inhibitors for N-acylethanolamine-hydrolyzing acid amidase as promising tool against bladder cancer. Bioorg. Med. Chem. 2017, 25, 1242–1249. [Google Scholar] [CrossRef]
- Hamtiaux, L.; Hansoulle, L.; Dauguet, N.; Muccioli, G.G.; Gallez, B.; Lambert, D.M. Increasing antiproliferative properties of endocannabinoids in N1E-115 neuroblastoma cells through inhibition of their metabolism. PLoS ONE 2011, 6, e26823. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef]
- Winkler, K.; Ramer, R.; Dithmer, S.; Ivanov, I.; Merkord, J.; Hinz, B. Fatty acid amide hydrolase inhibitors confer anti-invasive and antimetastatic effects on lung cancer cells. Oncotarget 2016, 7, 15047–15064. [Google Scholar] [CrossRef]
- Ma, M.; Bai, J.; Ling, Y.; Chang, W.; Xie, G.; Li, R.; Wang, G.; Tao, K. Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol. Med. Rep. 2016, 3, 2850–2856. [Google Scholar] [CrossRef]
- Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Bifulco, M. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 5, 273–282. [Google Scholar] [CrossRef]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Rodrigues, T.; Sieglitz, F.; Bernardes, G.J. Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents. Chem. Soc. Rev. 2016, 45, 6130–6137. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Heinemann, K.; Merkord, J.; Rohde, H.; Salamon, A.; Linnebacher, M.; Hinz, B. COX-2 and PPAR-γ confer cannabidiol-induced apoptosis of human lung cancer cells. Mol. Cancer Ther. 2013, 12, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Reggio, P.H. An Update on Non-CB1, Non-CB2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid. Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Ferro, R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Biol Regul. 2016, 60, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Pérez-Gómez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB2-GPR55 receptor heteromers modulates cancer cell signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef]
- Scarlett, K.A.; White, E.Z.; Coke, C.J.; Carter, J.R.; Bryant, L.K.; Hinton, C.V. Agonist-induced CXCR4 and CB2 Heterodimerization Inhibits Gα13/RhoA-mediated Migration. Mol. Cancer Res. 2018, 16, 728–739. [Google Scholar] [CrossRef]
- Pérez-Gómez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; García-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martín-Villar, E.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef]
- Pesce, M.; D’Alessandro, A.; Borrelli, O.; Gigli, S.; Seguella, L.; Cuomo, R.; Esposito, G.; Sarnelli, G. Endocannabinoid-related compounds in gastrointestinal diseases. J. Cell Mol. Med. 2018, 22, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, S.B.; Palmqvist, R.; Henriksson, M.L.; Dahlin, A.M.; Edin, S.; Jacobsson, S.O.; Öberg, Å.; Fowler, C.J. High tumour cannabinoid CB1 receptor immunoreactivity negatively impacts disease-specific survival in stage II microsatellite stable colorectal cancer. PLoS ONE 2011, 8, e23003. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, H.; Li, Y.; Li, L.; Qiu, Y.; Ren, J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol Rep. 2015, 1, 447–454. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Gomez, I.; Martin, P.; Sánchez, A.; Román, L.; Tejerina, E.; Bonilla, F.; Merino, A.G.; de Herreros, A.G.; Provencio, M.; et al. Cannabinoids receptor type 2, CB2, expression correlates with human colon cancer progression and predicts patient survival. Oncoscience 2015, 2, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor alpha-mediated ceramide de novo synthesis in colon cancer cells. Clin. Cancer Res. 2008, 23, 7691–7700. [Google Scholar] [CrossRef]
- Hasenoehrl, C.; Feuersinger, D.; Sturm, E.M.; Bärnthaler, T.; Heitzer, E.; Graf, R.; Grill, M.; Pichler, M.; Beck, S.; Butcher, L.; et al. G protein-coupled receptor GPR55 promotes colorectal cancer and has opposing effects to cannabinoid receptor 1. Int. J. Cancer 2018, 1, 121–132. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; DuBois, R.N. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008, 15, 6468–6476. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.K.; Kang, W.K.; Park, J.M.; Ahn, H.J.; Kim, S.W.; Taek, O.S.; Choi, K.Y. Expression of the cannabinoid type I receptor and prognosis following surgery in colorectal cancer. Oncol. Lett. 2013, 3, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Suk, K.T.; Mederacke, I.; Gwak, G.Y.; Cho, S.W.; Adeyemi, A.; Friedman, R.; Schwabe, R.F. Opposite roles of cannabinoid receptors 1 and 2 in hepatocarcinogenesis. Gut 2016, 10, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Tutino, V.; Caruso, M.G.; De Nunzio, V.; Lorusso, D.; Veronese, N.; Gigante, I.; Notarnicola, M.; Giannelli, G. Down-Regulation of Cannabinoid Type 1 (CB1) Receptor and its Downstream Signaling Pathways in Metastatic Colorectal Cancer. Cancers 2019, 5, 708. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Martín-Sabroso, C.; Torres-Suárez, A.I. Insights into the effects of the endocannabinoid system in cancer: A review. Br. J. Pharmacol. 2018, 13, 2566–2580. [Google Scholar]
- Ortega, A.; García-Hernández, V.M.; Ruiz-García, E.; Meneses-García, A.; Herrera-Gómez, A.; Aguilar-Ponce, J.L.; Montes-Servín, E.; Prospero-García, O.; Del Angel, S.A. Comparing the effects of endogenous and synthetic cannabinoid receptor agonists on survival of gastric cancer cells. Life Sci. 2016, 165, 56–62. [Google Scholar] [CrossRef] [PubMed]
- DeMorrow, S.; Francis, H.; Gaudio, E.; Venter, J.; Franchitto, A.; Kopriva, S.; Onori, P.; Mancinelli, R.; Frampton, G.; Coufal, M.; et al. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the non canonicalWnt signaling pathway. Am. J. Physio. Gastrointest. Liver Physiol. 2008, 6, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ramirez, J.C.; Frampton, G.A.; Golden, L.E.; Quinn, M.A.; Pae, H.Y.; Horvat, D.; Liang, L.; DeMorrow, S. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab. Invest. 2011, 7, 1007–1017. [Google Scholar] [CrossRef]
- Proto, M.C.; Gazzerro, P.; Di Croce, L.; Santoro, A.; Malfitano, A.M.; Pisanti, S.; Laezza, C.; Bifulco, M. Interaction of endocannabinoid system and steroid hormones in the control of colon cancer cell growth. J. Cell Physiol. 2012, 1, 250–528. [Google Scholar] [CrossRef]
- Fiore, D.; Proto, M.C.; Pisanti, S.; Picardi, P.; Pagano Zottola, A.C.; Butini, S.; Gemma, S.; Casagni, A.; Laezza, C.; Vitale, M.; et al. Antitumor effect of pyrrolo-1,5-benzoxazepine-15 and its synergistic effect with Oxaliplatin and 5-FU in colorectal cancer cells. Cancer Biol. Ther. 2016, 8, 849–858. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.; Qin, Y.; Pan, Z.; Li, M.; Liu, X.; Chen, X.; Qu, G.; Zhou, L.; Xu, M.; Zheng, Q.; et al. Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells. Biomolecules 2019, 8, 302. [Google Scholar] [CrossRef]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stančić, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br. J. Pharmacol. 2016, 1, 142–154. [Google Scholar] [CrossRef]
- Aviello, G.; Romano, B.; Borrelli, F.; Capasso, R.; Gallo, L.; Piscitelli, F.; Di Marzo, V.; Izzo, A.A. Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J. Mol. Med. 2012, 8, 925–934. [Google Scholar] [CrossRef]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Honarmand, M.; Namazi, F.; Mohammadi, A.; Nazifi, S. Can cannabidiol inhibit angiogenesis in colon cancer? Comp. Clin. Path. 2019, 28, 165–172. [Google Scholar] [CrossRef]
- Greenhough, A.; Patsos, H.A.; Williams, A.C.; Paraskeva, C. The cannabinoid delta (9)-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signaling and induces BAD-mediated apoptosis in colorectal cancer cells. Int. J. Cancer 2007, 10, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Morell, C.; Rodríguez-Henche, N.; Diaz-Laviada, I. Involvement of PPARgamma in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death. Dis. 2013, 5, e618. [Google Scholar] [CrossRef] [PubMed]
- Raup-Konsavage, W.M.; Johnson, M.; Legare, C.A.; Yochum, G.S.; Morgan, D.J.; Vrana, K.E. Synthetic cannabinoid activity against colorectal cancer cells. Cannabis. Cannabinoid. Res. 2018, 1, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Pisanti, S.; Grimaldi, C.; Izzo, A.A.; Borrelli, F.; Proto, M.C.; Malfitano, A.M.; Gazzerro, P.; Laezza, C.; Bifulco, M. Rimonabant inhibits human colon cancer cell growth and reduces the formation of precancerous lesions in the mouse colon. Int. J. Cancer 2009, 5, 996–1003. [Google Scholar] [CrossRef]
- Proto, M.C.; Fiore, D.; Piscopo, C.; Franceschelli, S.; Bizzarro, V.; Laezza, C.; Lauro, G.; Feoli, A.; Tosco, A.; Bifulco, G.; et al. Inhibition of Wnt/β-Catenin pathway and Histone acetyltransferase activity by Rimonabant: A therapeutic target for colon cancer. Sci. Rep. 2017, 7, 11678. [Google Scholar] [CrossRef]
- Fiore, D.; Ramesh, P.; Proto, M.C.; Piscopo, C.; Franceschelli, S.; Anzelmo, S.; Medema, J.P.; Bifulco, M.; Gazzerro, P. Rimonabant Kills Colon Cancer Stem Cells without Inducing Toxicity in Normal Colon Organoids. Front. Pharmacol. 2018, 8, 949. [Google Scholar] [CrossRef]
- Gazzerro, P.; Malfitano, A.M.; Proto, M.C.; Santoro, A.; Pisanti, S.; Caruso, M.G.; Notarnicola, M.; Messa, C.; Laezza, C.; Misso, G.; et al. Synergistic inhibition of human colon cancer cell growth by the cannabinoid CB1 receptor antagonist rimonabant and oxaliplatin. Oncol. Rep. 2010, 1, 171–175. [Google Scholar]
- Xian, X.S.; Park, H.; Cho, Y.K.; Lee, I.S.; Kim, S.W.; Choi, M.G.; Chung, I.S.; Han, K.H.; Park, J.M. Effect of a synthetic cannabinoid agonist on the proliferation and invasion of gastric cancer cells. J. Cell Biochem. 2010, 2, 321–332. [Google Scholar] [CrossRef]
- Xian, X.S.; Park, H.; Choi, M.G.; Park, J.M. Cannabinoid receptor agonist as an alternative drug in 5-fluorouracil-resistant gastric cancer cells. Anticancer Res. 2013, 6, 2541–2547. [Google Scholar]
- Tashkin, D.P.; Roth, M.D. Pulmonary effects of inhaled cannabis smoke. Am. J. Drug Alcohol Abuse 2019, 45, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. Impact of Cannabis, Cannabinoids, and Endocannabinoids in the Lungs. Front. Pharmacol. 2016, 7, 317. [Google Scholar] [CrossRef] [PubMed]
- Staiano, R.I.; Loffredo, S.; Borriello, F.; Iannotti, F.A.; Piscitelli, F.; Orlando, P.; Secondo, A.; Granata, F.; Lepore, M.T.; Fiorelli, A.; et al. Human lung-resident macrophages express CB1 and CB2 receptors whose activation inhibits the release of angiogenic and lymphangiogenic factors. J. Leukoc. Biol. 2016, 99, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014, 9, 2475–2486. [Google Scholar] [CrossRef]
- Preet, A.; Ganju, R.K.; Groopman, J.E. Delta9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene 2008, 27, 339–346. [Google Scholar] [CrossRef]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. BiochemPharmacol 2014, 92, 312–325. [Google Scholar] [CrossRef]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef]
- Ramer, R.; Rohde, A.; Merkord, J.; Rohde, H.; Hinz, B. Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lung cancer cells. Pharm. Res. 2010, 27, 2162–2174. [Google Scholar] [CrossRef]
- Ravi, J.; Elbaz, M.; Wani, N.A.; Nasser, M.W.; Ganju, R.K. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol. Carcinog. 2016, 55, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. ∆9-Tetrahydrocannabinol Inhibits Cell Cycle Progression in Human Breast Cancer Cells through Cdc2 Regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, MW.; Ravi, J.; Wani, NA.; Ahirwar, DK.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer 2007, 6, 2921–2927. [Google Scholar] [CrossRef]
- Elbaz, M.; Ahirwar, D.; Xiaoli, Z.; Zhou, X.; Lustberg, M.; Nasser, M.W.; Shilo, K.; Ganju, R.K. TRPV2 is a novel biomarker and therapeutic target in triple negative breast cancer. Oncotarget 2016, 9, 33459–33470. [Google Scholar] [CrossRef]
- Laezza, C.; Pisanti, S.; Crescenzi, E.; Bifulco, M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006, 580, 6076–6082. [Google Scholar] [CrossRef]
- Laezza, C.; Pisanti, S.; Malfitano, A.M.; Bifulco, M. The anandamide analog, Met-F-AEA, controls human breast cancer cell migration via the RHOA/RHO kinase signaling pathway. Endocr. Relat. Cancer. 2008, 15, 965–974. [Google Scholar] [CrossRef]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, AM.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, F.; Ostad, S.N.; Aliebrahimi, S.; Daman, Z. Anti-invasion Effects of Cannabinoids Agonist and Antagonist on Human Breast Cancer Stem Cells. Iran. J. Pharm. Res. 2017, 16, 1479–1486. [Google Scholar] [PubMed]
- Pisanti, S.; Borselli, C.; Oliviero, O.; Laezza, C.; Gazzerro, P.; Bifulco, M. Antiangiogenic activity of the endocannabinoid anandamide: Correlation to its tumor-suppressor efficacy. J. Cell. Physiol. 2007, 211, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Benito, S.; Moreno, E.; Seijo-Vila, M.; Tundidor, I.; Andradas, C.; Caffarel, M.M.; Caro-Villalobos, M.; Urigüen, L.; Diez-Alarcia, R.; Moreno-Bueno, G.; et al. Therapeutic targeting of HER2-CB2R heteromers in HER2-positive breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Mukhtar, H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005, 65, 1635–1641. [Google Scholar] [CrossRef]
- Mimeault, M.; Pommery, N.; Wattez, N.; Bailly, C.; Hénichart, J.P. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: Implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate 2003, 56, 1–12. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Endsley, MP.; Isbell, M.A.; Falck, J.R.; Iwamoto, Y.; Hillard, CJ.; Campbell, W.B. 2-arachidonoylglycerol: A novel inhibitor of androgen-independent prostate cancer cell invasion. Cancer Res. 2004, 64, 8826–8830. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Gomez-Granados, A.D.; Tang, A.T.; Pfeiffer, A.W.; Williams, C.L.; Campbell, W.B. Cannabinoid receptor type 1 (CB1) activation inhibits small GTPase RhoA activity and regulates motility of prostate carcinoma cells. Endocrinology 2012, 153, 29. [Google Scholar] [CrossRef]
- Morell, C.; Bort, A.; Vara, D.; Ramos-Torres, A.; Rodríguez-Henche, N.; Díaz-Laviada, I. The cannabinoid WIN 55,212-2 prevents neuroendocrine differentiation of LNCaP prostate cancer cells. Prostate Cancer Prostatic Dis. 2016, 19, 248–257. [Google Scholar] [CrossRef]
- Orellana-Serradell, O.; Poblete, C.E.; Sanchez, C.; Castellón, E.A.; Gallegos, I.; Huidobro, C.; Llanos, MN.; Contreras, H.R. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol. Rep. 2015, 33, 1599–1608. [Google Scholar] [CrossRef]
- Sharma, M.; Hudson, J.; Adomat, H.; Guns, E.; Cox, M. In vitro anticancer activity of plant-derived cannabidiol on prostate cancer cell lines. Pharmacol. Pharm. 2014, 5, 806–820. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Schiano Moriello, A.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2013, 168, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Vasisth, G.; Kapoor, A. Systematic review of the potential role of cannabinoids as antiproliferative agents for urological cancers. Can. Urol. Assoc. J. 2017, 11, E138–E142. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Silva, F.J.; Suárez, J.; Baixeras, E.; Cobo, N.; Bautista, D.; Cuesta-Muñoz, A.L.; Fuentes, E.; Juan-Pico, P.; Castro, M.J.; Milman, G.; et al. Presence of functional cannabinoid receptors in human endocrine pancreas. Diabetologia 2008, 51, 476–487. [Google Scholar]
- Gonzalez-Mariscal, I.; Krzysik-Walker, S.M.; Doyle, M.E.; Liu, Q.R.; Cimbro, R.; Calyo, S.S.C.; Ghosh, S.; Cieśla, L.; Moaddel, R.; Carlson, O.D.; et al. Human CB1 receptor isoforms, present in hepatocytes and B-cells, are involved in regulating metabolism. Sci. Rep. 2016, 6, 33302. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Doyle, M.E.; Liu, Z.; Lao, Q.; Shin, Y.K.; Carlson, O.D.; Kim, H.S.; Thomas, S.; Napora, J.K.; Lee, E.K.; et al. Cannabinoids inhibit insulin receptor signaling in pancreatic beta-cells. Diabetes 2011, 60, 1198–1209. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 2012, 142, 1218–1228. [Google Scholar] [CrossRef]
- Linari, G.; Agostini, S.; Amadoro, G.; Ciotti, M.T.; Florenzano, F.; Improta, G.; Petrella, C.; Severini, C.; Broccardo, M. Involvement of cannabinoid CB1- and CB2-receptors in the modulation of exocrine pancreatic secretion. Pharmacol. Res. 2009, 59, 207–214. [Google Scholar] [CrossRef]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Yang, L.; Wang, B.; Cui, L.; Li, C.; Zhuo, Y.; Zhang, L.; Zhang, S.; Zhang, Q.; Wang, X. The role of 2-arachidonoylglycerol in the regulation of the tumor-immune microenvironment in murine models of pancreatic cancer. Biomed. Pharmacother. 2019, 115, 108952. [Google Scholar] [CrossRef] [PubMed]
- Fogli, S.; Nieri, P.; Chicca, A.; Adinolfi, B.; Mariotti, V.; Iacopetti, P.; Breschi, M.C.; Pellegrini, S. Cannabinoid derivatives induce cell death in pancreatic MIA PaCa-2 cells via a receptor-independent mechanism. FEBS Lett. 2006, 580, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Lakiotaki, E.; Giaginis, C.; Tolia, M.; Alexandrou, P.; Delladetsima, I.; Giannopoulou, I.; Kyrgias, G.; Patsouris, E.; Theocharis, S. Clinical Significance of Cannabinoid Receptors CB1 and CB2 Expression in Human Malignant and Benign Thyroid Lesions. Biomed. Res. Int. 2015, 2015, 839403. [Google Scholar] [CrossRef]
- Cozzolino, R.; Calì, G.; Bifulco, M.; Laccetti, P. A metabolically stable analogue of anandamide, Met-F-AEA, inhibits human thyroid carcinoma cell lines by activation of apoptosis. Invest. New Drugs. 2010, 28, 115–123. [Google Scholar] [CrossRef]
- Shi, Y.; Zou, M.; Baitei, E.Y.; Alzahrani, A.S.; Parhar, R.S.; Al-Makhalafi, Z.; Al-Mohanna, F.A. Cannabinoid 2 receptor induction by IL-12 and its potential as a therapeutic target for the treatment of anaplastic thyroid carcinoma. Cancer Gene Ther. 2008, 15, 101–107. [Google Scholar] [CrossRef]
- Kushchayeva, Y.; Jensen, K.; Burman, K.D.; Vasko, V. Repositioning therapy for thyroid cancer: New insights on established medications. Endocr. Relat. Cancer 2014, 21, R183–R194. [Google Scholar] [CrossRef]
- López-Valero, I.; Saiz-Ladera, C.; Torres, S.; Hernández-Tiedra, S.; García-Taboada, E.; Rodríguez-Fornés, F.; Barba, M.; Dávila, D.; Salvador-Tormo, N.; Guzmán, M.; et al. Targeting Glioma Initiating Cells with A combined therapy of cannabinoids and temozolomide. Biochem. Pharmacol. 2018, 157, 266–274. [Google Scholar] [CrossRef]
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Sandalcioglu, I.E.; Karsak, M. Cannabinoids in Glioblastoma Therapy: New Applications for Old Drugs. Front. Mol. Neurosci. 2018, 11, 159. [Google Scholar] [CrossRef]
- Rocha, F.C.; Dos Santos Júnior, J.G.; Stefano, S.C.; da Silveira, D.X. Systematic review of the literature on clinical and experimental trials on the antitumor effects of cannabinoids in gliomas. J. Neurooncol. 2014, 116, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Massi, P.; Valenti, M.; Solinas, M.; Parolaro, D. Molecular mechanisms involved in the antitumor activity of cannabinoids on gliomas: Role for oxidative stress. Cancers 2010, 2, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Marcu, J.P.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Horowitz, M.P.; Lee, J.; Pakdel, A.; Allison, J.; Limbad, C.; Moore, D.H.; et al. Cannabidiol enhances the inhibitory effects of delta9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol. Cancer Ther. 2010, 1, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Dennis, J.L.; Dalgleish, A.G.; Liu, W.M. Inhibiting Heat Shock Proteins Can Potentiate the Cytotoxic Effect of Cannabidiol in Human Glioma Cells. Anticancer Res. 2015, 35, 5827–5837. [Google Scholar] [PubMed]
- Galanti, G.; Fisher, T.; Kventsel, I.; Shoham, J.; Gallily, R.; Mechoulam, R.; Lavie, G.; Amariglio, N.; Rechavi, G.; Toren, A. Delta 9-tetrahydrocannabinol inhibits cell cycle progression by downregulation of E2F1 in human glioblastoma multiforme cells. Acta Oncol. 2008, 47, 1062–1070. [Google Scholar] [CrossRef]
- Blázquez, C.; Casanova, M.L.; Planas, A.; Gómez Del Pulgar, T.; Villanueva, C.; Fernández-Aceñero, M.J.; Aragonés, J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003, 17, 529–531. [Google Scholar] [CrossRef]
- Blázquez, C.; González-Feria, L.; Alvarez, L.; Haro, A.; Casanova, M.L.; Guzmán, M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004, 64, 5617–5623. [Google Scholar] [CrossRef]
- Solinas, M.; Massi, P.; Cantelmo, A.R.; Cattaneo, M.G.; Cammarota, R.; Bartolini, D.; Cinquina, V.; Valenti, M.; Vicentini, L.M.; Noonan, D.M.; et al. Cannabidiol inhibits angiogenesis by multiple mechanisms. Br. J. Pharmacol. 2012, 167, 1218–1231. [Google Scholar] [CrossRef]
- Ciaglia, E.; Torelli, G.; Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Malfitano, A.M.; Fiore, D.; Pagano Zottola, A.C.; Proto, M.C.; et al. Cannabinoid receptor CB1 regulates STAT3 activity and its expression dictates the responsiveness to SR141716 treatment in human glioma patients’ cells. Oncotarget 2015, 6, 15464–15481. [Google Scholar] [CrossRef]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013, 73, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Massi, P.; Cinquina, V.; Valenti, M.; Bolognini, D.; Gariboldi, M.; Monti, E.; Rubino, T.; Parolaro, D. Cannabidiol, a non-psychoactive cannabinoid compound, inhibits proliferation and invasion in U87-MG and T98G glioma cells through a multitarget effect. PLoS ONE 2013, 8, e76918. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Salazar, M.; Carracedo, A.; Lorente, M.; Egia, A.; González-Feria, L.; Haro, A.; Velasco, G.; Guzmán, M. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008, 68, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulam, R.; Alvarez, L.; Guzmán, M.; Galve-Roperh, I. Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef]
- Schultz, S.; Beyer, M. GW Pharmaceuticals Achieves Positive Results in Phase 2 Proof of Concept Study in Glioma. 2017. Available online: http://ir.gwpharm.com/static-files/cde942fe-555c-4b2f-9cc9-f34d24c7ad27 (accessed on 12 October 2019).
- Schultz, S. GW Pharmaceuticals Plc Investor Presentation—February 2018. Available online: http://ir.gwpharm.com/static-files/e7afbad8-ab2c-4c8a-8e21-b9d3a7d36c70 (accessed on 12 October 2019).
- Forte, I.M.; Indovina, P.; Iannuzzi, C.A.; Cirillo, D.; Di Marzo, D.; Barone, D.; Capone, F.; Pentimalli, F.; Giordano, A. Targeted therapy based on p53 reactivation reduces both glioblastoma cell growth and resistance to temozolomide. Int. J. Oncol. 2019, 54, 2189–2199. [Google Scholar] [CrossRef]
- Aparicio-Blanco, J.; Romero, I.A.; Male, D.K.; Slowing, K.; García-García, L.; Torres-Suárez, A.I. Cannabidiol Enhances the Passage of Lipid Nanocapsules across the Blood-Brain Barrier Both In Vitro and In Vivo. Mol. Pharm. 2019, 16, 1999–2010. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The Endocannabinoid System: A Target for Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 747. https://doi.org/10.3390/ijms21030747
Laezza C, Pagano C, Navarra G, Pastorino O, Proto MC, Fiore D, Piscopo C, Gazzerro P, Bifulco M. The Endocannabinoid System: A Target for Cancer Treatment. International Journal of Molecular Sciences. 2020; 21(3):747. https://doi.org/10.3390/ijms21030747
Chicago/Turabian StyleLaezza, Chiara, Cristina Pagano, Giovanna Navarra, Olga Pastorino, Maria Chiara Proto, Donatella Fiore, Chiara Piscopo, Patrizia Gazzerro, and Maurizio Bifulco. 2020. "The Endocannabinoid System: A Target for Cancer Treatment" International Journal of Molecular Sciences 21, no. 3: 747. https://doi.org/10.3390/ijms21030747
APA StyleLaezza, C., Pagano, C., Navarra, G., Pastorino, O., Proto, M. C., Fiore, D., Piscopo, C., Gazzerro, P., & Bifulco, M. (2020). The Endocannabinoid System: A Target for Cancer Treatment. International Journal of Molecular Sciences, 21(3), 747. https://doi.org/10.3390/ijms21030747