Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function



Abstract
1. Introduction
2. HDL Metabolism, Structure, and Composition
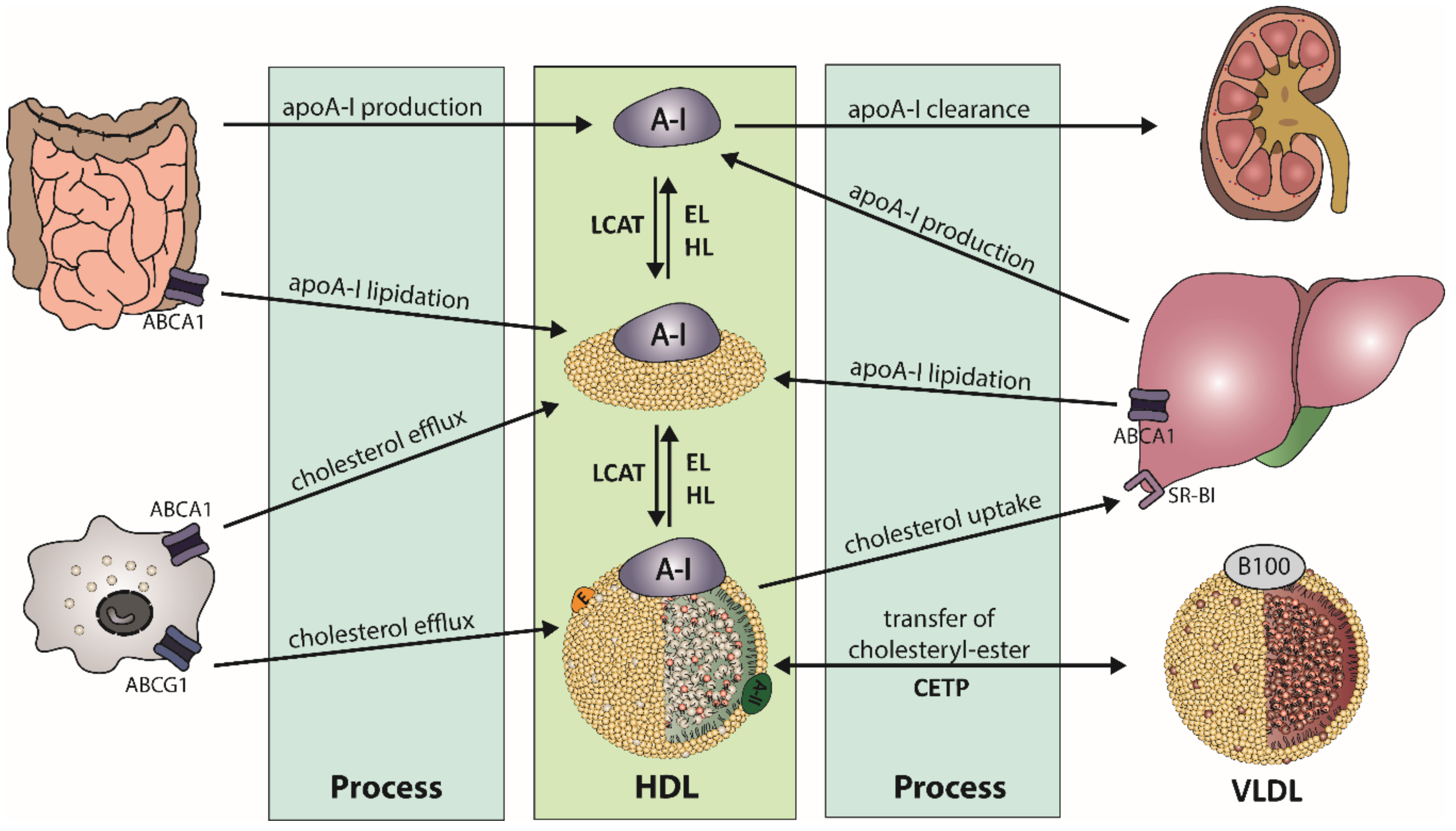
2.1. HDL Metabolism
2.2. HDL Structure and Composition
2.3. HDL Subclasses
2.4. Important Functions of HDL
3. HDL-C-Raising Therapies and Cardiovascular Outcome
4. Obesity Alters HDL-C Levels
5. Obesity, HDL, and Cardiovascular Risk
5.1. Obesity Leads to a Shift in HDL Subclass Distribution
5.2. Obesity Affects HDL Function
5.3. Adiponectin and HDL
5.4. Obesity and HDL-Associated Sphingosine-1-Phosphate (S1P)
6. Bariatric Surgery Improves HDL Levels and Function
7. Effects of Pharmacological Anti-Obesity Interventions on HDL Levels and Function
8. Effects of Dietary Approaches on HDL Levels and Function
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Knight, J.A. Diseases and Disorders Associated with Excess Body Weight. Ann. Clin. Lab. Sci. 2011, 41, 107–121. [Google Scholar] [PubMed]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Steg, P.G.; Ravisy, J.; Lorgis, L.; Laurent, Y.; Sicard, P.; Janin-Manificat, L.; Beer, J.C.; Makki, H.; Lagrost, A.C.; et al. Relation Between Body Mass Index, Waist Circumference, and Death After Acute Myocardial Infarction. Circulation 2008, 118, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 587–591. [Google Scholar] [CrossRef]
- Narkiewicz, K. Obesity and hypertension—The issue is more complex than we thought. Nephrol. Dial. Transplant. 2006, 21, 264–267. [Google Scholar] [CrossRef]
- Bays, H.E.; Toth, P.P.; Kris-Etherton, P.M.; Abate, N.; Aronne, L.J.; Brown, W.V.; Gonzalez-Campoy, J.M.; Jones, S.R.; Kumar, R.; La Forge, R.; et al. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 304–383. [Google Scholar] [CrossRef]
- Rashid, S.; Genest, J. Effect of obesity on high-density lipoprotein metabolism. Obesity 2007, 15, 2875–2888. [Google Scholar] [CrossRef]
- Woudberg, N.J.; Lecour, S.; Goedecke, J.H. HDL Subclass Distribution Shifts with Increasing Central Adiposity. Available online: https://www.hindawi.com/journals/jobe/2019/2107178/ (accessed on 10 August 2020).
- Wang, H.; Peng, D.-Q. New insights into the mechanism of low high-density lipoprotein cholesterol in obesity. Lipids Health Dis. 2011, 10, 176. [Google Scholar] [CrossRef]
- Rader, D.J.; Tall, A.R. Is it time to revise the HDL cholesterol hypothesis? Nat. Med. 2012, 18, 1344–1346. [Google Scholar] [CrossRef]
- Parks, J.S.; Chung, S.; Shelness, G.S. Hepatic ABC transporters and triglyceride metabolism. Curr. Opin. Lipidol. 2012, 23, 196–200. [Google Scholar] [CrossRef]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Norum, K.R.; Remaley, A.T.; Miettinen, H.E.; Strøm, E.H.; Balbo, B.E.P.; Sampaio, C.A.T.L.; Wiig, I.; Kuivenhoven, J.A.; Calabresi, L.; Tesmer, J.J.; et al. Lecithin:cholesterol acyltransferase: Symposium on 50 years of biomedical research from its discovery to latest findings. J. Lipid Res. 2020, 61, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Sorci-Thomas, M.G.; Bhat, S.; Thomas, M.J. Activation of lecithin: Cholesterol acyltransferase by HDL ApoA-I central helices. Clin. Lipidol. 2009, 4, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Schaefer, E.J.; Anthanont, P.; Asztalos, B.F. HDL metabolism, composition, function and deficiency. Curr. Opin. Lipidol. 2014, 25, 194–199. [Google Scholar] [CrossRef]
- Kozarsky, K.F.; Donahee, M.H.; Rigotti, A.; Iqbal, S.N.; Edelman, E.R.; Krieger, M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature 1997, 387, 414–417. [Google Scholar] [CrossRef]
- Duong, M.; Psaltis, M.; Rader, D.J.; Marchadier, D.; Barter, P.J.; Rye, K.-A. Evidence that hepatic lipase and endothelial lipase have different substrate specificities for high-density lipoprotein phospholipids. Biochemistry 2003, 42, 13778–13785. [Google Scholar] [CrossRef]
- Albers, J.J.; Vuletic, S.; Cheung, M.C. Role of Plasma Phospholipid Transfer Protein in Lipid and Lipoprotein Metabolism. Biochim. Biophys. Acta 2012, 1821, 345–357. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Toth, P.P.; Barter, P.J.; Rosenson, R.S.; Boden, W.E.; Chapman, M.J.; Cuchel, M.; D’Agostino, R.B.; Davidson, M.H.; Davidson, W.S.; Heinecke, J.W.; et al. High-density lipoproteins: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 484–525. [Google Scholar] [CrossRef]
- Kannel, W.B.; Dawber, T.R.; Friedman, G.D.; Glennon, W.E.; Mcnamara, P.M. Risk Factors in coronary heart disease. An evaluation of several serum lipids as predictors of coronary heart disease; The Framingham Study. Ann. Intern. Med. 1964, 61, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [PubMed]
- Birner-Gruenberger, R.; Schittmayer, M.; Holzer, M.; Marsche, G. Understanding high-density lipoprotein function in disease: Recent advances in proteomics unravel the complexity of its composition and biology. Prog. Lipid Res. 2014, 56, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [CrossRef] [PubMed]
- Kostner, G.; Alaupovic, P. Composition and structure of plasma lipoproteins. Separation and quantification of the lipoprotein families occurring in the high density lipoproteins of human plasma. Biochemistry 1972, 11, 3419–3428. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef]
- Fernández-Hernando, C. Antiatherogenic Properties of High-Density Lipoprotein–Enriched MicroRNAs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, e13–e14. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Antiatherogenic small, dense HDL--guardian angel of the arterial wall? Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 144–153. [Google Scholar] [CrossRef]
- Duriez, P.; Fruchart, J.C. High-density lipoprotein subclasses and apolipoprotein A-I. Clin. Chim. Acta Int. J. Clin. Chem. 1999, 286, 97–114. [Google Scholar] [CrossRef]
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and Anti-Inflammatory Role of Paraoxonase 1: Implication in Arteriosclerosis Diseases. N. Am. J. Med. Sci. 2012, 4, 523–532. [Google Scholar] [CrossRef]
- Serna, J.; García-Seisdedos, D.; Alcázar, A.; Lasunción, M.Á.; Busto, R.; Pastor, Ó. Quantitative lipidomic analysis of plasma and plasma lipoproteins using MALDI-TOF mass spectrometry. Chem. Phys. Lipids 2015, 189, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Yetukuri, L.; Söderlund, S.; Koivuniemi, A.; Seppänen-Laakso, T.; Niemelä, P.S.; Hyvönen, M.; Taskinen, M.-R.; Vattulainen, I.; Jauhiainen, M.; Oresic, M. Composition and lipid spatial distribution of HDL particles in subjects with low and high HDL-cholesterol. J. Lipid Res. 2010, 51, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J. Lipid Res. 2009, 50, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Jonas, A.; Kézdy, K.E.; Wald, J.H. Defined apolipoprotein A-I conformations in reconstituted high density lipoprotein discs. J. Biol. Chem. 1989, 264, 4818–4824. [Google Scholar]
- Woudberg, N.J.; Pedretti, S.; Lecour, S.; Schulz, R.; Vuilleumier, N.; James, R.W.; Frias, M.A. Pharmacological Intervention to Modulate HDL: What Do We Target? Front. Pharmacol. 2018, 8, 989. [Google Scholar] [CrossRef]
- De la Llera-Moya, M.; Drazul-Schrader, D.; Asztalos, B.F.; Cuchel, M.; Rader, D.J.; Rothblat, G.H. The Ability to Promote Efflux via ABCA1 Determines the Capacity of Serum Specimens with Similar HDL-C to Remove Cholesterol from Macrophages. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 796–801. [Google Scholar] [CrossRef]
- Davidson, W.S.; Silva, R.A.G.D.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef]
- Nofer, J.R.; Van Der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef]
- Argraves, K.M.; Gazzolo, P.J.; Groh, E.M.; Wilkerson, B.A.; Matsuura, B.S.; Twal, W.O.; Hammad, S.M.; Argraves, W.S. High Density Lipoprotein-associated Sphingosine 1-Phosphate Promotes Endothelial Barrier Function. J. Biol. Chem. 2008, 283, 25074–25081. [Google Scholar] [CrossRef]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Kastelein, J.P.; Hayden, M.R. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J. Lipid Res. 2001, 42, 1717–1726. [Google Scholar] [PubMed]
- Tang, C.; Oram, J.F. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2009, 1791, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldán, Á.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131. [Google Scholar] [CrossRef]
- Rothblat, G.H.; Phillips, M.C. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr. Opin. Lipidol. 2010, 21, 229. [Google Scholar] [CrossRef]
- Phillips, M.C.; Johnson, W.J.; Rothblat, G.H. Mechanisms and Consequences of Cellular Cholesterol Exchange and Transfer. Available online: https://pubmed.ncbi.nlm.nih.gov/3297153/ (accessed on 14 October 2020).
- Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules 2020, 10, 1348. [Google Scholar] [CrossRef]
- Assmann, G.; Gotto, A.M. HDL Cholesterol and Protective Factors in Atherosclerosis. Circulation 2004, 109, III-8–III-14. [Google Scholar] [CrossRef]
- Nofer, J.-R.; Assmann, G. Atheroprotective effects of high-density lipoprotein-associated lysosphingolipids. Trends Cardiovasc. Med. 2005, 15, 265–271. [Google Scholar] [CrossRef]
- Nofer, J.-R.; Kehrel, B.; Fobker, M.; Levkau, B.; Assmann, G.; von Eckardstein, A. HDL and arteriosclerosis: Beyond reverse cholesterol transport. Atherosclerosis 2002, 161, 1–16. [Google Scholar] [CrossRef]
- Navab, M.; Ananthramaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Fonarow, G.C.; Vahabzadeh, K.; Hama, S.; Hough, G.; Kamranpour, N.; et al. The oxidation hypothesis of atherogenesis: The role of oxidized phospholipids and HDL. J. Lipid Res. 2004, 45, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Callegari, E.; Inoue, H.; Catapano, A.L. HDL3 Induces Cyclooxygenase-2 Expression and Prostacyclin Release in Human Endothelial Cells Via a p38 MAPK/CRE-Dependent Pathway: Effects on COX-2/PGI-Synthase Coupling. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.; Förster, W. Influence of human low density and high density lipoprotein cholesterol on the in vitro prostaglandin I2 synthetase activity. Biochim. Biophys. Acta BBA-Lipids Lipid Metab. 1980, 620, 352–355. [Google Scholar] [CrossRef]
- Drew, B.G.; Fidge, N.H.; Gallon-Beaumier, G.; Kemp, B.E.; Kingwell, B.A. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2004, 101, 6999–7004. [Google Scholar] [CrossRef]
- Li, X.-A.; Titlow, W.B.; Jackson, B.A.; Giltiay, N.; Nikolova-Karakashian, M.; Uittenbogaard, A.; Smart, E.J. High density lipoprotein binding to scavenger receptor, Class B, type I activates endothelial nitric-oxide synthase in a ceramide-dependent manner. J. Biol. Chem. 2002, 277, 11058–11063. [Google Scholar] [CrossRef]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef]
- Van Linthout, S.; Spillmann, F.; Lorenz, M.; Meloni, M.; Jacobs, F.; Egorova, M.; Stangl, V.; De Geest, B.; Schultheiss, H.P.; Tschope, C. Vascular-Protective Effects of High-Density Lipoprotein Include the Downregulation of the Angiotensin II Type 1 Receptor. Hypertension 2009, 53, 682–687. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, H.; Rizzo, A.N.; Zhang, W.; Mai, Y.; Ye, M. Endothelial nitric oxide synthase dimerization is regulated by heat shock protein 90 rather than by phosphorylation. PLoS ONE 2014, 9, e105479. [Google Scholar] [CrossRef]
- Panzenböck, U.; Stocker, R. Formation of methionine sulfoxide-containing specific forms of oxidized high-density lipoproteins. Biochim. Biophys. Acta BBA-Proteins Proteom. 2005, 1703, 171–181. [Google Scholar] [CrossRef]
- Garner, B.; Waldeck, A.R.; Witting, P.K.; Rye, K.A.; Stocker, R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J. Biol. Chem. 1998, 273, 6088–6095. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Billecke, S.; Sorenson, R.; Bisgaier, C.; Newton, R.; Rosenblat, M.; Erogul, J.; Hsu, C.; Dunlop, C.; La Du, B. Paraoxonase active site required for protection against LDL oxidation involves its free sulfhydryl group and is different from that required for its arylesterase/paraoxonase activities: Selective action of human paraoxonase allozymes Q and R. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Goulinet, S.; Chapman, M.J. Plasma LDL and HDL subspecies are heterogenous in particle content of tocopherols and oxygenated and hydrocarbon carotenoids. Relevance to oxidative resistance and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 786–796. [Google Scholar] [CrossRef]
- Navab, M.; Imes, S.S.; Hama, S.Y.; Hough, G.P.; Ross, L.A.; Bork, R.W.; Valente, A.J.; Berliner, J.A.; Drinkwater, D.C.; Laks, H. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J. Clin. Investig. 1991, 88, 2039–2046. [Google Scholar] [CrossRef]
- Cockerill, G.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1987–1994. [Google Scholar] [CrossRef]
- Calabresi, L.; Franceschini, G.; Sirtori, C.R.; De Palma, A.; Saresella, M.; Ferrante, P.; Taramelli, D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem. Biophys. Res. Commun. 1997, 238, 61–65. [Google Scholar] [CrossRef]
- Bursill, C.A.; Castro, M.L.; Beattie, D.T.; Nakhla, S.; van der Vorst, E.; Heather, A.K.; Barter, P.J.; Rye, K.-A. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1773–1778. [Google Scholar] [CrossRef]
- Wilson, P.W.; Abbott, R.D.; Castelli, W.P. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arterioscler. Off. J. Am. Heart Assoc. Inc. 1988, 8, 737–741. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L.; Wang, N. The failure of torcetrapib: Was it the molecule or the mechanism? Arterioscler. Thromb. Vasc. Biol. 2007, 27, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tardif, J.-C.; Nicholls, S.J.; Revkin, J.H.; Shear, C.L.; Duggan, W.T.; Ruzyllo, W.; Bachinsky, W.B.; Lasala, G.P.; Lasala, G.P.; et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N. Engl. J. Med. 2007, 356, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.P.; van Leuven, S.I.; Burgess, L.; Evans, G.W.; Kuivenhoven, J.A.; Barter, P.J.; Revkin, J.H.; Grobbee, D.E.; Riley, W.A.; Shear, C.L.; et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 1620–1630. [Google Scholar] [CrossRef]
- Marsche, G.; Saemann, M.D.; Heinemann, A.; Holzer, M. Inflammation alters HDL composition and function: Implications for HDL-raising therapies. Pharmacol. Ther. 2013, 137, 341–351. [Google Scholar] [CrossRef]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and reduces VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef]
- Sacks, F.M.; Rudel, L.L.; Conner, A.; Akeefe, H.; Kostner, G.; Baki, T.; Rothblat, G.; de la Llera-Moya, M.; Asztalos, B.; Perlman, T.; et al. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J. Lipid Res. 2009, 50, 894–907. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Smits, L.P.; Stefanutti, C.; Soran, H.; Kwok, S.; de Graaf, J.; Gaudet, D.; Keyserling, C.H.; Klepp, H.; Frick, J.; et al. The effect of an apolipoprotein A-I–containing high-density lipoprotein–mimetic particle (CER-001) on carotid artery wall thickness in patients with homozygous familial hypercholesterolemia: The Modifying Orphan Disease Evaluation (MODE) study. Am. Heart J. 2015, 169, 736–742.e1. [Google Scholar] [CrossRef]
- Parolini, C.; Marchesi, M.; Lorenzon, P.; Castano, M.; Balconi, E.; Miragoli, L.; Chaabane, L.; Morisetti, A.; Lorusso, V.; Martin, B.J.; et al. Dose-Related Effects of Repeated ETC-216 (Recombinant Apolipoprotein A-IMilano/1-Palmitoyl-2-Oleoyl Phosphatidylcholine Complexes) Administrations on Rabbit Lipid-Rich Soft Plaques: In Vivo Assessment by Intravascular Ultrasound and Magnetic Resonance Imaging. J. Am. Coll. Cardiol. 2008, 51, 1098–1103. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Grégoire, J.; L’Allier, P.L.; Ibrahim, R.; Lespérance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.-A.; et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. JAMA 2007, 297, 1675–1682. [Google Scholar] [CrossRef]
- Waksman, R.; Torguson, R.; Kent, K.M.; Pichard, A.D.; Suddath, W.O.; Satler, L.F.; Martin, B.D.; Perlman, T.J.; Maltais, J.-A.B.; Weissman, N.J.; et al. A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J. Am. Coll. Cardiol. 2010, 55, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Martinez, I.; Sourlas, A.; Bouza, K.V.; Campos, F.N.; Torres, V.; Montan, P.D.; Guzman, E. High-density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease. Drugs Context 2018, 7, 212525. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Christodoulidis, G.; Cheng, J.; Lerakis, S.; Vittorio, T.J. High-Density Lipoprotein Functionality in Coronary Artery Disease. Am. J. Med. Sci. 2014, 347, 504–508. [Google Scholar] [CrossRef]
- Ashen, M.D.; Blumenthal, R.S. Clinical practice. Low HDL cholesterol levels. N. Engl. J. Med. 2005, 353, 1252–1260. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Queipo-Ortuño, M.I.; Fernandez-Garcia, D.; Gomez-Huelgas, R.; Tinahones, F.J.; Cardona, F. Adipose Tissue Gene Expression of Factors Related to Lipid Processing in Obesity. PLoS ONE 2011, 6, e24783. [Google Scholar] [CrossRef]
- Walton, R.G.; Zhu, B.; Unal, R.; Spencer, M.; Sunkara, M.; Morris, A.J.; Charnigo, R.; Katz, W.S.; Daugherty, A.; Howatt, D.A.; et al. Increasing Adipocyte Lipoprotein Lipase Improves Glucose Metabolism in High Fat Diet-induced Obesity. J. Biol. Chem. 2015, 290, 11547–11556. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef]
- Taskinen, M.-R.; Nikkilä, E.A. Lipoprotein lipase of adipose tissue and skeletal muscle in human obesity: Response to glucose and to semistarvation. Metabolism 1981, 30, 810–817. [Google Scholar] [CrossRef]
- Feng, R.; Luo, C.; Li, C.; Du, S.; Okekunle, A.P.; Li, Y.; Chen, Y.; Zi, T.; Niu, Y. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: A case—Control study. Lipids Health Dis. 2017, 16, 165. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N. Insulin resistance and cardiovascular disease. J. Clin. Investig. 2000, 106, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Uffelman, K.D.; Lewis, G.F. The mechanism of HDL lowering in hypertriglyceridemic, insulin-resistant states. J. Diabetes Complicat. 2002, 16, 24–28. [Google Scholar] [CrossRef]
- Carr, M.C.; Hokanson, J.E.; Zambon, A.; Deeb, S.S.; Barrett, P.H.; Purnell, J.Q.; Brunzell, J.D. The contribution of intraabdominal fat to gender differences in hepatic lipase activity and low/high density lipoprotein heterogeneity. J. Clin. Endocrinol. Metab. 2001, 86, 2831–2837. [Google Scholar] [CrossRef]
- Chatterjee, C.; Sparks, D.L. Hepatic Lipase, High Density Lipoproteins, and Hypertriglyceridemia. Am. J. Pathol. 2011, 178, 1429–1433. [Google Scholar] [CrossRef]
- Blades, B.; Vega, G.L.; Grundy, S.M. Activities of lipoprotein lipase and hepatic triglyceride lipase in postheparin plasma of patients with low concentrations of HDL cholesterol. Arterioscler. Thromb. J. Vasc. Biol. 1993, 13, 1227–1235. [Google Scholar] [CrossRef]
- Rashid, S.; Barrett, P.H.R.; Uffelman, K.D.; Watanabe, T.; Adeli, K.; Lewis, G.F. Lipolytically Modified Triglyceride-Enriched HDLs Are Rapidly Cleared from the Circulation. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 483–487. [Google Scholar] [CrossRef][Green Version]
- Lönnqvist, F.; Nordfors, L.; Jansson, M.; Thörne, A.; Schalling, M.; Arner, P. Leptin secretion from adipose tissue in women. Relationship to plasma levels and gene expression. J. Clin. Investig. 1997, 99, 2398–2404. [Google Scholar] [CrossRef]
- Singh, P.; Peterson, T.E.; Sert-Kuniyoshi, F.H.; Glenn, J.A.; Davison, D.E.; Romero-Corral, A.; Pusalavidyasagar, S.; Jensen, M.D.; Somers, V.K. Leptin signaling in adipose tissue: Role in lipid accumulation and weight gain. Circ. Res. 2012, 111, 599–603. [Google Scholar] [CrossRef]
- Wu, D.M.; Shen, M.H.; Chu, N.F. Relationship between plasma leptin levels and lipid profiles among school children in Taiwan--the Taipei Children Heart Study. Eur. J. Epidemiol. 2001, 17, 911–916. [Google Scholar] [CrossRef]
- Rainwater, D.L.; Comuzzie, A.G.; VandeBerg, J.L.; Mahaney, M.C.; Blangero, J. Serum leptin levels are independently correlated with two measures of HDL. Atherosclerosis 1997, 132, 237–243. [Google Scholar] [CrossRef]
- Lundåsen, T.; Liao, W.; Angelin, B.; Rudling, M. Leptin induces the hepatic high density lipoprotein receptor scavenger receptor B type I (SR-BI) but not cholesterol 7alpha-hydroxylase (Cyp7a1) in leptin-deficient (ob/ob) mice. J. Biol. Chem. 2003, 278, 43224–43228. [Google Scholar] [CrossRef] [PubMed]
- Dullaart, R.P.F.; de Vries, R.; Dallinga-Thie, G.M.; van Tol, A.; Sluiter, W.J. Plasma cholesteryl ester transfer protein mass and phospholipid transfer protein activity are associated with leptin in type 2 diabetes mellitus. Biochim. Biophys. Acta 2007, 1771, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Radeau, T.; Lau, P.; Robb, M.; McDonnell, M.; Ailhaud, G.; McPherson, R. Cholesteryl ester transfer protein (CETP) mRNA abundance in human adipose tissue: Relationship to cell size and membrane cholesterol content. J. Lipid Res. 1995, 36, 2552–2561. [Google Scholar] [PubMed]
- Bamba, V.; Rader, D.J. Obesity and Atherogenic Dyslipidemia. Gastroenterology 2007, 132, 2181–2190. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hirose, H.; Saito, I.; Tomita, M.; Taniyama, M.; Matsubara, K.; Okazaki, Y.; Ishii, T.; Nishikai, K.; Saruta, T. Correlation of the adipocyte-derived protein adiponectin with insulin resistance index and serum high-density lipoprotein-cholesterol, independent of body mass index, in the Japanese population. Clin. Sci. Lond. Engl. 2002, 103, 137–142. [Google Scholar] [CrossRef]
- Ryo, M.; Nakamura, T.; Kihara, S.; Kumada, M.; Shibazaki, S.; Takahashi, M.; Nagai, M.; Matsuzawa, Y.; Funahashi, T. Adiponectin as a biomarker of the metabolic syndrome. Circ. J. Off. J. Jpn. Circ. Soc. 2004, 68, 975–981. [Google Scholar] [CrossRef]
- Sattar, N.; Wannamethee, G.; Sarwar, N.; Tchernova, J.; Cherry, L.; Wallace, A.; Danesh, J.; Whincup, P. Adiponectin and Coronary Heart Disease. Circulation 2006, 114, 623–629. [Google Scholar] [CrossRef]
- Cnop, M.; Havel, P.J.; Utzschneider, K.M.; Carr, D.B.; Sinha, M.K.; Boyko, E.J.; Retzlaff, B.M.; Knopp, R.H.; Brunzell, J.D.; Kahn, S.E. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: Evidence for independent roles of age and sex. Diabetologia 2003, 46, 459–469. [Google Scholar] [CrossRef]
- Martin, L.J.; Woo, J.G.; Daniels, S.R.; Goodman, E.; Dolan, L.M. The relationships of adiponectin with insulin and lipids are strengthened with increasing adiposity. J. Clin. Endocrinol. Metab. 2005, 90, 4255–4259. [Google Scholar] [CrossRef]
- Baratta, R.; Amato, S.; Degano, C.; Farina, M.G.; Patanè, G.; Vigneri, R.; Frittitta, L. Adiponectin relationship with lipid metabolism is independent of body fat mass: Evidence from both cross-sectional and intervention studies. J. Clin. Endocrinol. Metab. 2004, 89, 2665–2671. [Google Scholar] [CrossRef]
- Després, J.P.; Couillard, C.; Gagnon, J.; Bergeron, J.; Leon, A.S.; Rao, D.C.; Skinner, J.S.; Wilmore, J.H.; Bouchard, C. Race, visceral adipose tissue, plasma lipids, and lipoprotein lipase activity in men and women: The Health, Risk Factors, Exercise Training, and Genetics (HERITAGE) family study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.C.; Ayyobi, A.F.; Murdoch, S.J.; Deeb, S.S.; Brunzell, J.D. Contribution of hepatic lipase, lipoprotein lipase, and cholesteryl ester transfer protein to LDL and HDL heterogeneity in healthy women. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Lamarche, B.; Uffelman, K.D.; Heatherington, A.C.; Honig, M.A.; Szeto, L.W.; Barrett, P.H. Clearance of postprandial and lipolytically modified human HDL in rabbits and rats. J. Lipid Res. 1997, 38, 1771–1778. [Google Scholar] [PubMed]
- Rashid, S.; Trinh, D.K.; Uffelman, K.D.; Cohn, J.S.; Rader, D.J.; Lewis, G.F. Expression of human hepatic lipase in the rabbit model preferentially enhances the clearance of triglyceride-enriched versus native high-density lipoprotein apolipoprotein A-I. Circulation 2003, 107, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Murdoch, S.; Uffelman, K.; Naples, M.; Szeto, L.; Albers, A.; Adeli, K.; Brunzell, J.D. Hepatic lipase mRNA, protein, and plasma enzyme activity is increased in the insulin-resistant, fructose-fed Syrian golden hamster and is partially normalized by the insulin sensitizer rosiglitazone. Diabetes 2004, 53, 2893–2900. [Google Scholar] [CrossRef]
- Ishida, T.; Choi, S.; Kundu, R.K.; Hirata, K.-I.; Rubin, E.M.; Cooper, A.D.; Quertermous, T. Endothelial lipase is a major determinant of HDL level. J. Clin. Investig. 2003, 111, 347–355. [Google Scholar] [CrossRef]
- Jin, W.; Millar, J.S.; Broedl, U.; Glick, J.M.; Rader, D.J. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J. Clin. Investig. 2003, 111, 357–362. [Google Scholar] [CrossRef]
- Ma, K.; Cilingiroglu, M.; Otvos, J.D.; Ballantyne, C.M.; Marian, A.J.; Chan, L. Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc. Natl. Acad. Sci. USA 2003, 100, 2748–2753. [Google Scholar] [CrossRef]
- Maugeais, C.; Tietge, U.J.F.; Broedl, U.C.; Marchadier, D.; Cain, W.; McCoy, M.G.; Lund-Katz, S.; Glick, J.M.; Rader, D.J. Dose-dependent acceleration of high-density lipoprotein catabolism by endothelial lipase. Circulation 2003, 108, 2121–2126. [Google Scholar] [CrossRef]
- Jaye, M.; Lynch, K.J.; Krawiec, J.; Marchadier, D.; Maugeais, C.; Doan, K.; South, V.; Amin, D.; Perrone, M.; Rader, D.J. A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 1999, 21, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Badellino, K.O.; Wolfe, M.L.; Reilly, M.P.; Rader, D.J. Endothelial lipase concentrations are increased in metabolic syndrome and associated with coronary atherosclerosis. PLoS Med. 2006, 3, e22. [Google Scholar] [CrossRef] [PubMed]
- De Lemos, A.S.; Wolfe, M.L.; Long, C.J.; Sivapackianathan, R.; Rader, D.J. Identification of genetic variants in endothelial lipase in persons with elevated high-density lipoprotein cholesterol. Circulation 2002, 106, 1321–1326. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castro, A.M.; Macedo-de la Concha, L.E.; Pantoja-Meléndez, C.A. Low-grade inflammation and its relation to obesity and chronic degenerative diseases. Rev. Méd. Hosp. Gen. México 2017, 80, 101–105. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Chung, S.; Cuffe, H.; Marshall, S.M.; McDaniel, A.L.; Ha, J.H.; Kavanagh, K.; Hong, C.; Tontonoz, P.; Temel, R.E.; Parks, J.S. Dietary Cholesterol Promotes Adipocyte Hypertrophy and Adipose Tissue Inflammation in Visceral, but Not in Subcutaneous, Fat in Monkeys. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1880–1887. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Nikkilä, E.A.; Miettinen, T.A. Regulation of cholesterol synthesis and storage in fat cells. J. Lipid Res. 1975, 16, 211–223. [Google Scholar]
- Farkas, J.; Angel, A.; Avigan, M.I. Studies on the compartmentation of lipid in adipose cells. II. Cholesterol accumulation and distribution in adipose tissue components. J. Lipid Res. 1973, 14, 344–356. [Google Scholar]
- Schreibman, P.H.; Dell, R.B. Human adipocyte cholesterol. Concentration, localization, synthesis, and turnover. J. Clin. Investig. 1975, 55, 986–993. [Google Scholar] [CrossRef]
- Krause, B.R.; Hartman, A.D. Adipose tissue and cholesterol metabolism. J. Lipid Res. 1984, 25, 97–110. [Google Scholar]
- McGillicuddy, F.C.; Reilly, M.P. Adipose tissue modulation of HDL. Clin. Lipidol. 2010, 5, 601–606. [Google Scholar] [CrossRef]
- Zhang, Y.; McGillicuddy, F.C.; Hinkle, C.C.; O’Neill, S.; Glick, J.M.; Rothblat, G.H.; Reilly, M.P. Adipocyte Modulation of High-Density Lipoprotein Cholesterol. Circulation 2010, 121, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Woudberg, N.J.; Goedecke, J.H.; Blackhurst, D.; Frias, M.; James, R.; Opie, L.H.; Lecour, S. Association between ethnicity and obesity with high-density lipoprotein (HDL) function and subclass distribution. Lipids Health Dis. 2016, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Woudberg, N.J.; Mendham, A.E.; Katz, A.A.; Goedecke, J.H.; Lecour, S. Exercise intervention alters HDL subclass distribution and function in obese women. Lipids Health Dis. 2018, 17, 232. [Google Scholar] [CrossRef]
- Davidson, W.S.; Heink, A.; Sexmith, H.; Dolan, L.M.; Gordon, S.M.; Otvos, J.D.; Melchior, J.T.; Elder, D.A.; Khoury, J.; Geh, E.; et al. Obesity is associated with an altered HDL subspecies profile among adolescents with metabolic disease. J. Lipid Res. 2017, 58, 1916–1923. [Google Scholar] [CrossRef]
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.; Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis Modalities and HDL Composition and Function. J. Am. Soc. Nephrol. 2015, 26, 2267–2276. [Google Scholar] [CrossRef]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia Alters HDL Composition and Function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. Ther. 2012, 10, 353–361. [Google Scholar] [CrossRef]
- Trieb, M.; Horvath, A.; Birner-Gruenberger, R.; Spindelboeck, W.; Stadlbauer, V.; Taschler, U.; Curcic, S.; Stauber, R.E.; Holzer, M.; Pasterk, L.; et al. Liver disease alters high-density lipoprotein composition, metabolism and function. Biochim. Biophys. Acta 2016, 1861, 630–638. [Google Scholar] [CrossRef]
- Holzer, M.; Wolf, P.; Curcic, S.; Birner-Gruenberger, R.; Weger, W.; Inzinger, M.; El-Gamal, D.; Wadsack, C.; Heinemann, A.; Marsche, G. Psoriasis alters HDL composition and cholesterol efflux capacity. J. Lipid Res. 2012, 53, 1618–1624. [Google Scholar] [CrossRef]
- Trieb, M.; Wolf, P.; Knuplez, E.; Weger, W.; Schuster, C.; Peinhaupt, M.; Holzer, M.; Trakaki, A.; Eichmann, T.; Lass, A.; et al. Abnormal composition and function of high-density lipoproteins in atopic dermatitis patients. Allergy 2019, 74, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Trakaki, A.; Sturm, G.J.; Pregartner, G.; Scharnagl, H.; Eichmann, T.O.; Trieb, M.; Knuplez, E.; Holzer, M.; Stadler, J.T.; Heinemann, A.; et al. Allergic rhinitis is associated with complex alterations in high-density lipoprotein composition and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef]
- Perségol, L.; Vergès, B.; Foissac, M.; Gambert, P.; Duvillard, L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 2006, 49, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Sasahara, T.; Nestel, P.; Fidge, N.; Sviridov, D. Cholesterol transport between cells and high density lipoprotein subfractions from obese and lean subjects. J. Lipid Res. 1998, 39, 544–554. [Google Scholar]
- Marsche, G.; Zelzer, S.; Meinitzer, A.; Kern, S.; Meissl, S.; Pregartner, G.; Weghuber, D.; Almer, G.; Mangge, H. Adiponectin Predicts High-Density Lipoprotein Cholesterol Efflux Capacity in Adults Irrespective of Body Mass Index and Fat Distribution. J. Clin. Endocrinol. Metab. 2017, 102, 4117–4123. [Google Scholar] [CrossRef]
- Talbot, C.P.J.; Plat, J.; Joris, P.J.; Konings, M.; Kusters, Y.H.A.M.; Schalkwijk, C.G.; Ritsch, A.; Mensink, R.P. HDL cholesterol efflux capacity and cholesteryl ester transfer are associated with body mass, but are not changed by diet-induced weight loss: A randomized trial in abdominally obese men. Atherosclerosis 2018, 274, 23–28. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Nigro, E.; Scudiero, O.; Monaco, M.L.; Palmieri, A.; Mazzarella, G.; Costagliola, C.; Bianco, A.; Daniele, A. New Insight into Adiponectin Role in Obesity and Obesity-Related Diseases. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Arita, Y.; Nishida, M.; Matsuyama, A.; Okamoto, Y.; Ishigami, M.; Kuriyama, H.; Kishida, K.; Nishizawa, H.; et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001, 103, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, F.; Oku, H.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.; Maeda, N.; Tsujii, K.; Ishigami, M.; et al. Adiponectin accelerates reverse cholesterol transport by increasing high density lipoprotein assembly in the liver. Biochem. Biophys. Res. Commun. 2007, 358, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Oku, H.; Matsuura, F.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.; Maeda, N.; Ohama, T.; Ishigami, M.; et al. Adiponectin deficiency suppresses ABCA1 expression and ApoA-I synthesis in the liver. FEBS Lett. 2007, 581, 5029–5033. [Google Scholar] [CrossRef] [PubMed]
- Tsubakio-Yamamoto, K.; Matsuura, F.; Koseki, M.; Oku, H.; Sandoval, J.C.; Inagaki, M.; Nakatani, K.; Nakaoka, H.; Kawase, R.; Yuasa-Kawase, M.; et al. Adiponectin prevents atherosclerosis by increasing cholesterol efflux from macrophages. Biochem. Biophys. Res. Commun. 2008, 375, 390–394. [Google Scholar] [CrossRef]
- Liang, B.; Wang, X.; Guo, X.; Yang, Z.; Bai, R.; Liu, M.; Xiao, C.; Bian, Y. Adiponectin upregulates ABCA1 expression through liver X receptor alpha signaling pathway in RAW 264.7 macrophages. Int. J. Clin. Exp. Pathol. 2015, 8, 450–457. [Google Scholar]
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPARγ-LXR-ABCA1 Pathway in Macrophages Is Involved in Cholesterol Efflux and Atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Vergès, B.; Petit, J.M.; Duvillard, L.; Dautin, G.; Florentin, E.; Galland, F.; Gambert, P. Adiponectin is an important determinant of apoA-I catabolism. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1364–1369. [Google Scholar] [CrossRef]
- Hafiane, A.; Daskalopoulou, S.S. Adiponectin’s mechanisms in high-density lipoprotein biogenesis and cholesterol efflux. Metabolism 2020, 113, 154393. [Google Scholar] [CrossRef]
- Posadas-Sánchez, R.; Posadas-Romero, C.; Mendoza-Pérez, E.; Caracas-Portilla, N.A.; Cardoso-Saldaña, G.; Medina-Urrutia, A.; Jorge-Galarza, E.; Juárez-Rojas, J.G. Cholesterol efflux and metabolic abnormalities associated with low high-density-lipoprotein-cholesterol and high triglycerides in statin-treated coronary men with low-density lipoprotein-cholesterol <70 mg/dl. Am. J. Cardiol. 2012, 109, 636–641. [Google Scholar] [CrossRef]
- Wang, M.; Wang, D.; Zhang, Y.; Wang, X.; Liu, Y.; Xia, M. Adiponectin increases macrophages cholesterol efflux and suppresses foam cell formation in patients with type 2 diabetes mellitus. Atherosclerosis 2013, 229, 62–70. [Google Scholar] [CrossRef]
- Clarenbach, J.; Vega, G.; Adams-Huet, B.; Considine, R.; Ricks, M.; Sumner, A. Variability in Postheparin Hepatic Lipase Activity Is Associated with Plasma Adiponectin Levels in African Americans. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2007, 55, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; von Eynatten, M.; Schiekofer, S.; Nawroth, P.P.; Dugi, K.A. Low Plasma Adiponectin Levels Are Associated With Increased Hepatic Lipase Activity In Vivo: Response to Kobayashi et al. Diabetes Care 2006, 29, 181. [Google Scholar] [CrossRef][Green Version]
- Magge, S.N.; Stettler, N.; Koren, D.; Levitt Katz, L.E.; Gallagher, P.R.; Mohler, E.R.; Rader, D.J. Adiponectin is associated with favorable lipoprotein profile, independent of BMI and insulin resistance, in adolescents. J. Clin. Endocrinol. Metab. 2011, 96, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Otvos, J.D.; Flyvbjerg, A.; Miserez, A.R.; Frystyk, J.; Sinnreich, R.; Kark, J.D. Adiponectin and Lipoprotein Particle Size. Diabetes Care 2009, 32, 1317–1319. [Google Scholar] [CrossRef]
- Tsubakio-Yamamoto, K.; Sugimoto, T.; Nishida, M.; Okano, R.; Monden, Y.; Kitazume-Taneike, R.; Yamashita, T.; Nakaoka, H.; Kawase, R.; Yuasa-Kawase, M.; et al. Serum adiponectin level is correlated with the size of HDL and LDL particles determined by high performance liquid chromatography. Metabolism 2012, 61, 1763–1770. [Google Scholar] [CrossRef]
- Shin, M.J.; Kim, O.Y. Plasma adiponectin is associated with less atherogenic lipoprotein phenotype. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 770–775. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kusunoki, M.; Murase, Y.; Kawashiri, M.; Higashikata, T.; Miwa, K.; Katsuda, S.; Takata, M.; Asano, A.; Nohara, A.; et al. Relationship of Lipoprotein Lipase and Hepatic Triacylglycerol Lipase Activity to Serum Adiponectin Levels in Japanese Hyperlipidemic Men. Horm. Metab. Res. 2005, 37, 505–509. [Google Scholar] [CrossRef]
- Von Eynatten, M.; Schneider, J.G.; Humpert, P.M.; Rudofsky, G.; Schmidt, N.; Barosch, P.; Hamann, A.; Morcos, M.; Kreuzer, J.; Bierhaus, A.; et al. Decreased plasma lipoprotein lipase in hypoadiponectinemia: An association independent of systemic inflammation and insulin resistance. Diabetes Care 2004, 27, 2925–2929. [Google Scholar] [CrossRef]
- Van Greevenbroek, M.M.J.; Schalkwijk, C.G.; Stehouwer, C.D.A. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: Causes and consequences. Neth. J. Med. 2013, 71, 174–187. [Google Scholar]
- Monteiro, R.; Azevedo, I. Chronic Inflammation in Obesity and the Metabolic Syndrome. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef]
- Kaser, S.; Tatarczyk, T.; Stadlmayr, A.; Ciardi, C.; Ress, C.; Tschoner, A.; Sandhofer, A.; Paulweber, B.; Ebenbichler, C.F.; Patsch, J.R. Effect of obesity and insulin sensitivity on adiponectin isoform distribution. Eur. J. Clin. Investig. 2008, 38, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.K.; Ciaraldi, T.P.; Henry, R.R.; Wittgrove, A.C.; Phillips, S.A. Adipose tissue depot and cell size dependency of adiponectin synthesis and secretion in human obesity. Adipocyte 2013, 2, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Blachnio-Zabielska, A.U.; Koutsari, C.; Tchkonia, T.; Jensen, M.D. Sphingolipid content of human adipose tissue: Relationship to adiponectin and insulin resistance. Obesity 2012, 20, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar] [CrossRef]
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef]
- Gazit, S.L.; Mariko, B.; Thérond, P.; Decouture, B.; Xiong, Y.; Couty, L.; Bonnin, P.; Baudrie, V.; Le Gall, S.M.; Dizier, B.; et al. Platelet and Erythrocyte Sources of S1P Are Redundant for Vascular Development and Homeostasis, but Both Rendered Essential After Plasma S1P Depletion in Anaphylactic Shock. Circ. Res. 2016, 119, e110–e126. [Google Scholar] [CrossRef]
- Lee, M.J.; Van Brocklyn, J.R.; Thangada, S.; Liu, C.H.; Hand, A.R.; Menzeleev, R.; Spiegel, S.; Hla, T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 1998, 279, 1552–1555. [Google Scholar] [CrossRef]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS ONE 2013, 8, e72449. [Google Scholar] [CrossRef]
- Majumdar, I.; Mastrandrea, L.D. Serum sphingolipids and inflammatory mediators in adolescents at risk for metabolic syndrome. Endocrine 2012, 41, 442–449. [Google Scholar] [CrossRef]
- Green, C.L.; Mitchell, S.E.; Derous, D.; Wang, Y.; Chen, L.; Han, J.-D.J.; Promislow, D.E.L.; Lusseau, D.; Douglas, A.; Speakman, J.R. The effects of graded levels of calorie restriction: IX. Global metabolomic screen reveals modulation of carnitines, sphingolipids and bile acids in the liver of C57BL/6 mice. Aging Cell 2017, 16, 529–540. [Google Scholar] [CrossRef]
- Silva, V.R.R.; Micheletti, T.O.; Pimentel, G.D.; Katashima, C.K.; Lenhare, L.; Morari, J.; Mendes, M.C.S.; Razolli, D.S.; Rocha, G.Z.; de Souza, C.T.; et al. Hypothalamic S1P/S1PR1 axis controls energy homeostasis. Nat. Commun. 2014, 5, 4859. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Federspiel, C.K.; Borup, A.; Christensen, P.M.; Madsen, A.N.; Heine, M.; Nielsen, C.H.; Kjaer, A.; Holst, B.; Heeren, J.; et al. The Apolipoprotein M/S1P Axis Controls Triglyceride Metabolism and Brown Fat Activity. Cell Rep. 2018, 22, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Dahlbäck, B. Lean ApoM-/-Mice with Hyperactive Brown Adipose Tissue. Trends Endocrinol. Metab. 2018, 29, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Frej, C.; Mendez, A.J.; Ruiz, M.; Castillo, M.; Hughes, T.A.; Dahlbäck, B.; Goldberg, R.B. A Shift in ApoM/S1P Between HDL-Particles in Women With Type 1 Diabetes Mellitus Is Associated with Impaired Anti-Inflammatory Effects of the ApoM/S1P Complex. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1194–1205. [Google Scholar] [CrossRef]
- Brolin, R.E. Bariatric surgery and long-term control of morbid obesity. JAMA 2002, 288, 2793–2796. [Google Scholar] [CrossRef]
- Steinbrook, R. Surgery for severe obesity. N. Engl. J. Med. 2004, 350, 1075–1079. [Google Scholar] [CrossRef]
- Kuno, T.; Tanimoto, E.; Morita, S.; Shimada, Y.J. Effects of Bariatric Surgery on Cardiovascular Disease: A Concise Update of Recent Advances. Front. Cardiovasc. Med. 2019, 6. [Google Scholar] [CrossRef]
- Benraoune, F.; Litwin, S.E. Reductions in Cardiovascular Risk after Bariatric Surgery. Curr. Opin. Cardiol. 2011, 26, 555–561. [Google Scholar] [CrossRef]
- Lorkowski, S.W.; Brubaker, G.; Rotroff, D.M.; Kashyap, S.R.; Bhatt, D.L.; Nissen, S.E.; Schauer, P.R.; Aminian, A.; Smith, J.D. Bariatric Surgery Improves HDL Function Examined by ApoA1 Exchange Rate and Cholesterol Efflux Capacity in Patients with Obesity and Type 2 Diabetes. Biomolecules 2020, 10, 551. [Google Scholar] [CrossRef]
- Raffaelli, M.; Guidone, C.; Callari, C.; Iaconelli, A.; Bellantone, R.; Mingrone, G. Effect of gastric bypass versus diet on cardiovascular risk factors. Ann. Surg. 2014, 259, 694–699. [Google Scholar] [CrossRef]
- Christou, N.V.; Sampalis, J.S.; Liberman, M.; Look, D.; Auger, S.; McLean, A.P.H.; MacLean, L.D. Surgery decreases long-term mortality, morbidity, and health care use in morbidly obese patients. Ann. Surg. 2004, 240, 416–424. [Google Scholar] [CrossRef]
- Williams, D.B.; Hagedorn, J.C.; Lawson, E.H.; Galanko, J.A.; Safadi, B.Y.; Curet, M.J.; Morton, J.M. Gastric bypass reduces biochemical cardiac risk factors. Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 2007, 3, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Zlabek, J.A.; Grimm, M.S.; Larson, C.J.; Mathiason, M.A.; Lambert, P.J.; Kothari, S.N. The effect of laparoscopic gastric bypass surgery on dyslipidemia in severely obese patients. Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 2005, 1, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.M.; Austrheim-Smith, I.T.; Stanhope, K.L.; Van Loan, M.D.; Ali, M.R.; Wolfe, B.M.; Havel, P.J. Circulating concentrations of high-molecular-weight adiponectin are increased following Roux-en-Y gastric bypass surgery. Diabetologia 2006, 49, 2552–2558. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-P.; Krzyzanowska, K.; Möhlig, M.; Spranger, J.; Pfeiffer, A.F.H.; Schernthaner, G. Effects of marked weight loss on plasma levels of adiponectin, markers of chronic subclinical inflammation and insulin resistance in morbidly obese women. Int. J. Obes. 2005, 29, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, C.D.; Lissner, L.; Wedel, H.; Sjöström, L. Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: The SOS Intervention Study. Obes. Res. 1999, 7, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Schauer, P.R.; Kashyap, S.R.; Wolski, K.; Brethauer, S.A.; Kirwan, J.P.; Pothier, C.E.; Thomas, S.; Abood, B.; Nissen, S.E.; Bhatt, D.L. Bariatric Surgery versus Intensive Medical Therapy in Obese Patients with Diabetes. N. Engl. J. Med. 2012, 366, 1567–1576. [Google Scholar] [CrossRef]
- Schauer, P.R.; Bhatt, D.L.; Kirwan, J.P.; Wolski, K.; Brethauer, S.A.; Navaneethan, S.D.; Aminian, A.; Pothier, C.E.; Kim, E.S.H.; Nissen, S.E.; et al. Bariatric surgery versus intensive medical therapy for diabetes—3-year outcomes. N. Engl. J. Med. 2014, 370, 2002–2013. [Google Scholar] [CrossRef]
- Schauer, P.R.; Bhatt, D.L.; Kirwan, J.P.; Wolski, K.; Aminian, A.; Brethauer, S.A.; Navaneethan, S.D.; Singh, R.P.; Pothier, C.E.; Nissen, S.E.; et al. Bariatric Surgery versus Intensive Medical Therapy for Diabetes—5-Year Outcomes. N. Engl. J. Med. 2017, 376, 641–651. [Google Scholar] [CrossRef]
- Mingrone, G.; Panunzi, S.; De Gaetano, A.; Guidone, C.; Iaconelli, A.; Nanni, G.; Castagneto, M.; Bornstein, S.; Rubino, F. Bariatric-metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomised controlled trial. Lancet Lond. Engl. 2015, 386, 964–973. [Google Scholar] [CrossRef]
- Lorkowski, S.W.; Brubaker, G.; Li, L.; Li, X.S.; Hazen, S.L.; Smith, J.D. A Novel Cell-Free Fluorescent Assay for HDL Function: Low Apolipoprotein A1 Exchange Rate Associated with Increased Incident Cardiovascular Events. J. Appl. Lab. Med. 2020, 5, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Heffron, S.P.; Lin, B.-X.; Parikh, M.; Scolaro, B.; Adelman, S.J.; Collins, H.L.; Berger, J.S.; Fisher, E.A. Changes in High-Density Lipoprotein Cholesterol Efflux Capacity After Bariatric Surgery Are Procedure Dependent. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zvintzou, E.; Skroubis, G.; Chroni, A.; Petropoulou, P.-I.; Gkolfinopoulou, C.; Sakellaropoulos, G.; Gantz, D.; Mihou, I.; Kalfarentzos, F.; Kypreos, K.E. Effects of bariatric surgery on HDL structure and functionality: Results from a prospective trial. J. Clin. Lipidol. 2014, 8, 408–417. [Google Scholar] [CrossRef]
- Longitudinal Assessment of Bariatric Surgery (LABS) Consortium; Flum, D.R.; Belle, S.H.; King, W.C.; Wahed, A.S.; Berk, P.; Chapman, W.; Pories, W.; Courcoulas, A.; McCloskey, C.; et al. Perioperative safety in the longitudinal assessment of bariatric surgery. N. Engl. J. Med. 2009, 361, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, S.; Reis, F.; Ferreira, C.; Nunes, S.; Viana, S.; Catarino, A.; Rocha-Pereira, P.; Belo, L.; Monteiro, L.; Catarino, C.; et al. Weight loss achieved by bariatric surgery modifies high-density lipoprotein subfractions and low-density lipoprotein oxidation towards atheroprotection. Clin. Biochem. 2019, 63, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kjellmo, C.A.; Karlsson, H.; Nestvold, T.K.; Ljunggren, S.; Cederbrant, K.; Marcusson-Ståhl, M.; Mathisen, M.; Lappegård, K.T.; Hovland, A. Bariatric surgery improves lipoprotein profile in morbidly obese patients by reducing LDL cholesterol, apoB, and SAA/PON1 ratio, increasing HDL cholesterol, but has no effect on cholesterol efflux capacity. J. Clin. Lipidol. 2018, 12, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Osto, E.; Doytcheva, P.; Corteville, C.; Bueter, M.; Dörig, C.; Stivala, S.; Buhmann, H.; Colin, S.; Rohrer, L.; Hasballa, R.; et al. Rapid and body weight-independent improvement of endothelial and high-density lipoprotein function after Roux-en-Y gastric bypass: Role of glucagon-like peptide-1. Circulation 2015, 131, 871–881. [Google Scholar] [CrossRef]
- Ding, S.; Huang, H.; Xu, Y.; Zhu, H.; Zhong, C. MiR-222 in Cardiovascular Diseases: Physiology and Pathology. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Taïbi, F.; Metzinger-Le Meuth, V.; Massy, Z.A.; Metzinger, L. miR-223: An inflammatory oncomiR enters the cardiovascular field. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2014, 1842, 1001–1009. [Google Scholar] [CrossRef]
- Ho, J.H.; Ong, K.L.; Torres, L.F.C.; Liu, Y.; Adam, S.; Iqbal, Z.; Dhage, S.; Ammori, B.J.; Syed, A.A.; Rye, K.-A.; et al. High-density lipoprotein-associated miRNA is increased following Roux-en-Y gastric bypass surgery for severe obesity. J. Lipid Res. 2020, jlr.RA120000963. [Google Scholar] [CrossRef]
- MacLean, P.S.; Wing, R.R.; Davidson, T.; Epstein, L.; Goodpaster, B.; Hall, K.D.; Levin, B.E.; Perri, M.G.; Rolls, B.J.; Rosenbaum, M.; et al. NIH working group report: Innovative research to improve maintenance of weight loss. Obesity 2015, 23, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Mauer, E.A.; Shukla, A.P.; Rathi, S.; Aronne, L.J. Low adoption of weight loss medications: A comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity 2016, 24, 1955–1961. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Cho, H.-J.; Kang, H.-C.; Youn, B.-B.; Lee, K.-R. Effects on Weight Reduction and Safety of Short-Term Phentermine Administration in Korean Obese People. Yonsei Med. J. 2006, 47, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Bessesen, D.H.; Van Gaal, L.F. Progress and challenges in anti-obesity pharmacotherapy. Lancet Diabetes Endocrinol. 2018, 6, 237–248. [Google Scholar] [CrossRef]
- Davidson, M.H.; Tonstad, S.; Oparil, S.; Schwiers, M.; Day, W.W.; Bowden, C.H. Changes in cardiovascular risk associated with phentermine and topiramate extended-release in participants with comorbidities and a body mass index ≥27 kg/m(2). Am. J. Cardiol. 2013, 111, 1131–1138. [Google Scholar] [CrossRef][Green Version]
- Garvey, W.T.; Ryan, D.H.; Look, M.; Gadde, K.M.; Allison, D.B.; Peterson, C.A.; Schwiers, M.; Day, W.W.; Bowden, C.H. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): A randomized, placebo-controlled, phase 3 extension study. Am. J. Clin. Nutr. 2012, 95, 297–308. [Google Scholar] [CrossRef]
- Mittendorfer, B.; Ostlund, R.E.; Patterson, B.W.; Klein, S. Orlistat Inhibits Dietary Cholesterol Absorption. Obes. Res. 2001, 9, 599–604. [Google Scholar] [CrossRef]
- Berne, C.; Orlistat Swedish Type 2 diabetes Study Group. A randomized study of orlistat in combination with a weight management programme in obese patients with Type 2 diabetes treated with metformin. Diabet. Med. J. Br. Diabet. Assoc. 2005, 22, 612–618. [Google Scholar] [CrossRef]
- Hanefeld, M.; Sachse, G. The effects of orlistat on body weight and glycaemic control in overweight patients with type 2 diabetes: A randomized, placebo-controlled trial. Diabetes Obes. Metab. 2002, 4, 415–423. [Google Scholar] [CrossRef]
- Torgerson, J.S.; Hauptman, J.; Boldrin, M.N.; Sjöström, L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: A randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004, 27, 155–161. [Google Scholar] [CrossRef]
- Kelley, D.E.; Bray, G.A.; Pi-Sunyer, F.X.; Klein, S.; Hill, J.; Miles, J.; Hollander, P. Clinical efficacy of orlistat therapy in overweight and obese patients with insulin-treated type 2 diabetes: A 1-year randomized controlled trial. Diabetes Care 2002, 25, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Hollander, P.A.; Elbein, S.C.; Hirsch, I.B.; Kelley, D.; McGill, J.; Taylor, T.; Weiss, S.R.; Crockett, S.E.; Kaplan, R.A.; Comstock, J.; et al. Role of Orlistat in the Treatment of Obese Patients With Type 2 Diabetes: A 1-year randomized double-blind study. Diabetes Care 1998, 21, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.M.; Leiter, L.; Hollander, P.; Wadden, T.; Anderson, J.W.; Doyle, M.; Foreyt, J.; Aronne, L.; Klein, S. Effect of Orlistat in Overweight and Obese Patients With Type 2 Diabetes Treated With Metformin. Diabetes Care 2002, 25, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A.; Mora, S.; Kolovou, G.; Baum, H.; Bruckert, E.; Watts, G.F.; Sypniewska, G.; Wiklund, O.; Borén, J.; et al. Fasting is not routinely required for determination of a lipid profile: Clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur. Heart J. 2016, 37, 1944–1958. [Google Scholar] [CrossRef]
- Langsted, A.; Freiberg, J.J.; Nordestgaard, B.G. Fasting and nonfasting lipid levels: Influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation 2008, 118, 2047–2056. [Google Scholar] [CrossRef]
- Fidler, M.C.; Sanchez, M.; Raether, B.; Weissman, N.J.; Smith, S.R.; Shanahan, W.R.; Anderson, C.M.; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: The BLOSSOM trial. J. Clin. Endocrinol. Metab. 2011, 96, 3067–3077. [Google Scholar] [CrossRef]
- O’Neil, P.M.; Smith, S.R.; Weissman, N.J.; Fidler, M.C.; Sanchez, M.; Zhang, J.; Raether, B.; Anderson, C.M.; Shanahan, W.R. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: The BLOOM-DM study. Obesity 2012, 20, 1426–1436. [Google Scholar] [CrossRef]
- Tuccinardi, D.; Farr, O.M.; Upadhyay, J.; Oussaada, S.M.; Mathew, H.; Paschou, S.A.; Perakakis, N.; Koniaris, A.; Kelesidis, T.; Mantzoros, C.S. Lorcaserin treatment decreases body weight and improves cardiometabolic risk factors of obese adults: A 6-month-long, randomized, placebo-controlled, double-blind clinical trial. Diabetes Obes. Metab. 2019, 21, 1487–1492. [Google Scholar] [CrossRef]
- Burcelin, R.; Gourdy, P. Harnessing glucagon-like peptide-1 receptor agonists for the pharmacological treatment of overweight and obesity. Obes. Rev. 2017, 18, 86–98. [Google Scholar] [CrossRef]
- Christou, G.A.; Katsiki, N.; Kiortsis, D.N. The Current Role of Liraglutide in the Pharmacotherapy of Obesity. Curr. Vasc. Pharmacol. 2016, 14, 201–207. [Google Scholar] [CrossRef]
- Engelbrechtsen, L.; Lundgren, J.; Wewer Albrechtsen, N.J.; Mahendran, Y.; Iepsen, E.W.; Finocchietto, P.; Jonsson, A.E.; Madsbad, S.; Holst, J.J.; Vestergaard, H.; et al. Treatment with liraglutide may improve markers of CVD reflected by reduced levels of apoB. Obes. Sci. Pract. 2017, 3, 425–433. [Google Scholar] [CrossRef] [PubMed]
- D’Innocenzo, S.; Biagi, C.; Lanari, M. Obesity and the Mediterranean Diet: A Review of Evidence of the Role and Sustainability of the Mediterranean Diet. Nutrients 2019, 11, 1306. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D. Dietary and Policy Priorities for Cardiovascular Disease, Diabetes, and Obesity–A Comprehensive Review. Circulation 2016, 133, 187–225. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L. Calorie restriction and cardiometabolic health. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Snel, M.; Jonker, J.T.; Hammer, S.; Lamb, H.J.; de Roos, A.; Meinders, A.E.; Pijl, H.; Romijn, J.A.; Smit, J.W.A.; et al. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases plasma CETP and increases apolipoprotein AI levels without improving the cholesterol efflux properties of HDL. Diabetes Care 2011, 34, 2576–2580. [Google Scholar] [CrossRef] [PubMed]
- Maroofi, M.; Nasrollahzadeh, J. Effect of intermittent versus continuous calorie restriction on body weight and cardiometabolic risk markers in subjects with overweight or obesity and mild-to-moderate hypertriglyceridemia: A randomized trial. Lipids Health Dis. 2020, 19, 216. [Google Scholar] [CrossRef]
- Liang, K.-W.; Lee, W.-J.; Lee, I.-T.; Lee, W.-L.; Lin, S.-Y.; Hsu, S.-L.; Wan, C.-J.; Yu, C.-Y.; Tsai, I.-C.; Fu, C.-P.; et al. Persistent elevation of paraoxonase-1 specific enzyme activity after weight reduction in obese non-diabetic men with metabolic syndrome. Clin. Chim. Acta 2011, 412, 1835–1841. [Google Scholar] [CrossRef]
- Kotani, K.; Sakane, N.; Sano, Y.; Tsuzaki, K.; Matsuoka, Y.; Egawa, K.; Yoshimura, M.; Horikawa, C.; Kitagawa, Y.; Kiso, Y.; et al. Changes on the physiological lactonase activity of serum paraoxonase 1 by a diet intervention for weight loss in healthy overweight and obese women. J. Clin. Biochem. Nutr. 2009, 45, 329–334. [Google Scholar] [CrossRef][Green Version]
- Shoji, T.; Nishizawa, Y.; Koyama, H.; Hagiwara, S.; Aratani, H.; Sasao, K.; Kishimoto, H.; Tanishita, H.; Morii, H. High-density-lipoprotein metabolism during a very-low-calorie diet. Am. J. Clin. Nutr. 1992, 56, 297S–298S. [Google Scholar] [CrossRef]
- Hietanen, E.; Laitinen, M.; Ahonen, E.; Marniemi, J.; Nousiainen, U. Changes in Serum Lipids and Lecithin: Cholesterol Acyltransferase Enzyme during 1 Week Weight Reduction of Women on a Low-Calorie Diet. Enzyme 1983, 29, 160–166. [Google Scholar] [CrossRef]
- Varady, K.A.; Bhutani, S.; Church, E.C.; Klempel, M.C. Short-term modified alternate-day fasting: A novel dietary strategy for weight loss and cardioprotection in obese adults. Am. J. Clin. Nutr. 2009, 90, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Varady, K.A.; Bhutani, S.; Klempel, M.C.; Kroeger, C.M.; Trepanowski, J.F.; Haus, J.M.; Hoddy, K.K.; Calvo, Y. Alternate day fasting for weight loss in normal weight and overweight subjects: A randomized controlled trial. Nutr. J. 2013, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Stekovic, S.; Hofer, S.J.; Tripolt, N.; Aon, M.A.; Royer, P.; Pein, L.; Stadler, J.T.; Pendl, T.; Prietl, B.; Url, J.; et al. Alternate Day Fasting Improves Physiological and Molecular Markers of Aging in Healthy, Non-obese Humans. Cell Metab. 2019, 30, 462–476.e6. [Google Scholar] [CrossRef] [PubMed]
- Bardagjy, A.S.; Steinberg, F.M. Relationship between HDL Functional Characteristics and Cardiovascular Health and Potential Impact of Dietary Patterns: A Narrative Review. Nutrients 2019, 11, 1231. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Hernáez, Á.; Castañer, O.; Elosua, R.; Pintó, X.; Estruch, R.; Salas-Salvadó, J.; Corella, D.; Arós, F.; Serra-Majem, L.; Fiol, M.; et al. Mediterranean Diet Improves High-Density Lipoprotein Function in High-Cardiovascular-Risk Individuals. Circulation 2017, 135, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.V.; Li, L.; Byun, J.; Guo, Y.; Michailidis, G.; Jaiswal, M.; Chen, Y.E.; Pop-Busui, R.; Pennathur, S. Therapeutic Lifestyle Changes Improve HDL Function by Inhibiting Myeloperoxidase-Mediated Oxidation in Patients With Metabolic Syndrome. Diabetes Care 2018. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDL Subclass | Size | Shape | Abundant Components | Important Functions |
|---|---|---|---|---|
| Pre-β HDL | 9.6 nm diameter, 4.7 nm thickness | discoidal | ApoA-I, phospholipids | ABCA1-Cholesterol efflux |
| HDL3 | 7.5 nm, 175 kDa | spherical | Protein:lipid ratio 55:45 PON1, ApoA-II, ApoM, S1P | Anti-oxidative activity Anti-inflammatory activity ABCA1-Cholesterol efflux |
| HDL2 | 10 nm, 350 kDa | spherical | Protein:lipid ratio 40:60 | ABCG1- Cholesterol Efflux |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stadler, J.T.; Marsche, G. Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function. Int. J. Mol. Sci. 2020, 21, 8985. https://doi.org/10.3390/ijms21238985
Stadler JT, Marsche G. Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function. International Journal of Molecular Sciences. 2020; 21(23):8985. https://doi.org/10.3390/ijms21238985
Chicago/Turabian StyleStadler, Julia T., and Gunther Marsche. 2020. "Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function" International Journal of Molecular Sciences 21, no. 23: 8985. https://doi.org/10.3390/ijms21238985
APA StyleStadler, J. T., & Marsche, G. (2020). Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function. International Journal of Molecular Sciences, 21(23), 8985. https://doi.org/10.3390/ijms21238985