Super-Resolution Imaging of Tight and Adherens Junctions: Challenges and Open Questions


Abstract
1. Introduction
1.1. Adherens Junctions and Tight Junctions
1.2. Resolution Limit
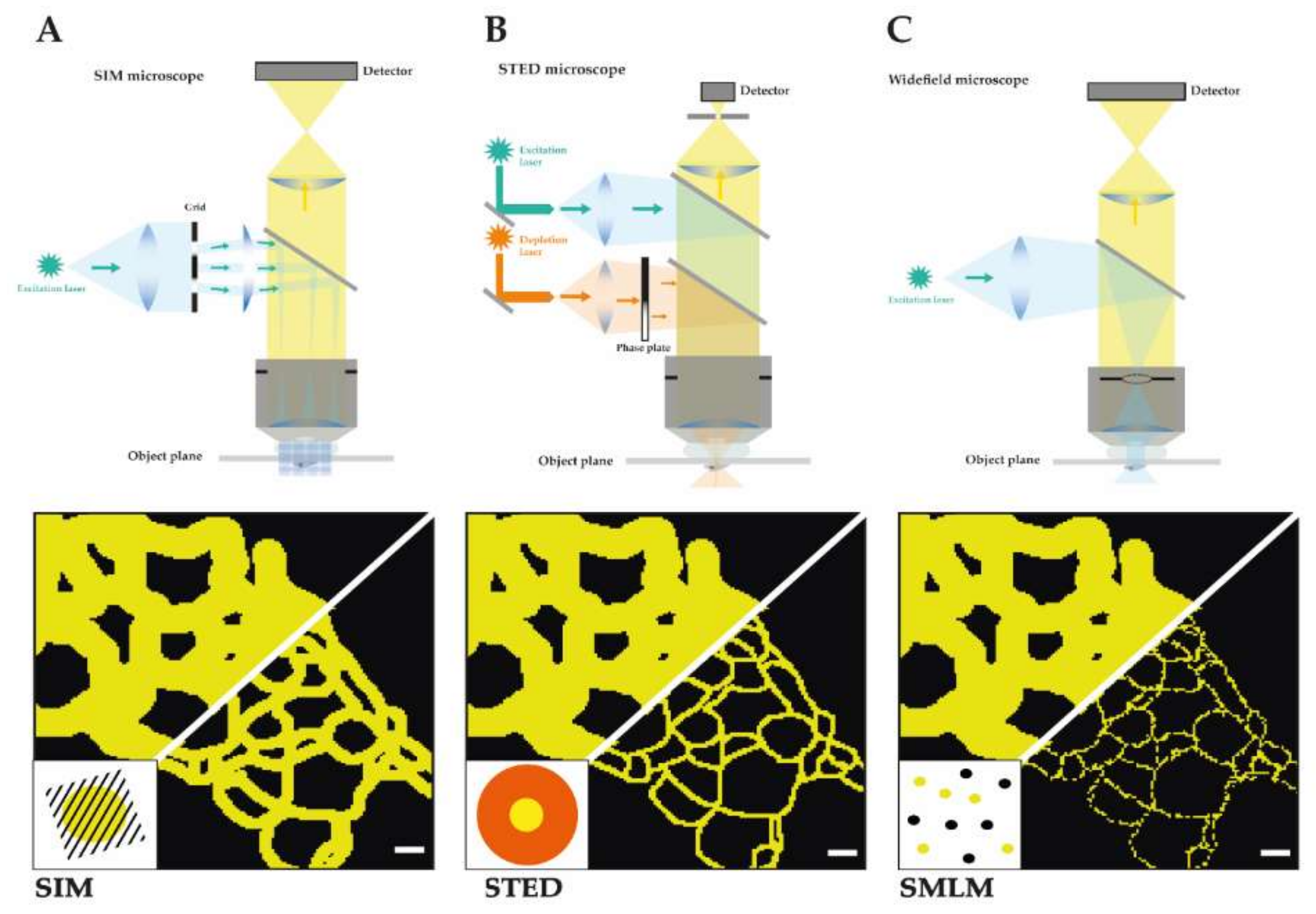
1.3. Structured Illumination Microscopy (SIM)
1.4. Stimulated Emission Depletion (STED)
1.5. Single Molecule Localization Microscopy (SMLM)
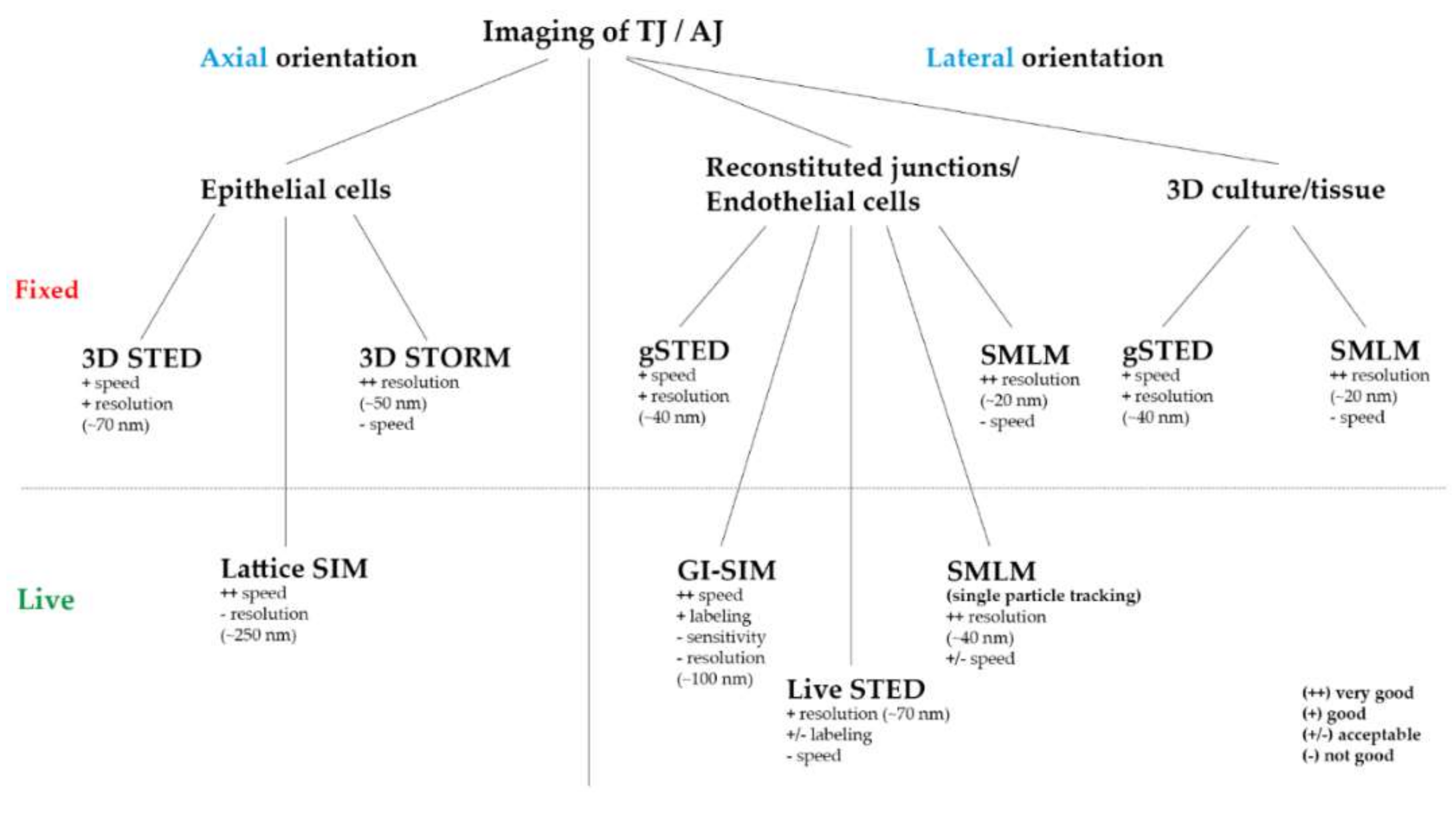
2. Results
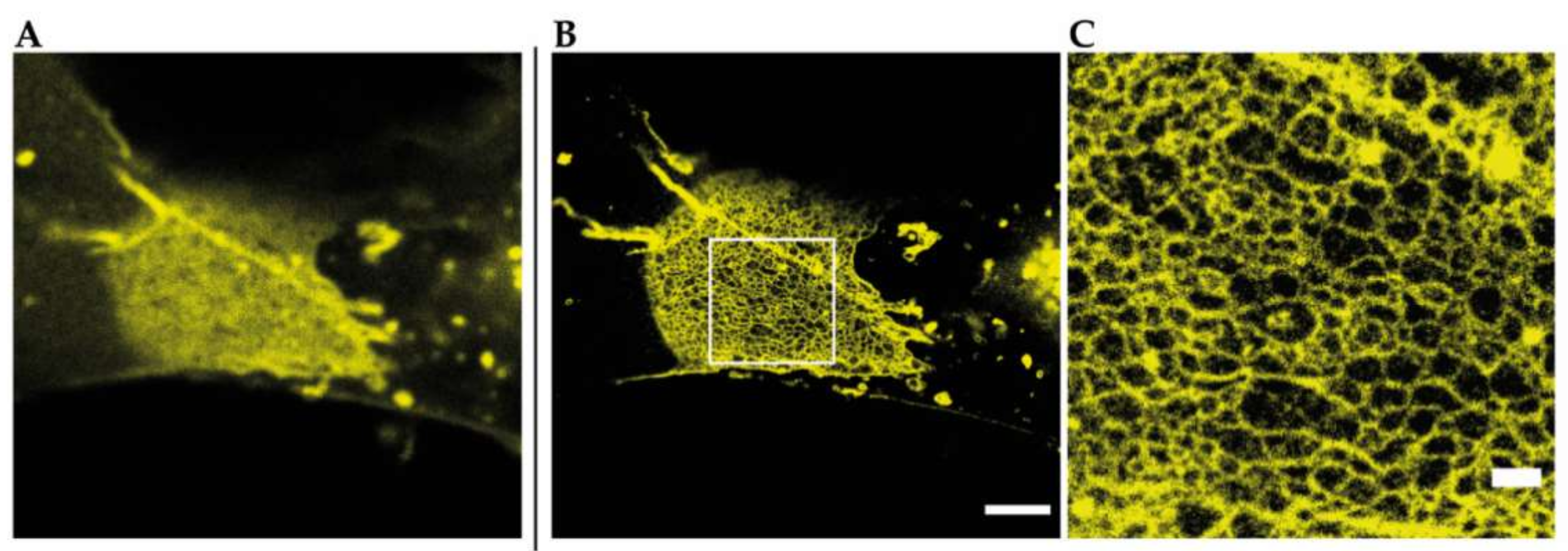
2.1. Super-Resolution Imaging of Adherens Junctions Reveals Nanoscale Clustering and Stratified Intracellular Organization
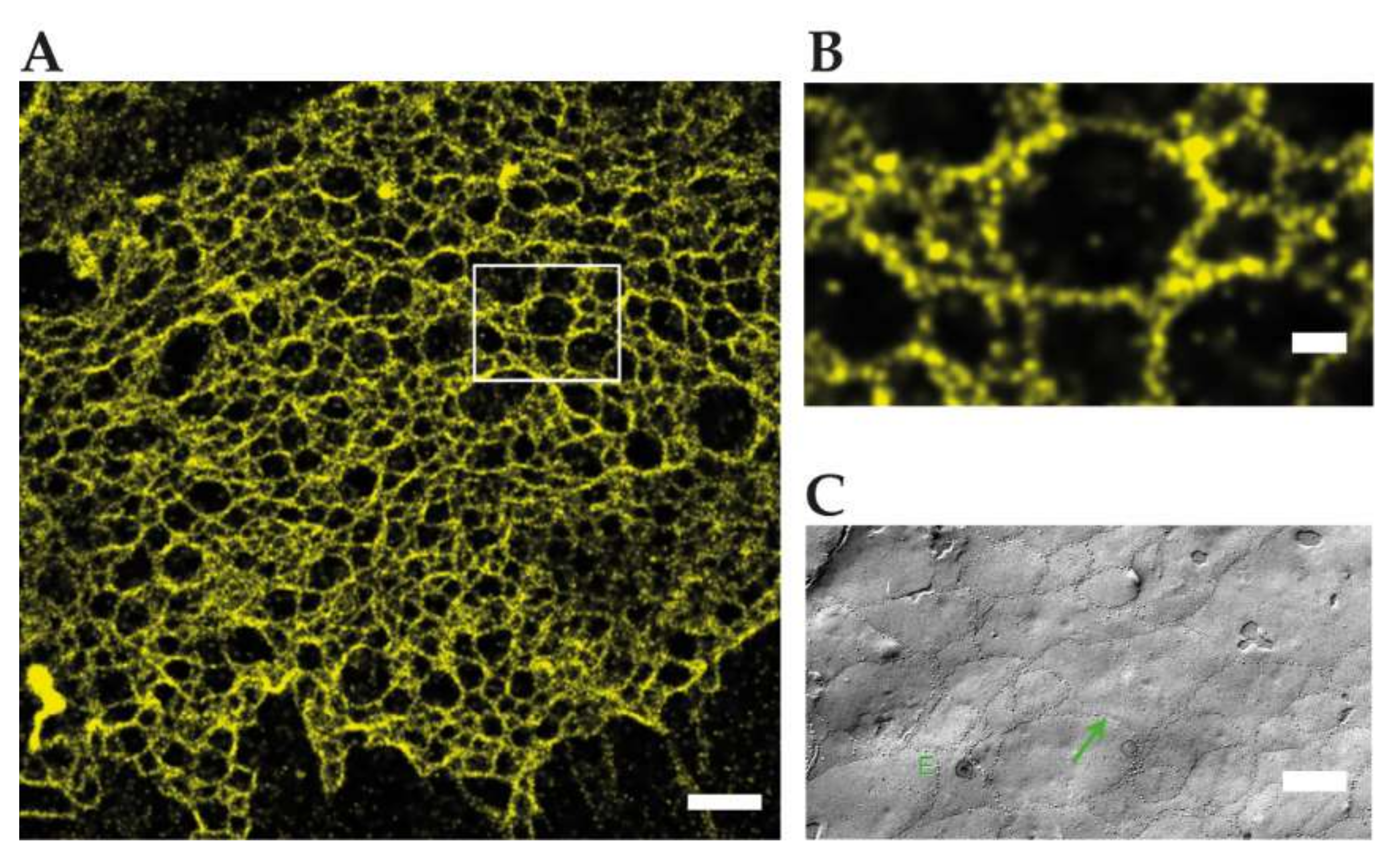
2.2. Super-Resolution Imaging Shows Claudin Meshworks, Strand Dynamics, and Molecular Composition of the TJ
3. Discussion and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AJ | Adherens junction |
| dSTORM | Direct stochastic optical reconstruction microscopy |
| E-cad | E-cad |
| eGFP | Enhanced green fluorescent protein |
| EM | Electron microscopy |
| FA | Focal adhesion |
| FCS | Fluorescence correlation spectroscopy |
| FFEM | Freeze fracture electron microscopy |
| fr | Frame |
| FRAP | Fluorescence recovery after photobleaching |
| FRET | Fluorescence resonance energy transfer |
| gSTED | Gated stimulated emission depletion |
| JAM-A | Junctional adhesion molecule A |
| kW | Kilowatt |
| min | Minute |
| ms | Millisecond |
| NA | Numerical aperture |
| nm | Nanometer |
| PALM | Photoactivated localization microscopy |
| PSF | Point spread function |
| s | Second |
| SIM | Structured illumination microscopy |
| SMLM | Single molecule localization microscopy |
| SRM | Super-resolution microscopy |
| TAMP | Tight junction-associated MARVEL protein |
| TEM | Transmission electron microscopy |
| TJ (bTJ, tTJ) | Tight junction (bicellular TJ, tricellular TJ) |
| YFP | Yellow fluorescent protein |
| ZO-1, -2, -3 | Zonula occludens protein 1, 2, or 3 |
References
- Günzel, D.; Fromm, M. Claudins and other tight junction proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Farquahr, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [PubMed]
- Staehelin, L.A. Further observations on the fine structure of freeze-cleaved tight junctions. J. Cell Sci. 1973, 13, 763–786. [Google Scholar]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (Zonula Occludens) in a variety of epithelia. J. Cell Biol. 1986, 103, 755–766. [Google Scholar] [CrossRef]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Martìn-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998, 142, 117–127. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, J.R. Tight Junction–associated MARVEL Proteins MarvelD3, Tricellulin, and Occludin Have Distinct but Overlapping Functions. Mol. Biol. Cell 2010, 21, 1200–1213. [Google Scholar] [CrossRef]
- Cording, J.; Berg, J.; Kading, N.; Bellmann, C.; Tscheik, C.; Westphal, J.K.; Milatz, S.; Gunzel, D.; Wolburg, H.; Piontek, J.; et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J. Cell Sci. 2013, 126, 554–564. [Google Scholar] [CrossRef]
- Jesaitis, L.A.; Goodenough, D.A. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J. Cell Biol. 1994, 124, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Gonzalez-Mariscal, L.; Matter, K.; Cereijido, M.; Anderson, J.M. Assembly of the tight junction: The role of diacylglycerol. J. Cell Biol. 1993, 123, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Tietgens, A.J.; Anderson, J.M. Visualizing the dynamic coupling of claudin strands to the actin cytoskeleton through ZO-1. Mol. Biol. Cell 2017, 28, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tilney, L.G. Interactions between actin filaments and between actin filaments and membranes in quick-frozen and deeply etched hair cells of the chick ear. J. Cell Biol. 1982, 95, 249–261. [Google Scholar] [CrossRef]
- Madara, J.L.; Pappenheimer, J.R. Structural basis for physiological regulation of paracellular pathways in intestinal epithelia. J. Membr. Biol. 1987, 100, 149–164. [Google Scholar] [CrossRef]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.; Schulzke, J.D.; Amasheh, S.; Günzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef]
- Rosenthal, R.; Günzel, D.; Piontek, J.; Krug, S.M.; Ayala-Torres, C.; Hempel, C.; Theune, D.; Fromm, M. Claudin-15 forms a water channel through the tight junction with distinct function compared to claudin-2. Acta Physiol. 2019, 2018, 1–15. [Google Scholar] [CrossRef]
- Amasheh, S.; Meiri, N.; Gitter, A.H.; Schöneberg, T.; Mankertz, J.; Schulzke, J.D.; Fromm, M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J. Cell Sci. 2002, 115, 4969–4976. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Fanning, A.S.; Anderson, J.M. Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am. J. Physiol. Ren. Physiol. 2003, 285, 1078–1084. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Rogan, S.; Yu, A.; Vidal, L.S.; Holmes, J.; Anderson, J.M. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am. J. Physiol. Ren. Physiol. 2006, 291, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.; Günzel, D.; Krug, S.M.; Amasheh, S.; Rosenthal, R.; Schulzke, J.D.; Fromm, A.; Conrad, M.P. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell. Mol. Life Sci. 2012, 69, 2765–2778. [Google Scholar]
- Hou, J.; Renigunta, A.; Konrad, M.; Gomes, A.S.; Schneeberger, E.E.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J. Clin. Investig. 2008, 118, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Renigunta, A.; Yang, J.; Waldegger, S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. USA 2010, 107, 18010–18015. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Günzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Michael, F. Tricellulin Forms a Barrier to Macromolecules in Tricellular Tight Junctions without Affecting Ion Permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar] [CrossRef]
- Higashi, T.; Tokuda, S.; Kitajiri, S.I.; Masuda, S.; Nakamura, H.; Oda, Y.; Furuse, M. Analysis of the ‘angulin’ proteins LSR, ILDR1 and ILDR2—Tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J. Cell Sci. 2013, 126, 3797. [Google Scholar] [CrossRef]
- Krug, S.M.; Bojarski, C.; Fromm, A.; Lee, I.M.; Dames, P.; Richter, J.F.; Turner, J.R.; Fromm, M.; Schulzke, J.D. Tricellulin is regulated via interleukin-13-receptor α2, affects macromolecule uptake, and is decreased in ulcerative colitis. Mucosal Immunol. 2018, 11, 345–356. [Google Scholar] [CrossRef]
- Huber, A.H.; Weis, W.I.; West, C.D. The Structure of the ß-Catenin/E-Cadherin Complex and the Molecular Basis of Diverse Ligand Recognition by ß-Catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef]
- Pokutta, S.; Weis, W.I. Structure of the Dimerization and ß-Catenin-Binding Region of a-Catenin. Mol. Cell 2000, 5, 533–543. [Google Scholar] [CrossRef]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. α-Catenin Is a Molecular Switch that Binds E-Cadherin-β-Catenin and Regulates Actin-Filament Assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Watabe-Uchida, M.; Uchida, N.; Imamura, Y.; Nagafuchi, A.; Fujimoto, K.; Uemura, T.; Vermeulen, S.; Van Roy, F.; Adamson, E.D.; Takeichi, M. α-Catenin-Vinculin Interaction Functions to Organize the Apical Junctional Complex in Epithelial Cells. J. Cell Biol. 1998, 142, 847–857. [Google Scholar] [CrossRef]
- Weiss, E.E.; Kroemker, M.; Rüdiger, A.; Jockusch, B.M.; Rüdiger, M. Vinculin Is Part of the Cadherin–Catenin Junctional Complex: Complex Formation between α-Catenin and Vinculin. J. Cell Biol. 1998, 141, 755–764. [Google Scholar] [CrossRef]
- Kobielak, A.; Fuchs, E.; Medical, H.H. α-Catenin: At the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 2004, 5, 614–625. [Google Scholar] [CrossRef]
- Nanes, B.A.; Chiasson-MacKenzie, C.; Lowery, A.M.; Ishiyama, N.; Faundez, V.; Ikura, M.; Vincent, P.A.; Kowalczyk, A.P. P120-Catenin Binding Masks an Endocytic Signal Conserved in Classical Cadherins. J. Cell Biol. 2012, 199, 365–380. [Google Scholar] [CrossRef]
- Lecuit, T.; Yap, A.S. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat. Cell Biol. 2015, 17, 533–539. [Google Scholar] [CrossRef]
- Kaufmann, R.; Piontek, J.; Grüll, F.; Kirchgessner, M.; Rossa, J.; Blasig, I.E.; Cremer, C. Visualization and Quantitative Analysis of Reconstituted Tight Junctions Using Localization Microscopy. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Schlingmann, B.; Overgaard, C.E.; Molina, S.A.; Lynn, K.S.; Mitchell, L.A.; Dorsainvil White, S.; Mattheyses, A.L.; Guidot, D.M.; Capaldo, C.T.; Koval, M. Regulation of claudin/zonula occludens-1 complexes by hetero-claudin interactions. Nat. Commun. 2016, 7, 12276. [Google Scholar] [CrossRef]
- Quang, B.A.T.; Mani, M.; Markova, O.; Lecuit, T.; Lenne, P.F. Principles of E-cadherin supramolecular organization in vivo. Curr. Biol. 2013, 23, 2197–2207. [Google Scholar]
- Wu, Y.; Kanchanawong, P.; Zaidel-Bar, R. Actin-Delimited Adhesion-Independent Clustering of E-Cadherin Forms the Nanoscale Building Blocks of Adherens Junctions. Dev. Cell 2015, 32, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Bertocchi, C.; Wang, Y.; Ravasio, A.; Hara, Y.; Wu, Y.; Sailov, T.; Baird, M.A.; Davidson, M.W.; Zaidel-bar, R.; Toyama, Y.; et al. Nanoscale architecture of cadherin-based cell adhesions. Nat. Cell Biol. 2017, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. Mikrosk. Anat. 1873, 9, 413–418. [Google Scholar] [CrossRef]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Kim, D.; So, P.T.C. Extended resolution wide-field optical imaging: Internal reflection fluorescence microscopy. Opt. Lett. 2006, 31, 945–947. [Google Scholar] [CrossRef]
- Guo, M.; Chandris, P.; Giannini, J.P.; Trexler, A.J.; Fischer, R.; Chen, J.; Vishwasrao, H.D.; Rey-Suarez, I.; Wu, Y.; Wu, X.; et al. Single-shot super-resolution total internal reflection fluorescence microscopy. Nat. Methods 2018, 15, 425–428. [Google Scholar] [CrossRef]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing Intracellular Organelle and Cytoskeletal Interactions at Nanoscale Resolution on Millisecond Timescales. Cell 2018, 175, 1430–1442. [Google Scholar] [CrossRef]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef]
- Li, D.; Shao, L.; Chen, B.C.; Zhang, X.; Zhang, M.; Moses, B.; Milkie, D.E.; Beach, J.R.; Hammer, J.A.; Pasham, M.; et al. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 2015, 349, aab3500. [Google Scholar] [CrossRef]
- De Luca, G.M.R.; Breedijk, R.M.P.; Brandt, R.A.J.; Zeelenberg, C.H.C.; de Jong, B.E.; Timmermans, W.; Azar, L.N.; Hoebe, R.A.; Stallinga, S.; Manders, E.M.M. Re-scan confocal microscopy: Scanning twice for better resolution. Biomed. Opt. Express 2013, 4, 2644. [Google Scholar] [CrossRef] [PubMed]
- Huff, J. The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12, 1205. [Google Scholar] [CrossRef]
- Wegel, E.; Göhler, A.; Lagerholm, B.C.; Wainman, A.; Uphoff, S.; Kaufmann, R.; Dobbie, I.M. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Sci. Rep. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780. [Google Scholar] [CrossRef]
- Vicidomini, G.; Moneron, G.; Han, K.Y.; Westphal, V.; Ta, H.; Reuss, M.; Engelhardt, J.; Eggeling, C.; Hell, S.W. Sharper low-power STED nanoscopy by time gating. Nat. Methods 2011, 8, 571–575. [Google Scholar] [CrossRef]
- Curdt, F.; Herr, S.J.; Lutz, T.; Schmidt, R.; Engelhardt, J.; Sahl, S.J.; Hell, S.W. isoSTED nanoscopy with intrinsic beam alignment. Opt. Express 2015, 23, 30891–30903. [Google Scholar] [CrossRef]
- Grimm, J.B.; Muthusamy, A.K.; Liang, Y.; Brown, T.A.; Lemon, W.C.; Patel, R.; Lu, R.; Macklin, J.J.; Keller, P.J.; Ji, N.; et al. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987. [Google Scholar] [CrossRef]
- Bottanelli, F.; Kromann, E.B.; Allgeyer, E.S.; Erdmann, R.S.; Baguley, S.W.; Sirinakis, G.; Schepartz, A.; Baddeley, D.; Toomre, D.K.; Rothman, J.E.; et al. Two-colour live-cell nanoscale imaging of intracellular targets. Nat. Commun. 2015, 7, 1–5. [Google Scholar] [CrossRef]
- Maraspini, R.; Wang, C.-H.; Honigmann, A. Optimization of 2D and 3D cell culture to study membrane organization with STED microscopy. J. Phys. Appl. Phys. 2020, 53, 014001. [Google Scholar] [CrossRef]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N.G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.; Huang, B.; Dempsey, G.T.; Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 2007, 317, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Hosy, E.; Levet, F.; Constals, A.; Schulze, K.; Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E.; Tampe, R.; Choquet, D.; et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 2010, 99, 1303–1310. [Google Scholar] [CrossRef]
- Vaughan, J.C.; Jia, S.; Zhuang, X. Ultrabright photoactivatable fluorophores created by reductive caging. Nat. Methods 2012, 9, 1181–1184. [Google Scholar] [CrossRef]
- Jones, S.A.; Shim, S.H.; He, J.; Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 2011, 8, 499–505. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-Dimensional Super-Resolution Reconstruction Microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef]
- Juette, M.F.; Gould, T.J.; Lessard, M.D.; Mlodzianoski, M.J.; Nagpure, B.S.; Bennett, B.T.; Hess, S.T.; Bewersdorf, J. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 2008, 5, 527–529. [Google Scholar] [CrossRef]
- Shtengel, G.; Galbraith, J.A.; Galbraith, C.G.; Lippincott-Schwartz, J.; Gillette, J.M.; Manley, S.; Sougrat, R.; Waterman, C.M.; Kanchanawong, P.; Davidson, M.W.; et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 3125–3130. [Google Scholar] [CrossRef]
- Xu, K.; Babcock, H.P.; Zhuang, X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat. Methods 2012, 9, 185–188. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Sage, D.; Pham, T.A.; Babcock, H.; Lukes, T.; Pengo, T.; Chao, J.; Velmurugan, R.; Herbert, A.; Agrawal, A.; Colabrese, S.; et al. Super-resolution fight club: Assessment of 2D and 3D single-molecule localization microscopy software. Nat. Methods 2019, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Maître, J.L.; Berthoumieux, H.; Krens, S.F.G.; Salbreux, G.; Jülicher, F.; Paluch, E.; Heisenberg, C.P. Adhesion functions in cell sorting by mechanically coupling the cortices of adhering cells. Science 2012, 338, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.L.; Nelson, W.J.; Smith, S.J. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 1996, 135, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Case, L.B.; Baird, M.A.; Shtengel, G.; Campbell, S.L.; Hess, H.F.; Davidson, M.W.; Waterman, C.M. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 2015, 17, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Van Zanten, T.S.; Cambi, A.; Koopman, M.; Joosten, B.; Figdor, C.G.; Garcia-Parajo, M.F. Hotspots of GPI-anchored proteins and integrin nanoclusters function as nucleation sites for cell adhesion. Proc. Natl. Acad. Sci. USA 2009, 106, 18557–18562. [Google Scholar] [CrossRef]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; White, H.; Gillette, J.; Olenych, S.; Davidson, M.W.; Betzig, E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 20308–20313. [Google Scholar] [CrossRef]
- Grashoff, C.; Hoffman, B.D.; Brenner, M.D.; Zhou, R.; Parsons, M.; Yang, M.T.; McLean, M.A.; Sligar, S.G.; Chen, C.S.; Ha, T.; et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 2010, 466, 263–266. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsui, C.; Furuse, K.; Mimori-Kiyosue, Y.; Furuse, M.; Tsukita, S. Dynamic behavior of paired claudin strands within apposing plasma membranes. Proc. Natl. Acad. Sci. USA 2003, 100, 3971–3976. [Google Scholar] [CrossRef]
- Cording, J.; Arslan, B.; Staat, C.; Dithmer, S.; Krug, S.M.; Krüger, A.; Berndt, P.; Günther, R.; Winkler, L.; Blasig, I.E.; et al. Trictide, a tricellulin-derived peptide to overcome cellular barriers. Ann. N. Y. Acad. Sci. 2017, 1405, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Liss, V.; Barlag, B.; Nietschke, M.; Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lukinavičius, G.; Umezawa, K.; Olivier, N.; Honigmann, A.; Yang, G.; Plass, T.; Mueller, V.; Reymond, L.; Corrêa, I.R.; Luo, Z.G.; et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SIM | gSTED | SMLM | |
|---|---|---|---|
| Resolution limit | 100 nm (lateral) 250 nm (axial) | 40 nm (lateral) 70 nm (axial) | 20 nm (lateral) 50 nm (axial) |
| Multi-color | 4 colors | 2–4 colors | 2–4 colors |
| Sample preparation | Standard | Standard | Standard |
| Live imaging | Yes | Yes | Yes |
| Imaging speed | 10 ms–10 s | 10 s–5 min | 10 min |
| Illumination power | 10–100 W/cm2 | 1–100 kW/cm2 | 1–100 kW/cm2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonschior, H.; Haucke, V.; Lehmann, M. Super-Resolution Imaging of Tight and Adherens Junctions: Challenges and Open Questions. Int. J. Mol. Sci. 2020, 21, 744. https://doi.org/10.3390/ijms21030744
Gonschior H, Haucke V, Lehmann M. Super-Resolution Imaging of Tight and Adherens Junctions: Challenges and Open Questions. International Journal of Molecular Sciences. 2020; 21(3):744. https://doi.org/10.3390/ijms21030744
Chicago/Turabian StyleGonschior, Hannes, Volker Haucke, and Martin Lehmann. 2020. "Super-Resolution Imaging of Tight and Adherens Junctions: Challenges and Open Questions" International Journal of Molecular Sciences 21, no. 3: 744. https://doi.org/10.3390/ijms21030744
APA StyleGonschior, H., Haucke, V., & Lehmann, M. (2020). Super-Resolution Imaging of Tight and Adherens Junctions: Challenges and Open Questions. International Journal of Molecular Sciences, 21(3), 744. https://doi.org/10.3390/ijms21030744