Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect


Abstract
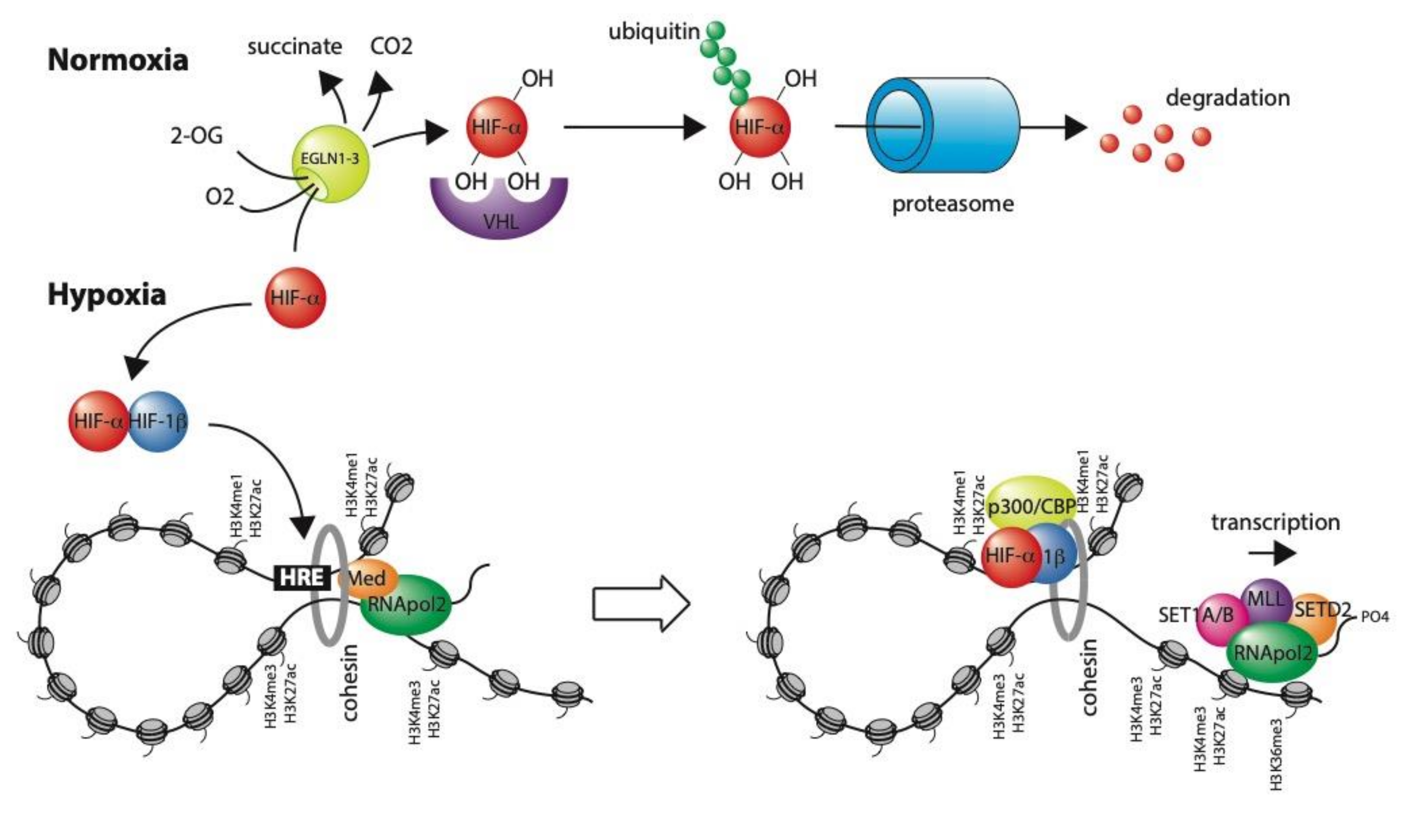
1. Introduction
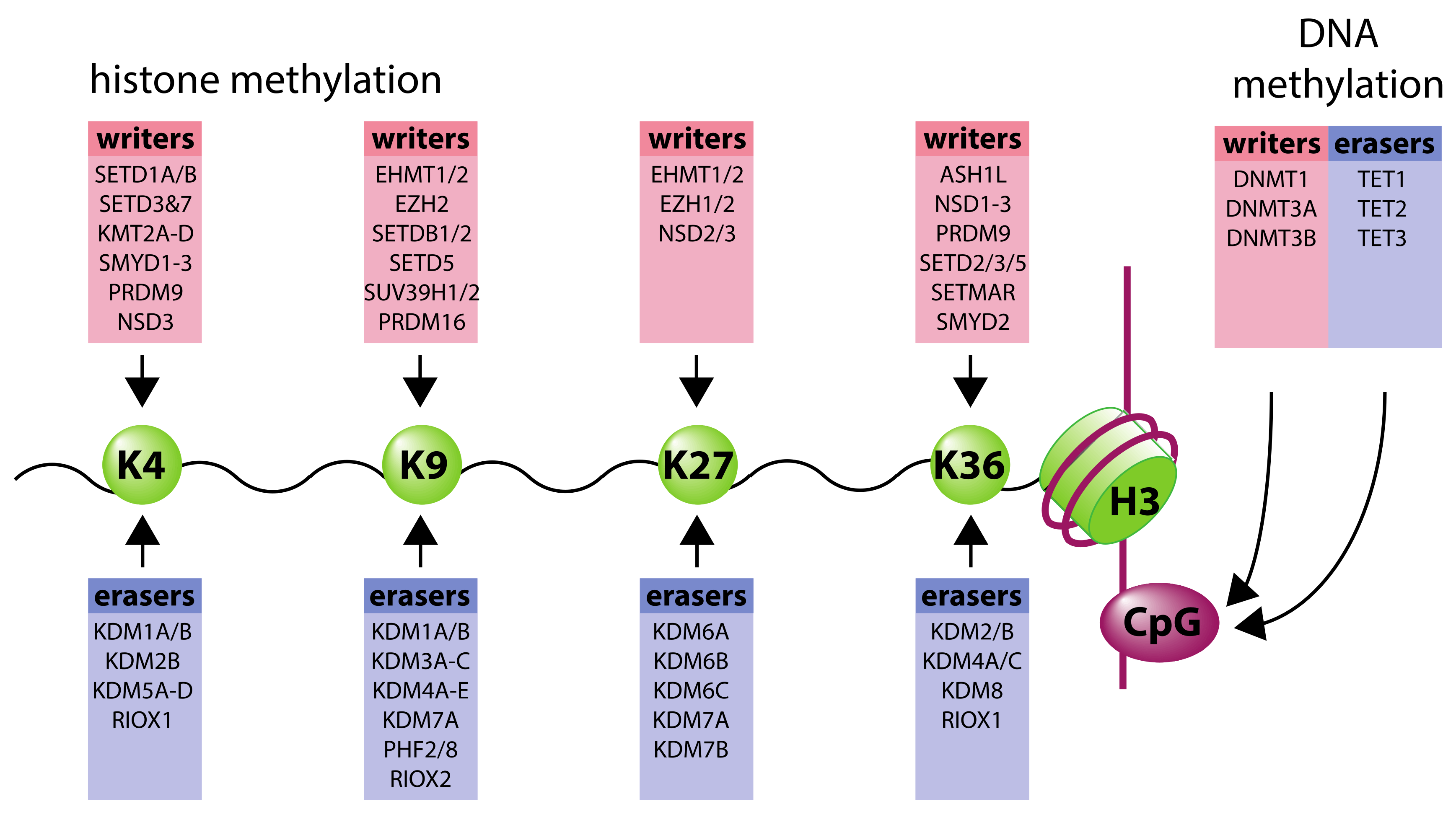
2. Organization of Chromatin Structure
3. Transcriptional Regulation in Hypoxia
4. Oxygen-Sensitivity of 2-Oxoglutarate-Dependent Epigenetic Modifiers
5. Oxygen-Dependent Changes in Enzyme Abundance
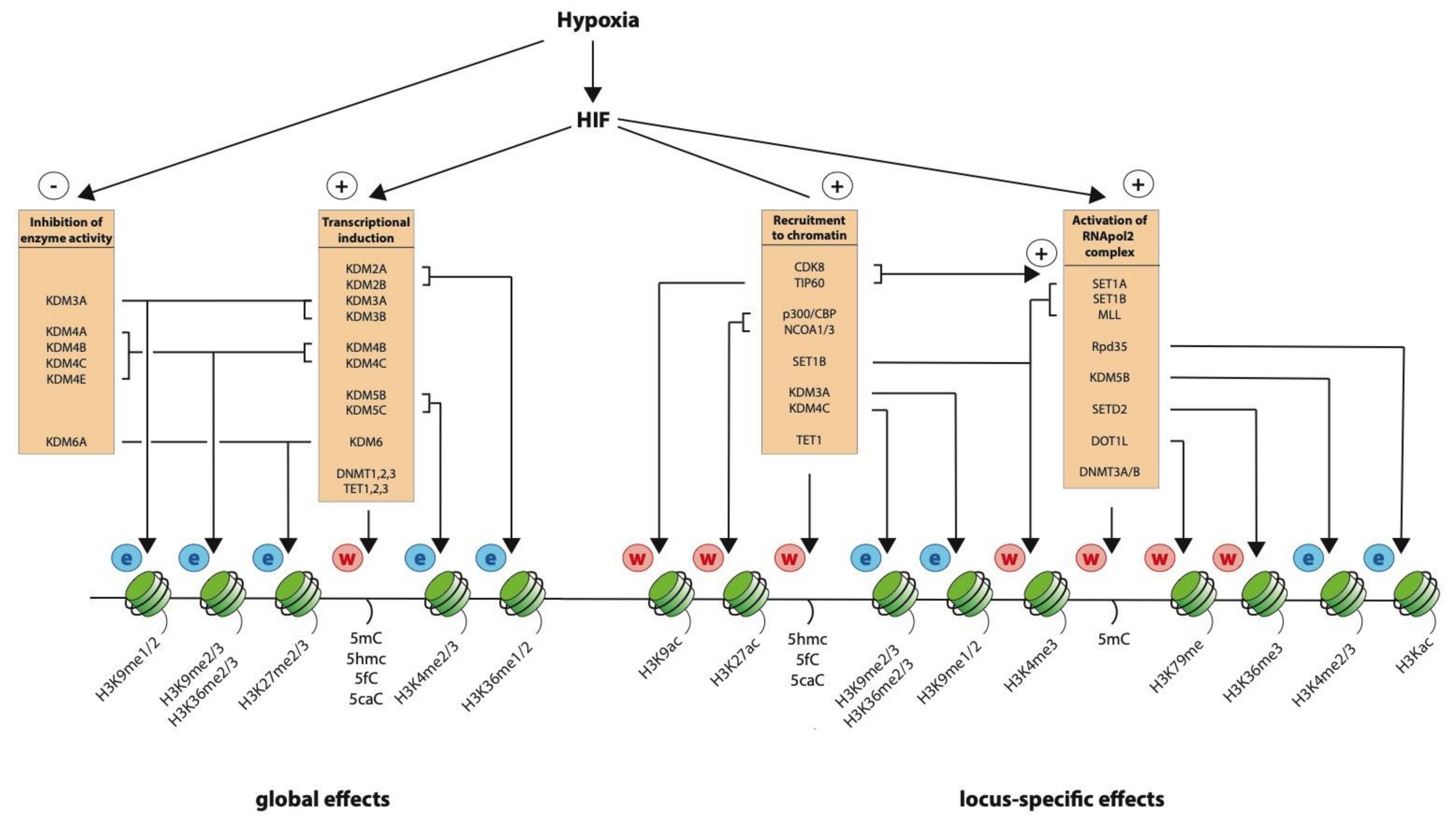
6. The Effect of Hypoxia on Global Histone Modification
7. Locus-Specific Changes in Chromatin
8. Recruitment of Histone Modifying Activity by HIF
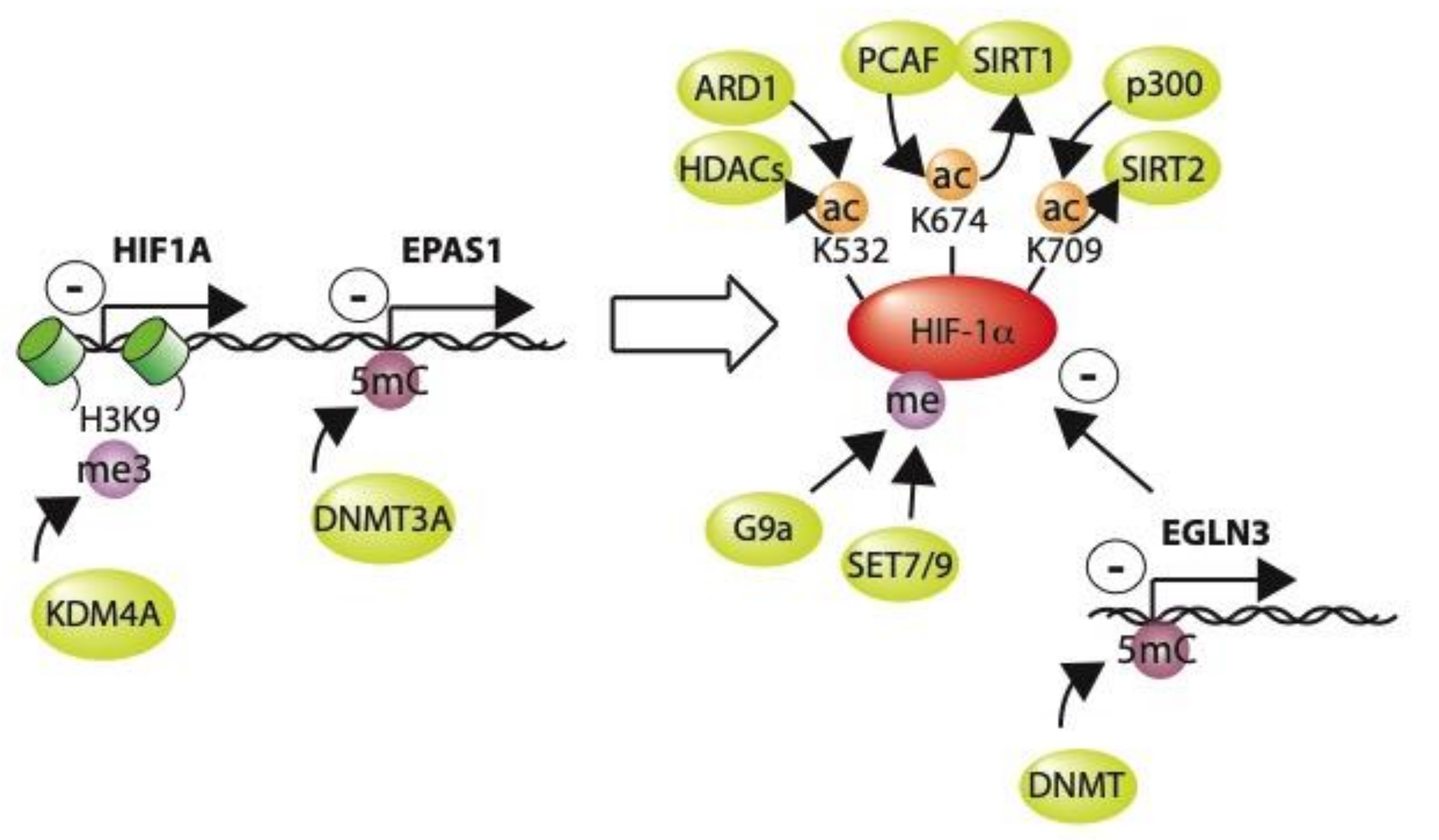
9. Post-Translational Modification of HIF by Epigenetic Modifiers
10. Changes in Histone Modification as a Consequence of Gene Activation
11. Epigenetic “Memory”
12. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2OG | 2-oxoglutarate |
| CBP | CREB-binding protein |
| ChIP | Chromatin immunoprecipitation |
| DNMT | DNA methyltransferase |
| EGLN/PHD | Egl-9 family hypoxia inducible factor / prolyl hydroxylase domain-containing protein |
| FIH | Factor inhibiting HIF |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HIF | Hypoxia-inducible factor |
| HMT | Histone methyltransferase |
| HRE | Hypoxia-response element |
| KDM | Lysine demethylase |
| PTM | Post-translational modification |
| TET | Ten-eleven-translocase |
| TSS | Transcriptional start site |
| VHL | Von Hippel-Lindau |
References
- Adriaens, M.E.; Prickaerts, P.; Chan-Seng-Yue, M.; Beucken, T.V.D.; Dahlmans, V.E.H.; Eijssen, L.; Beck, T.; Wouters, B.G.; Voncken, J.W.; Evelo, C.T. Quantitative analysis of ChIP-seq data uncovers dynamic and sustained H3K4me3 and H3K27me3 modulation in cancer cells under hypoxia. Epigenetics Chromatin 2016, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. The CBP co-activator is a histone acetyltransferase. Nat. Cell Biol. 1996, 384, 641–643. [Google Scholar] [CrossRef]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; E Wang, J.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial Cell–Specific regulation of Hypoxia-Inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef] [PubMed]
- Batie, M.; Druker, J.; D’Ignazio, L.; Rocha, S. KDM2 family members are regulated by HIF-1 in hypoxia. Cells 2017, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef]
- Benabdallah, N.S.; Bickmore, W.A. Regulatory domains and their mechanisms. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 45–51. [Google Scholar] [CrossRef]
- Benabdallah, N.S.; Williamson, I.; Illingworth, R.S.; Kane, L.; Boyle, S.; Sengupta, D.; Grimes, G.R.; Therizols, P.; Bickmore, W.A. Decreased enhancer-promoter proximity accompanying enhancer activation. Mol. Cell 2019, 76, 473–484.e7. [Google Scholar] [CrossRef]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [PubMed]
- Blackledge, N.P.; Klose, R. CpG island chromatin: A platform for gene regulation. Epigenetics 2011, 6, 147–152. [Google Scholar] [CrossRef]
- Busslinger, G.A.; Stocsits, R.R.; van der Lelij, P.; Axelsson, E.; Tedeschi, A.; Galjart, N.; Peters, J.M. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 2017, 544, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Carrero, P.; Okamoto, K.; Coumailleau, P.; O’Brien, S.; Tanaka, H.; Poellinger, L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1α. Mol. Cell. Biol. 2000, 20, 402–415. [Google Scholar] [CrossRef]
- Cascella, B.; Mirica, L.M. Kinetic analysis of iron-dependent histone demethylases: α-Ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry 2012, 51, 8699–8701. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, B.R.; Chen, S.T.; Hsieh, T.H.; Li, Y.J.; Kuo, M.Y.P. HDAC2 promotes cell migration/invasion abilities through HIF-1α stabilization in human oral squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 567–575. [Google Scholar] [CrossRef]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef]
- Choudhry, H.; Schödel, J.; Oikonomopoulos, S.; Camps, C.; Grampp, S.; Harris, A.L.; Ratcliffe, P.J.; Ragoussis, J.; Mole, D.R. Extensive regulation of the non-coding transcriptome by hypoxia: Role of HIF in releasing paused RNA pol2. EMBO Rep. 2013, 15, 70–76. [Google Scholar] [CrossRef]
- Costa, M.; Davidson, T.L.; Chen, H.; Ke, Q.; Zhang, P.; Yan, Y.; Huang, C.; Kluz, T. Nickel carcinogenesis: Epigenetics and hypoxia signaling. Mutat. Res. 2005, 592, 79–88. [Google Scholar] [CrossRef]
- Costa, M.; Yan, Y.; Zhao, D.; Salnikow, K. Molecular mechanisms of nickel carcinogenesis: Gene silencing by nickel delivery to the nucleus and gene activation/inactivation by nickel-induced cell signaling. J. Environ. Monit. 2003, 5, 222–223. [Google Scholar] [CrossRef]
- Dao, J.H.; Kurzeja, R.J.; Morachis, J.M.; Veith, H.; Lewis, J.; Yu, V.; Tegley, C.M.; Tagari, P. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal. Biochem. 2009, 384, 213–223. [Google Scholar] [CrossRef]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Dmitriev, R.I.; Papkovsky, D.B. In vitro ischemia decreases histone H4K16 acetylation in neural cells. FEBS Lett. 2014, 589, 138–144. [Google Scholar] [CrossRef]
- Dobrynin, G.; McAllister, T.E.; Leszczynska, K.B.; Ramachandran, S.; Krieg, A.J.; Kawamura, A.; Hammond, E.M. KDM4A regulates HIF-1 levels through H3K9me3. Sci. Rep. 2017, 7, 11094. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Ebert, B.L.; Bunn, H.F. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell. Biol. 1998, 18, 4089–4096. [Google Scholar] [CrossRef]
- Ehrismann, D.; Flashman, E.; Genn, D.N.; Mathioudakis, N.; Hewitson, K.S.; Ratcliffe, P.J.; Schofield, C.J. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 2006, 401, 227–234. [Google Scholar] [CrossRef]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.; Yu, G.; Rong, F.; Zhang, D.; Xu, C.; Du, J.; Li, Z.; Ouyang, G.; Xiao, W. TET is targeted for proteasomal degradation by the PHD-pVHL pathway to reduce DNA hydroxymethylation. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Fu, L.; Chen, L.; Yang, J.; Ye, T.; Chen, Y.; Fang, J. HIF-1α-induced histone demethylase JMJD2B contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis 2012, 33, 1664–1673. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef]
- Gates, L.A.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’: Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 Protein Regulates HIF1α Protein Lysine Acetylation and Cancer Cell Response to Hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1α Protein Stability Is Increased by Acetylation at Lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Gu, B.; Comerci, C.J.; McCarthy, D.G.; Saurabh, S.; Moerner, W.; Wysocka, J. Opposing Effects of Cohesin and Transcription on CTCF Organization Revealed by Super-resolution Imaging. Mol. Cell 2020. [Google Scholar] [CrossRef]
- Hampsey, M.; Reinberg, D. Tails of intrigue: Phosphorylation of RNA polymerase II mediates histone methylation. Cell 2003, 113, 429–432. [Google Scholar] [CrossRef][Green Version]
- Hancock, R.L.; Dunne, K.; Walport, L.J.; Flashman, E.; Kawamura, A. Epigenetic regulation by histone demethylases in hypoxia. Epigenomics 2015, 7, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.L.; Masson, N.; Dunne, K.; Flashman, E.; Kawamura, A. The activity of JmjC histone lysine demethylase KDM4A is highly sensitive to oxygen concentrations. ACS Chem. Biol. 2017, 12, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef]
- Huang, K.T.; Mikeska, T.; Dobrovic, A.; Fox, S.B. DNA methylation analysis of the HIF-1α prolyl hydroxylase domain genes PHD1, PHD2, PHD3 and the factor inhibiting HIF gene FIH in invasive breast carcinomas. Histopathology 2010, 57, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Islam, K.N.; Mendelson, C.R. Permissive effects of oxygen on cyclic AMP and interleukin-1 stimulation of surfactant protein a gene expression are mediated by epigenetic mechanisms. Mol. Cell. Biol. 2006, 26, 2901–2912. [Google Scholar] [CrossRef]
- Islam, S.; Leissing, T.M.; Chowdhury, R.; Hopkinson, R.J.; Schofield, C.J. 2-Oxoglutarate-dependent oxygenases. Annu. Rev. Biochem. 2018, 87, 585–620. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Jakubicková, L.; Biesová, Z.; Pastoreková, S.; Kettmann, R.; Pastorek, J. Methylation of the CA9 promoter can modulate expression of the tumor-associated carbonic anhydrase IX in dense carcinoma cell lines. Int. J. Oncol. 2005, 26, 1121–1127. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. 2008, 640, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1α stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [CrossRef]
- Kirmes, I.; Szczurek, A.; Prakash, K.; Charapitsa, I.; Heiser, C.; Musheev, M.U.; Schock, F.; Fornalczyk, K.; Ma, D.; Birk, U.; et al. A transient ischemic environment induces reversible compaction of chromatin. Genome Biol. 2015, 16, 246. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.Y.; Lu, G.; Ramkissoon, S.H.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.V.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1α enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 2009, 30, 344–353. [Google Scholar] [CrossRef]
- Lachance, G.; Uniacke, J.; Audas, T.E.; Holterman, C.E.; Franovic, A.; Payette, J.; Lee, S. DNMT3a epigenetic program regulates the HIF-2alpha oxygen-sensing pathway and the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 2014, 111, 7783–7788. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J., Jr.; Kaelin, W.G.; Godley, L.A.; Koivunen, P. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef]
- Laukka, T.; Myllykoski, M.; Looper, R.E.; Koivunen, P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J. Mol. Biol. 2018, 430, 3081–3092. [Google Scholar] [CrossRef]
- Lee, H.Y.; Choi, K.; Oh, H.; Park, Y.K.; Park, A.H. HIF-1-dependent induction of jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol. Cells 2014, 37, 43–50. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Chae, S.; Moon, Y.; Lee, H.Y.; Park, B.; Yang, E.G.; Hwang, D.; Park, H. Multi-dimensional histone methylations for coordinated regulation of gene expression under hypoxia. Nucleic Acids Res. 2017, 45, 11643–11657. [Google Scholar] [CrossRef]
- Li, Y.; Gruber, J.J.; Litzenburger, U.M.; Zhou, Y.; Miao, Y.R.; LaGory, E.L.; Li, A.M.; Hu, Z.; Yip, M.; Hart, L.S.; et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Lin, G.; Sun, W.; Haiyang, L.; Guo, J.; Liu, H.; Liang, J. Hypoxia induces the expression of TET enzymes in HepG2 cells. Oncol. Lett. 2017, 14, 6457–6462. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Geng, H.; Xue, C.; Beer, T.M.; Qian, D.Z. Functional regulation of hypoxia inducible factor-1α by SET9 lysine methyltransferase. Biochim. Biophys. Acta 2015, 1853, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia induces genomic DNA demethylation through the activation of HIF-1 and transcriptional upregulation of MAT2A in hepatoma cells. Mol. Cancer Ther. 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chenxi, X.; Xu, C.; Leng, X.; Cao, H.; Ouyang, G.; Xiaoqian, L. Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res. 2015, 43, 5081–5098. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chu, A.; Turker, M.S.; Glazer, P.M. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol. Cell. Biol. 2011, 31, 3339–3350. [Google Scholar] [CrossRef]
- Lu, Y.; Wajapeyee, N.; Turker, M.S.; Glazer, P.M. Silencing of the DNA mismatch repair gene MLH1 induced by hypoxic stress in a pathway dependent on the histone demethylase LSD1. Cell Rep. 2014, 8, 501–513. [Google Scholar] [CrossRef]
- Luo, W.; Chang, R.; Zhong, J.; Pandey, A.; Semenza, G.L. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, E3367–E3376. [Google Scholar] [CrossRef]
- Mariani, C.J.; VasanthaKumar, A.; Madzo, J.; Yesilkanal, A.; Bhagat, T.; Yu, Y.; Bhattacharyya, S.; Wenger, R.H.; Cohn, S.L.; Nanduri, J.; et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014, 7, 1343–1352. [Google Scholar] [CrossRef]
- Miar, A.; Arnaiz, E.; Bridges, E.M.; Beedie, S.; Cribbs, A.; Downes, D.J.; A Beagrie, R.; Rehwinkel, J.; Harris, A.L. Hypoxia induces transcriptional and translational downregulation of the type I interferon (IFN) pathway in multiple cancer cell types. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.I.; et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef]
- Murai, M.; Toyota, M.; Satoh, A.; Suzuki, H.; Akino, K.; Mita, H.; Sasaki, Y.; Ishida, T.; Shen, L.; Garcia-Manero, G.; et al. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br. J. Cancer 2005, 92, 1165–1172. [Google Scholar] [CrossRef]
- Musselman, C.A.; LaLonde, M.E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef]
- Niu, X.; Zhang, T.; Liao, L.; Zhou, L.; Lindner, D.J.; Zhou, M.; Rini, B.; Yan, Q.; Yang, H. The von Hippel–Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 2011, 31, 776–786. [Google Scholar] [CrossRef]
- Okami, J.; Simeone, D.M.; Logsdon, C.D. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 2004, 64, 5338–5346. [Google Scholar] [CrossRef] [PubMed]
- Olcina, M.M.; Foskolou, I.P.; Anbalagan, S.; Senra, J.M.; Pires, I.M.; Jiang, Y.; Ryan, A.J.; Hammond, E.M. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol. Cell 2013, 52, 758–766. [Google Scholar] [CrossRef]
- Olcina, M.M.; Leszczynska, K.B.; Senra, J.M.; Isa, N.F.; Harada, H.; Hammond, E.M. H3K9me3 facilitates hypoxia-induced p53-dependent apoptosis through repression of APAK. Oncogene 2015, 35, 793–799. [Google Scholar] [CrossRef]
- Osumek, J.E.; Revesz, A.; Morton, J.S.; Davidge, S.T.; Hardy, D.B. Enhanced trimethylation of histone H3 mediates impaired expression of hepatic glucose 6-phosphatase expression in offspring from rat dams exposed to hypoxia during pregnancy. Reprod. Sci. 2014, 21, 112–121. [Google Scholar] [CrossRef]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef]
- Place, T.L.; Fitzgerald, M.P.; Venkataraman, S.; Vorrink, S.U.; Case, A.J.; Teoh, M.L.T.; Domann, F.E. Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS ONE 2011, 6, e14617. [Google Scholar] [CrossRef]
- Platt, J.L.; Salama, R.; Smythies, J.; Choudhry, H.; Davies, J.; Hughes, J.R.; Ratcliffe, P.J.; Mole, D.R. Capture-C reveals preformed chromatin interactions between HIF -binding sites and distant promoters. EMBO Rep. 2016, 17, 1410–1421. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1[142] in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef]
- Prickaerts, P.; Adriaens, M.E.; Beucken, T.V.D.; Koch, E.; Dubois, L.J.; Dahlmans, V.E.H.; Gits, C.; Evelo, C.T.; Chan-Seng-Yue, M.A.; Wouters, B.G.; et al. Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenetics Chromatin 2016, 9, 46. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.W.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1α. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Shi, Z.; Bai, X.; Xia, Y.; Zheng, Y.; Xu, D.; Chen, F.; You, Y.; Fang, J.; et al. KDM3A senses oxygen availability to regulate PGC-1α-mediated mitochondrial biogenesis. Mol. Cell 2019, 76, 885–895. [Google Scholar] [CrossRef]
- Ross, S.E.; Bogdanovic, O. TET enzymes, DNA demethylation and pluripotency. Biochem. Soc. Trans. 2019, 47, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Ruas, J.L.; Poellinger, L.; Pereira, T.S. Role of CBP in regulating HIF-1-mediated activation of transcription. J. Cell Sci. 2005, 118, 301–311. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Rybnikova, E.; Samoilov, M. Current insights into the molecular mechanisms of hypoxic pre- and postconditioning using hypobaric hypoxia. Front. Neurosci. 2015, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Tarhonskaya, H.; Al-Qahtani, K.; Hopkinson, R.J.; Mccullagh, J.; Schofield, C.J.; Flashman, E. Investigations on the oxygen dependence of a 2-oxoglutarate histone demethylase. Biochem. J. 2012, 449, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Schmid, V.; LaFleur, V.N.; Lombardi, O.; Li, R.; Salama, R.; Colli, L.; Choudhry, H.; Chanock, S.; Ratcliffe, P.J.; Mole, D.R. Co-incidence of RCC-susceptibility polymorphisms with HIF cis-acting sequences supports a pathway tuning model of cancer. Sci. Rep. 2019, 9, 18768. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2010, 117, e207–e217. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Schorg, A.; Santambrogio, S.; Platt, J.L.; Schodel, J.; Lindenmeyer, M.T.; Cohen, C.D.; Schrodter, K.; Mole, D.R.; Wenger, R.H.; Hoogewijs, D. Destruction of a distal hypoxia response element abolishes trans-activation of the PAG1 gene mediated by HIF-independent chromatin looping. Nucleic Acids Res. 2015, 43, 5810–5823. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Seo, K.S.; Park, J.H.; Heo, J.Y.; Jing, K.; Han, J.; Min, K.N.; Kim, C.; Koh, G.Y.; Lim, K.; Kang, G.Y.; et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1α hydroxylation. Oncogene 2015, 34, 1354–1362. [Google Scholar] [CrossRef]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B.L. Induction of DNA Hypomethylation by Tumor Hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef]
- Skowronski, K.; Dubey, S.; Rodenhiser, D.I.; Coomber, B.L. Ischemia dysregulates DNA methyltransferases and p16INK4a methylation in human colorectal cancer cells. Epigenetics 2010, 5, 547–556. [Google Scholar] [CrossRef]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep. 2019, 20, e46401. [Google Scholar] [CrossRef]
- Spencer, T.E.; Jenster, G.; Burcin, M.M.; Allis, C.D.; Zhou, J.; Mizzen, C.A.; McKenna, N.J.; Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997, 389, 194–198. [Google Scholar] [CrossRef]
- Suzuki, N.; Vojnović, N.; Lee, K.L.; Yang, H.; Gradin, K.; Poellinger, L. HIF-dependent and reversible nucleosome disassembly in hypoxia-inducible gene promoters. Exp. Cell Res. 2018, 366, 181–191. [Google Scholar] [CrossRef]
- Tarhonskaya, H.; Chowdhury, R.; Leung, I.K.H.; Loik, N.D.; Mccullagh, J.S.O.; Claridge, T.D.W.; Schofield, C.J.; Flashman, E. Investigating the contribution of the active site environment to the slow reaction of hypoxia-inducible factor prolyl hydroxylase domain 2 with oxygen. Biochem. J. 2014, 463, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Tausendschön, M.; Dehne, N.; Brüne, B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine 2011, 53, 256–262. [Google Scholar] [CrossRef]
- Tausendschön, M.; Rehli, M.; Dehne, N.; Schmidl, C.; Döring, C.; Hansmann, M.L.; Brüne, B. Genome-wide identification of hypoxia-inducible factor-1 and -2 binding sites in hypoxic human macrophages alternatively activated by IL-10. Biochim. Biophys. Acta 2015, 1849, 10–22. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Tsai, Y.P.; Chen, H.F.; Chen, S.Y.; Cheng, W.C.; Wang, H.W.; Shen, Z.J.; Song, C.; Teng, D.S.C.; He, C.; Wu, K.J. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014, 15, 513. [Google Scholar] [CrossRef]
- Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. 2020, 20, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, R.; Wu, X.; Hankinson, O. Roles of coactivators in hypoxic induction of the erythropoietin gene. PLoS ONE 2010, 5, e10002. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Duan, Z.; Hu, W. Hypoxia-induced alterations of transcriptome and chromatin accessibility in HL-1 cells. IUBMB Life 2020, 72, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.C.; Banovich, N.E.; Sarkar, A.K.; Stephens, M.; Gilad, Y. Dynamic effects of genetic variation on gene expression revealed following hypoxic stress in cardiomyocytes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef]
- Wellmann, S.; Bettkober, M.; Zelmer, A.; Seeger, K.; Faigle, M.; Eltzschig, H.K.; Bührer, C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem. Biophys. Res. Commun. 2008, 372, 892–897. [Google Scholar] [CrossRef]
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Camenisch, G.; Gassmann, M. Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. Eur. J. Biochem. FEBS 1998, 253, 771–777. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef]
- Wu, M.Z.; Chen, S.F.; Nieh, S.; Benner, C.; Ger, L.P.; Jan, C.I.; Ma, L.; Chen, C.H.; Hishida, T.; Chang, H.T.; et al. Hypoxia drives breast tumor malignancy through a TET-TNFalpha-p38-MAPK signaling axis. Cancer Res. 2015, 75, 3912–3924. [Google Scholar] [CrossRef]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Xenaki, G.; Ontikatze, T.; Rajendran, R.; Stratford, I.J.; Dive, C.; Krstic-Demonacos, M.; Krstic-Demonacos, M. PCAF is an HIF-1α cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene 2008, 27, 5785–5796. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Kung, A.L. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009, 10, R113. [Google Scholar] [CrossRef]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef]
- Xu, X.H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018, 32, 6694–6705. [Google Scholar] [CrossRef]
- Yang, J.J.; Harris, A.L.; Davidoff, A.M. Hypoxia and hormone-mediated pathways converge at the histone demethylase KDM4B in cancer. Int. J. Mol. Sci. 2018, 19, 240. [Google Scholar] [CrossRef]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef]
- Yang, J.; Ledaki, I.; Turley, H.; Gatter, K.C.; Montero, J.C.M.; Li, J.L.; Harris, A.L. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. N. Y. Acad. Sci. 2009, 1177, 185–197. [Google Scholar] [CrossRef]
- Yin, H.; Blanchard, K.L. DNA methylation represses the expression of the human erythropoietin gene by two different mechanisms. Blood 2000, 95, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Y.; Jiang, S.; Liu, Y.; Huang, L.; Zhang, T.; Lu, G.; Gong, K.; Ji, X.; Shao, G. The effect of hypoxia preconditioning on DNA methyltransferase and PP1gamma in hippocampus of hypoxia preconditioned mice. High Alt. Med. Biol. 2014, 15, 483–490. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, H.; Chen, H.; Zavadil, J.; Kluz, T.; Arita, A.; Costa, M. Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res. 2010, 70, 4214–4221. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sensitivity of 2-OG-Dependent Dioxygenases to Oxygen | |||
|---|---|---|---|
| Enzyme | Reference | Km for Oxygen (mM) | Target |
| PHD2 | Hirsila, 2003 [49] | 250 | HIF |
| Ehrismann, 2007 [50] | 250 | ||
| Dao, 2009 [51] | 1746 ± 574 | ||
| Tarhonskaya, 2014 [52] | > 450 | ||
| FIH | Ehrismann, 2007 [50] | 90-237 | |
| C-P4H | Hirsila, 2003 [49] | 40 | collagen |
| KDM3A | Qian, 2019 [53] | 7.59% ± 0.80% | H3K9me1/2 |
| KDM4A | Cascella, 2012 [54] | 57 ± 10 | H3K9me2/3, H3K36me2/3, H1.4K26me2/3 |
| Hancock, 2017 [55] | 173 ± 23 | ||
| Chakraborty, 2019 [56] | 60 ± 20 | ||
| KDM4B | Chakraborty, 2019 [56] | 150 ± 40 | |
| KDM4C | Cascella, 2012 [54] | 158 ± 13 | |
| KDM4E | Cascella, 2012 [16] | 197 ± 16 | H3K9me2/3 |
| Sanchez-Fernandez, 2013 [57] | > 93 | ||
| KDM5A | Chakraborty, 2019 [56] | 90 ± 30 | H3K4me2/3 |
| KDM5B | Chakraborty, 2019 [56] | 40 ± 10 | |
| KDM5C | Chakraborty, 2019 [56] | 35 ± 10 | |
| KDM5D | Chakraborty, 2019 [56] | 25 ±5 | |
| KDM6A | Chakraborty, 2019 [56] | 200 ± 50 | H3K27me3 |
| KDM6B | Chakraborty, 2019 [56] | 25 ± 5 | |
| TET1 | Laukka, 2016 [58] | 30 | methylcytosine |
| Thienpont, 2016 [59] | 0.31% | ||
| TET2 | Laukka, 2016 [58] | 30 | |
| Thienpont, 2016 [59] | 0.53% | ||
| Chromatin Modifying Enzymes Targeted by HIF | Target | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KDM2A | ✔ | H3K36me1/2 | ||||||||||||||||
| KDM2B | ✔ | |||||||||||||||||
| KDM3A | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | H3K9me1/2 | |||||||||||
| KDM3B | ✔ | |||||||||||||||||
| KDM4B | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | H3K9me2/3, H3K36me2/3, H1.4K26me2/3 | |||||||||||
| KDM4C | ✔ | ✔ | ✔ | |||||||||||||||
| KDM5B | ✔ | ✔ | H3K4me2/3 | |||||||||||||||
| KDM5C | ✔ | |||||||||||||||||
| KDM6B | ✔ | H3K27me2/3 | ||||||||||||||||
| JMJD6 | ✔ | |||||||||||||||||
| PLU-1 | ✔ | |||||||||||||||||
| SMCX | ✔ | |||||||||||||||||
| RBP2 | ✔ | |||||||||||||||||
| KIAA1718 | ✔ | |||||||||||||||||
| TET1 | ✔ | ✔ | ✔ | ✔ | methylcytosine | |||||||||||||
| TET2 | ✔ | |||||||||||||||||
| TET3 | ✔ | ✔ | ||||||||||||||||
| DNMT1 | ✔ | ✔ | ✔ | cytosine | ||||||||||||||
| DNMT3A | ✔ | |||||||||||||||||
| DNMT3B | - | ✔ | ||||||||||||||||
| Pollard, 2008 [67] | Beyer, 2008 [68] | Wellman, 2009 [69] | Xia, 2009 [29] | Yang, 2009 [70] | Krieg, 2010 [71] | Fu, 2012 [72] | Niu, 2012 [73] | Lee, 2014 [74] | Batie, 2017 [76] | Liu, 2011 [40] | Watson, 2014 [77] | Xu, 2018 [78] | Mariana, 2014 [79] | Tsai, 2014 [80] | Wu, 2015 [81] | Lin, 2017 [82] | ||
| Global Changes to Histone Modifications in Hypoxia | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Cell Line(s) | % O2 | Time (h) | H2AK5 | H3K4 | H3K9 | H3K14 | H3K16 | H3K27 | H3K36 | H3K79 | H4 | H4K5 | H4K12 | H4R3 | |||||||||
| ac | ac | me1 | me2 | me3 | ac | me1 | me2 | me3 | ac | ac | ac | me2 | me3 | me2 | me3 | me2 | ac | ac | ac | me2 | ||||
| Costa, 2005 [86] | A549 | 0.5 | 1.5–9 | ↓ | ↓ | ↑ | ↑ | ↓ | ||||||||||||||||
| Chen, 2006 [87] | A549, HOS, HEK293, MES | 0.5 | 1.5–24 | ↓ | ↓ | ↑ | ↑ | |||||||||||||||||
| Islam, 2006 [89] | Fetal lung type II | 2 | 24 | ↓ | ↑ | |||||||||||||||||||
| Johnson, 2008 [90] | Hepa 1-6 | 0.2 | 48 | ↑ | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | |||||||||||
| Xia, 2009 [29] | HepG2 | 0.5–5 | 24 | ↑ | ↑ | ↑ | ↑ | |||||||||||||||||
| Zhou, 2010 [99] | Beas-2B, A549 | 1 | 6–48 | ↑ | ||||||||||||||||||||
| Tausendschon, 2011 [96] | RAW254.7 | 1–8 | 24 | ↑ | ↑ | ↑ | ||||||||||||||||||
| Wu, 2011 [97] | FADU, MCF-7 | 1 | 18 | ↓ | ↓ | ↑ | ↑ | - | - | ↓ | ↓ | |||||||||||||
| Olcina, 2013 [101] | RKO | <0.1, 2 | 6 to 18 | ↑ | ↑ | - | ||||||||||||||||||
| Watson, 2014 [77] | PwR-1E | 10% × 7wks, 3% × 4wks, then 1% × 3wks | ↓ | |||||||||||||||||||||
| Osumek, 2014 [94] | McA-RH777 | 1, 5 | 24 or 48 | ↑ | ||||||||||||||||||||
| Dmitriev, 2015 [100] | PC12 | 0 & no glucose | 1 to 9 | ↓ | ||||||||||||||||||||
| Olcina, 2016 [93] | RKO | <0.1, 2 | 6–48 | ↑ | ||||||||||||||||||||
| Prickaerts, 2016 [95] | MCF-7 | <0.2 | 8 or 24 | ↑ | ↑ | |||||||||||||||||||
| Dobrynin, 2017 [88] | RKO | 0.1, 2 | 24 | ↑ | ↑ | |||||||||||||||||||
| Hancock, 2017 [55] | U2OS | 0.1–5 | 24 | - | ↑ | ↑ | ↑ | |||||||||||||||||
| Lee, 2017 [91] | hADSC | <0.5, 1, 2 | 24 or 48 | ↑ | ↑ | ↑ | ||||||||||||||||||
| Batie, 2019 [66] | HeLa, HFF | 1 | 0.5–24 | ↑ | ↑ | ↑ | ↑ | ↑ | ||||||||||||||||
| Chakraborty, 2019 [56] | mHepa-1 c4 | 5 | 96 | ↑ | ↑ | |||||||||||||||||||
| Li, 2020 [92] | CHP134,SMS- KCNR, MEF | 0.5 | 6 or 24 | ↓ | ↑ | ↓ | ↑ | |||||||||||||||||
| Locus-Specific Changes to Histone Modifications in Hypoxia | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Cell Line(s) | % O2 | Time (h) | Gene Locus | H3K4 | H3K9 | H3K27 | Expression | ||||||
| ac | me1 | me2 | me3 | ac | me2 | me3 | ac | me3 | ||||||
| Islam, 2006 [89] | Human fetal | 2 | 24 | SP-A | ↑ | ↑ | ||||||||
| Chen, 2006 [87] | A549 | 0.5 | 6 | Dhfr, Mlh1 | ↑ | ↓ | ||||||||
| Cap43 | - | |||||||||||||
| Johnson, 2008 [90] | Hepa 1-6 | 0.2 | 48 | AFP, ALB | ↑ | ↓ | ↑ | ↓ | ↓ | |||||
| EGR1, VEGF | ↑ | ↑ | ↓ | ↓ | ↑ | |||||||||
| Brn3-b | - | ↑ | ↓ | |||||||||||
| Lu, 2011 [107] | MCF-7 or RKO | 0.01 | 12–72 | RAD51, BRCA1 | ↓ | ↓ | ↓ | ↑ | ↓ | |||||
| VEGF | ↑ | ↑ | ↑ | ↑ | ↑ | |||||||||
| Wu, 2011 [97] | FADU | 1 | 18 | CDH2, VIM | ↓ | ↑ | ↓ | ↑ | ||||||
| CDH1 | ↓ | ↑ | ↑ | ↑ | ↑ | ↓ | ||||||||
| JUP | ↓ | ↑ | ↑ | ↓ | ||||||||||
| MCF-7 | 1 | 18 | CDH1 | ↑ | ↑ | ↓ | ||||||||
| Tausendschon, 2011 [96] | RAW254.7 | 1 | 24 | Ccl2, Ccr1, Ccr5 | ↑ | ↑ | ↓ | |||||||
| ADM | - | - | ↑ | |||||||||||
| Choudhry, 2014 [41] | MCF-7 | 1 | 24 | ALDOA, ADM | ↑ | ↑ | ||||||||
| Lu, 2014 [108] | MCF-7 | 0.01 | 12–72 | MLH1 | ↓ | ↓ | ↓ | ↑ | ↓ | |||||
| Schorg, 2015 [109] | MCF-7 | 0.5 | 16 | PAG1 | ↑ | - | ↑ | ↑ | ||||||
| EGLN3 | ↑ | |||||||||||||
| 786-0 | 0.5 | 16 | PAG1 | ↑ | ↑ | ↑ | - | |||||||
| EGLN3 | ↑ | |||||||||||||
| Adriaens, 2016 & Prickaerts, 2016 [95,110] | MCF-7 | <0.2 | 8 or 24 | CCNA2, DPM1, NOL11, ATP2A3, FOXF1, IGFBP4 | ↑ | |||||||||
| ATF3, LPO, APLN, CYP1B1, SLC9A5 | ↑ | |||||||||||||
| GPRC5B, OPRL1 | ↑ | ↑ | ||||||||||||
| LOX | ↑ | ↓ | ||||||||||||
| Olcina, 2016 [93] | RKO | <0.1 | 6 | APAK | ↑ | ↓ | ||||||||
| Dobrynin, 2017 [88] | RKO | <0.1 | 24 | HIF-1A | ↑ | ↓ | ||||||||
| Lee, 2017 [91] | hADSC | <0.5 | 48 | SLC22A15, PFKP, MEF2D, RUSC2 | ↑ | ↑ | ||||||||
| PDE4C, PFKFB4, MT3, STC1 | ↑ | ↑ | ||||||||||||
| SEC22B, BZW2, HNRNPA3, LUM | ↑ | ↓ | ||||||||||||
| CC2D2A, HSD17B4 | ↓ | ↓ | ||||||||||||
| Suzuki, 2018 [42] | SK-N-BE(2)c | 1 | 4 or 24 | CA9, PGK1 | ↑ | ↑ | ||||||||
| Batie, 2019 [66] | HeLa | 1 | 1–24 | BNIP3L, KLF10, LOX, ENO1, STAG2, CA9 | ↑ | ↑ | ||||||||
| BAP1, KDM2B | - | - | ||||||||||||
| ACTB | ↑ | |||||||||||||
| Chakraborty, 2019 [56] | C2C12 | 2 | 96 | Actc1, Myl1, Myog, Myh1, Myom3, Igfn1, Mb | ↑ | |||||||||
| Adora1, Gjd2 | - | |||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kindrick, J.D.; Mole, D.R. Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. Int. J. Mol. Sci. 2020, 21, 8320. https://doi.org/10.3390/ijms21218320
Kindrick JD, Mole DR. Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. International Journal of Molecular Sciences. 2020; 21(21):8320. https://doi.org/10.3390/ijms21218320
Chicago/Turabian StyleKindrick, Jessica D., and David R. Mole. 2020. "Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect" International Journal of Molecular Sciences 21, no. 21: 8320. https://doi.org/10.3390/ijms21218320
APA StyleKindrick, J. D., & Mole, D. R. (2020). Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. International Journal of Molecular Sciences, 21(21), 8320. https://doi.org/10.3390/ijms21218320