Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression

Abstract
1. Introduction
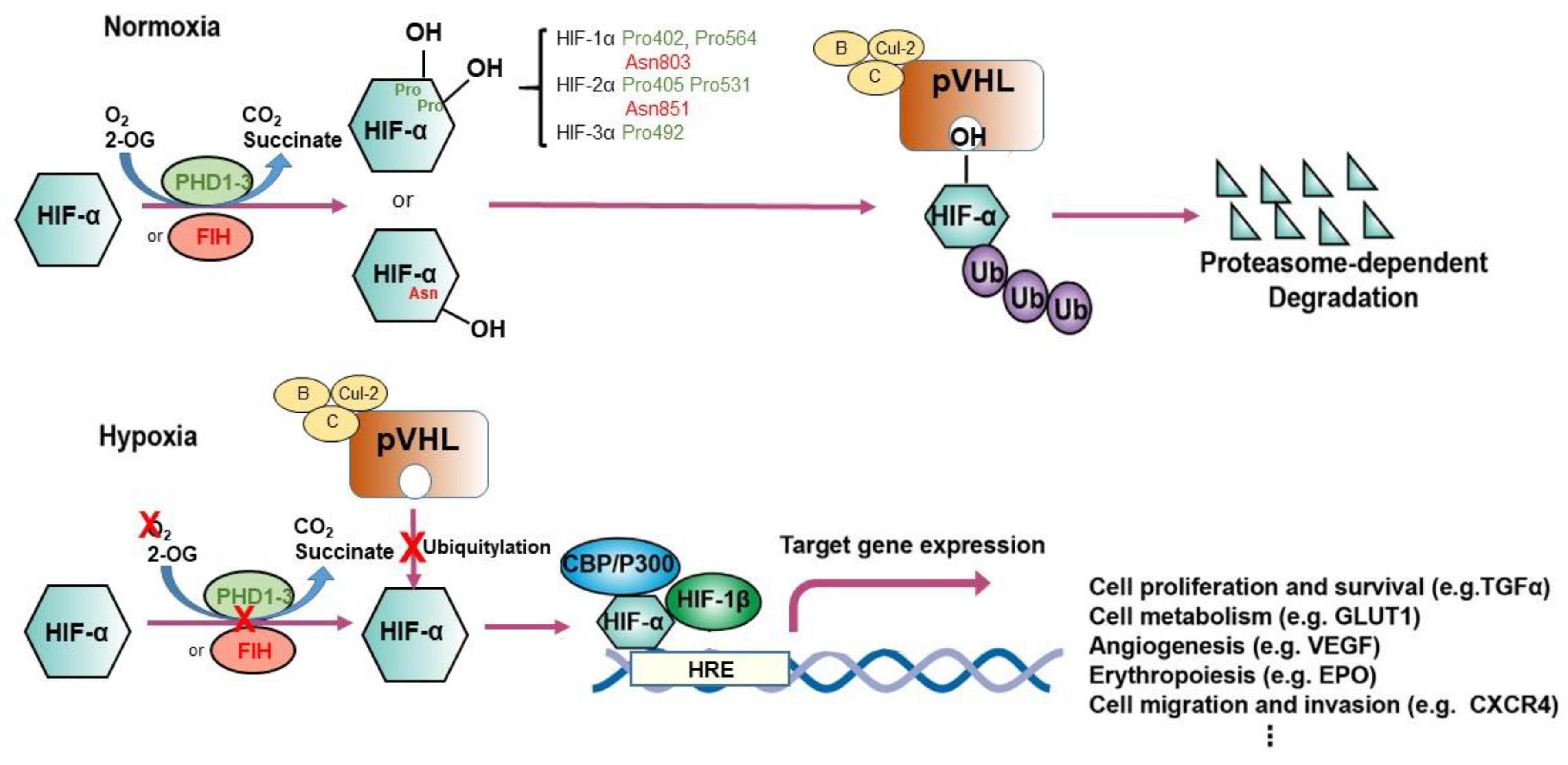
2. Canonical Hypoxia Signaling
2.1. HIF Transcription Factors: The Central Regulator of Oxygen Homeostasis
2.2. Prolyl Hydroxylation: The Adaptive Mediator of HIFs
2.3. pVHL: The Proteolysis Modulator of HIFs
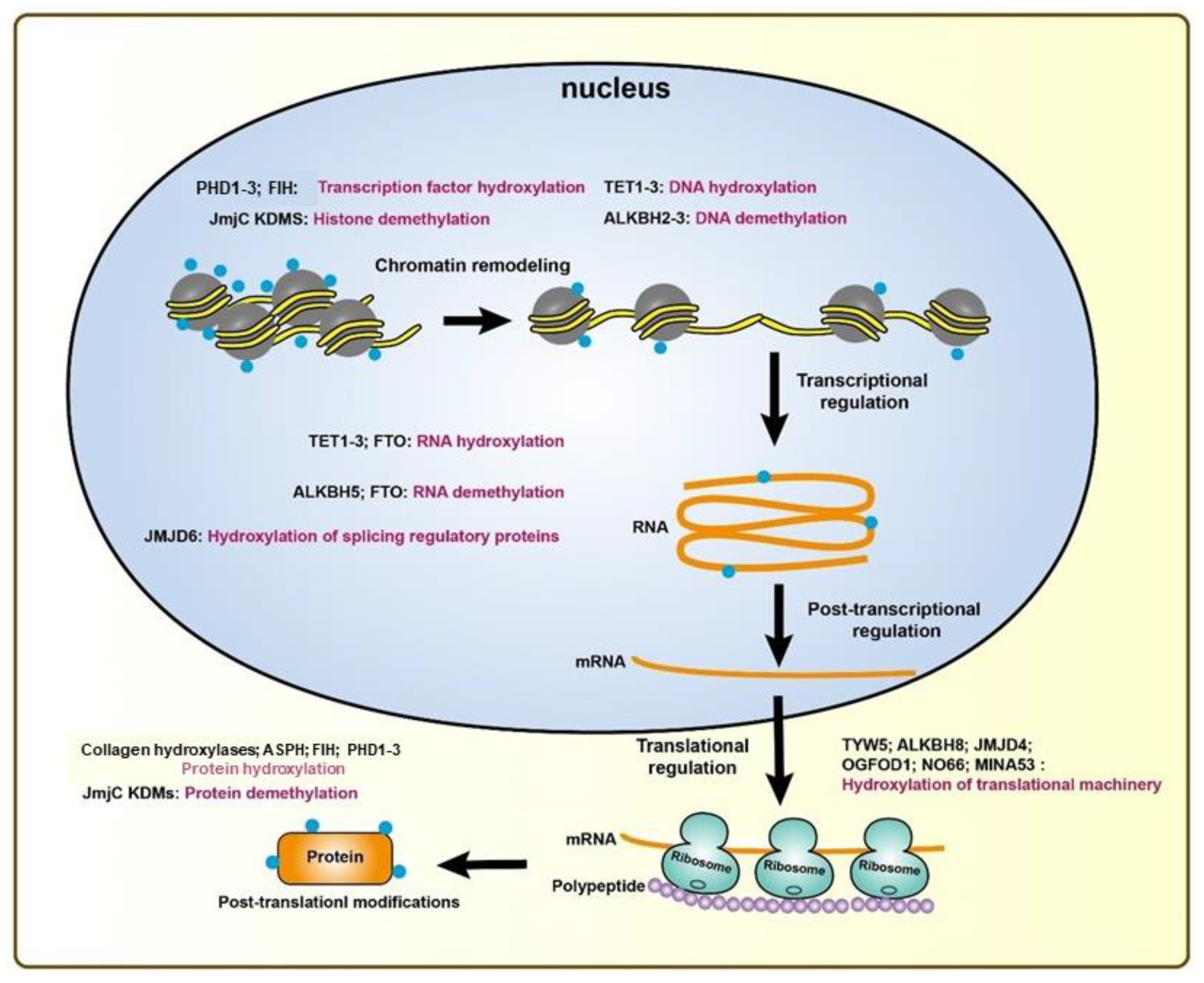
3. 2-Oxoglutarate-Dependent Enzymes in Gene Regulation and Cancer Progression
3.1. Proline and Asparagine Hydroxylases
3.2. DNA/RNA-Modifying Enzymes
3.3. JmjC Domain-containing Enzymes
4. Therapeutic Implications on Targeting Hypoxia and Oxygen Sensing Pathways in Cancer
4.1. Inhibitors for HIF-1α
4.2. Inhibitors for HIF-2α
4.3. Inhibitors for 2-OG-Dependent Enzymes
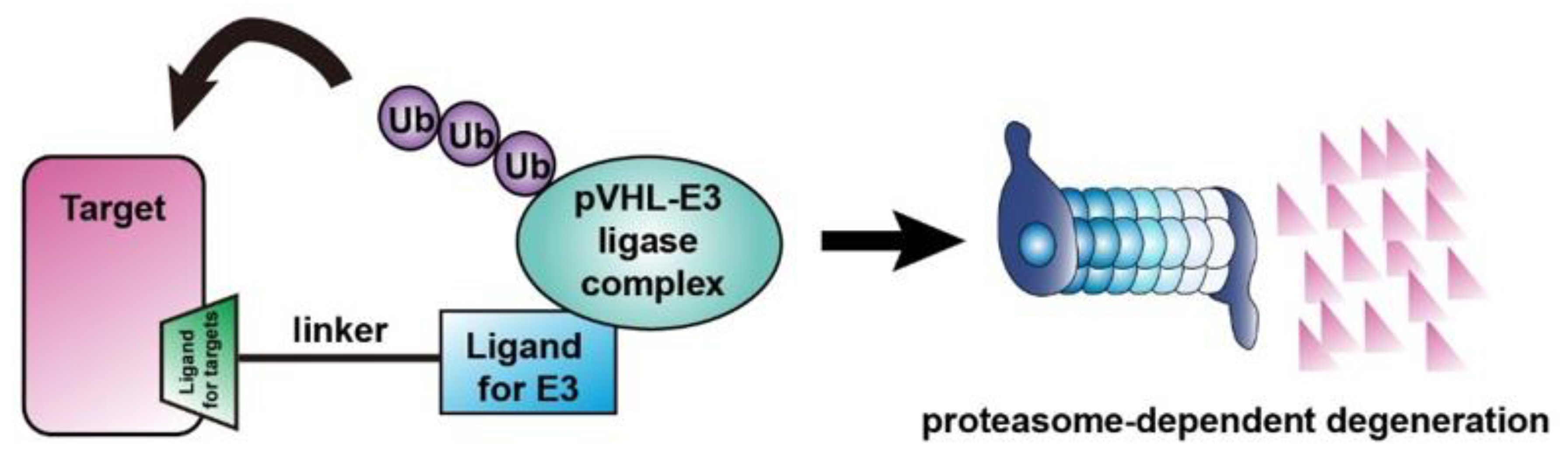
4.4. Targeting Proteins Using pVHL-Based PROTACs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-OG dependent | 2-oxoglutarate-dependent |
| 5caC | 5-carboxylcytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethycytosine |
| 5mC | 5-methylcytosine |
| ALDH2 | Aldehyde Dehydrogenase 2 |
| AML | Acute Myeloid Leukemia |
| ARNT | Aryl hydrocarbon Receptor Nuclear Translocator |
| Card9 | Caspase Recruitment Domain Family Member 9 |
| CBP | CREB-Binding Protein |
| ccRCC | Clear Cell Renal Cell Carcinoma |
| CEP68 | Centrosomal Protein 68 |
| CERKL | Ceramide Kinase-like Protein |
| CODD | C-terminal Oxygen-dependent Degradation Domain |
| CRBN | Cereblon |
| DEACM | Diethylamino Coumarin |
| DMNB | 4,5-dimethoxy-2-nitrobenzyl |
| EMT | Epithelial-Mesenchymal Transition |
| EPOR | Erythropoietin Receptor |
| FIH | Factor Inhibiting HIF |
| FLNA | Actin Cross-linker Filamin A |
| FTO | Fat mass and Obesity-associated protein |
| HIF | Hypoxia-Inducible Factor |
| HRE | Hypoxia-Responsive Elements |
| IP3R3 | Inositol-1,4,5-triphosphate Receptor type 3 |
| IPAS | Inhibitory PAS protein domain |
| JmjC | Jumonji C |
| KDM/JMJD | Histone Lysine Demethylase |
| LSD | Lysine Specific Demethylase |
| m6A | N6-Methyladenosine |
| NDRG3 | N-Myc Downstream-Regulated Gene 3 |
| NLR | NOD-Like Receptor |
| NODD | N-terminal Oxygen-dependent Degradation Domain |
| NPOM | 6-nitropiperonyloxymethyl |
| ODD | Oxygen-dependent Degradation Domain |
| PGC-1α | Proliferator-activated receptor-Gamma Coactivator-1 alpha |
| PHD | Prolyl Hydroxylase |
| PROTACs | PROteolysis-TArgeting Chimeras |
| pVHL | Von Hippel-Lindau protein |
| TBK1 | TANK Binding Kinase 1 |
| TDG | Thymine–DNA Glycosylase |
| TME | Tumor Micro-Environment |
| TET | Ten Eleven Translocation |
| Ub | Ubiquitylation |
| UPS | Ubiquitin-Proteasome System |
| VEGF | Vascular Endothelial Growth Factor |
| ZHX2 | Zinc fingers and Homeoboxes 2 |
References
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Batie, M.; Rocha, S. Gene transcription and chromatin regulation in hypoxia. Biochem. Soc. Trans. 2020, 48, 1121–1128. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Wielockx, B.; Grinenko, T.; Mirtschink, P.; Chavakis, T. Hypoxia Pathway Proteins in Normal and Malignant Hematopoiesis. Cells 2019, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.G.; Johnson, P.C.; Intaglietta, M. Oxygen Gradients in the Microcirculation. Physiol. Rev. 2003, 83, 933–963. [Google Scholar] [CrossRef] [PubMed]
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Zhang, Q.; Yan, Q.; Yang, H.; Wei, W. Oxygen sensing and adaptability won the 2019 Nobel Prize in Physiology or medicine. Genes Dis. 2019, 6, 328–332. [Google Scholar] [CrossRef]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Kuo, P.C. The tumor microenvironment. Surg. Oncol. 2012, 21, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The Clinical Significance of Hypoxia in Human Cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Zannella, V.E.; Pra, A.D.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming Metabolism with Metformin Improves Tumor Oxygenation and Radiotherapy Response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Mucaj, V.; Shay, J.E.S.; Simon, M.C. Effects of hypoxia and HIFs on cancer metabolism. Int. J. Hematol. 2012, 95, 464–470. [Google Scholar] [CrossRef]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nat. Cell Biol. 1996, 379, 88–91. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Haider, S.; McIntyre, A.; Van Stiphout, R.G.P.M.; Winchester, L.M.; Wigfield, S.; Harris, A.L.; Buffa, F.M. Genomic alterations underlie a pan-cancer metabolic shift associated with tumour hypoxia. Genome Biol. 2016, 17, 1–17. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; LaLonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451. [Google Scholar] [CrossRef]
- Krzywinska, E.; Stockmann, C. Hypoxia, Metabolism and Immune Cell Function. Biomedicines 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Losman, J.-A.; Kaelin, W.G. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kaelin, W.G. Role of VHL Gene Mutation in Human Cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Zhang, J. Understanding the Oxygen-Sensing Pathway and Its Therapeutic Implications in Diseases. Am. J. Pathol. 2020, 190, 1584–1595. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The von Hippel–Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873. [Google Scholar] [CrossRef]
- Okumura, F.; Uematsu, K.; Byrne, S.D.; Hirano, M.; Joo-Okumura, A.; Nishikimi, A.; Shuin, T.; Fukui, Y.; Nakatsukasa, K.; Kamura, T. Parallel Regulation of von Hippel-Lindau Disease by pVHL-Mediated Degradation of B-Myb and Hypoxia-Inducible Factor α. Mol. Cell. Biol. 2016, 36, 1803–1817. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide Association of Hypoxia-inducible Factor (HIF)-1α and HIF-2α DNA Binding with Expression Profiling of Hypoxia-inducible Transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.-H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA Binding, and Transactivation Properties of Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Rosenberger, C.; Mandriota, S.; Jürgensen, J.S.; Wiesener, M.S.; Hörstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.-U. Expression of Hypoxia-Inducible Factor-1 and -2 in Hypoxic and Ischemic Rat Kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef]
- Rossignol, F.; Vaché, C.; Clottes, E. Natural antisense transcripts of hypoxia-inducible factor 1alpha are detected in different normal and tumour human tissues. Gene 2002, 299, 135–140. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef]
- Greenald, D.; Jeyakani, J.; Pelster, B.; Sealy, I.; Mathavan, S.; Van Eeden, F.J. Genome-wide mapping of Hif-1α binding sites in zebrafish. BMC Genom. 2015, 16, 923. [Google Scholar] [CrossRef]
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Camenisch, G.; Gassmann, M. Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. JBIC J. Biol. Inorg. Chem. 1998, 253, 771–777. [Google Scholar] [CrossRef]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef]
- Hu, C.-J.; Iyer, S.; Sataur, A.; Covello, K.L.; Chodosh, L.A.; Simon, M.C. Differential Regulation of the Transcriptional Activities of Hypoxia-Inducible Factor 1 Alpha (HIF-1α) and HIF-2α in Stem Cells. Mol. Cell. Biol. 2006, 26, 3514–3526. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, C.; Zaborowska, Z.; Kurreck, J.; Erdmann, V.A.; Frei, U.; Wiesener, M.; Eckardt, K. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. FASEB J. 2004, 18, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2 regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA -binding specificities of the HIF -1α and HIF -2α transcription factors in chromatin. EMBO Rep. 2018, 20. [Google Scholar] [CrossRef]
- Platt, J.L.; Salama, R.; Smythies, J.; Choudhry, H.; Davies, J.; Hughes, J.R.; Ratcliffe, P.J.; Mole, D.R. Capture-C reveals preformed chromatin interactions between HIF -binding sites and distant promoters. EMBO Rep. 2016, 17, 1410–1421. [Google Scholar] [CrossRef]
- Lau, K.W.; Tian, Y.-M.; Raval, R.R.; Ratcliffe, P.J.; Pugh, C.W. Target gene selectivity of hypoxia-inducible factor-α in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br. J. Cancer 2007, 96, 1284–1292. [Google Scholar] [CrossRef]
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-Terminal Transactivation Domain Confers Target Gene Specificity of Hypoxia-inducible Factors HIF-1α and HIF-2α. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef]
- Ravenna, L.; Salvatori, L.; Russo, M.A. HIF3α: The little we know. FEBS J. 2015, 283, 993–1003. [Google Scholar] [CrossRef]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and Characterization of Hypoxia-Inducible Factor (HIF)-3α in Human Kidney: Suppression of HIF-Mediated Gene Expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS Domain Protein (IPAS) Is a Hypoxia-inducible Splicing Variant of the Hypoxia-inducible Factor-3α Locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186. [Google Scholar] [CrossRef]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.-J.; Duan, C. Hypoxia-Inducible Factor 3 Is an Oxygen-Dependent Transcription Activator and Regulates a Distinct Transcriptional Response to Hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Simon, M.C. From stem cells to cancer stem cells: HIF takes the stage. Curr. Opin. Cell Biol. 2012, 24, 232–235. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. et Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Physiol. 2015, 309, C775–C782. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79. [Google Scholar] [CrossRef]
- Masson, N.; Ratcliffe, P.J. Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014, 2, 3. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immun. 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef]
- Carroll, V.A.; Ashcroft, M. Role of Hypoxia-Inducible Factor (HIF)-1α versus HIF-2α in the Regulation of HIF Target Genes in Response to Hypoxia, Insulin-Like Growth Factor-I, or Loss of von Hippel-Lindau Function: Implications for Targeting the HIF Pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef]
- Yu, E.Z.; Li, Y.-Y.; Liu, X.-H.; Kagan, E.; McCarron, R.M. Antiapoptotic action of hypoxia-inducible factor-1α in human endothelial cells. Lab. Investig. 2004, 84, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Volm, M.; Koomägi, R. Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer Res. 2000, 20, 1527–1533. [Google Scholar]
- Srinivas, V.; Zhang, L.-P.; Zhu, X.-H.; Caro, J. Characterization of an Oxygen/Redox-Dependent Degradation Domain of Hypoxia-Inducible Factor α (HIF-α) Proteins. Biochem. Biophys. Res. Commun. 1999, 260, 557–561. [Google Scholar] [CrossRef]
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and Inhibitory Domains of Hypoxia-inducible Factor 1α: Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef]
- Pugh, C.W.; O’Rourke, J.F.; Nagao, M.; Gleadle, J.M.; Ratcliffe, P.J. Activation of Hypoxia-inducible Factor-1; Definition of Regulatory Domains within the α Subunit. J. Biol. Chem. 1997, 272, 11205–11214. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef]
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1 binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia Inducible Factor-α Binding and Ubiquitylation by the von Hippel-Lindau Tumor Suppressor Protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef]
- Maher, E.R.; Yates, J.R.W.; Harries, R.; Benjamin, C.; Moore, A.T.; Ferguson-Smith, M.A.; Harris, R. Clinical Features and Natural History of von Hippel-Lindau Disease. Qjm: Int. J. Med. 1990, 77, 1151–1163. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.-Y.; Huang, L.E.; Pavletich, N.P.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-la by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The von Hippel–Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 2005, 338, 627–638. [Google Scholar] [CrossRef]
- Kaelin, W.G. The von Hippel-Lindau Tumor Suppressor Protein: An Update; Academic Press: Waltham, MA, USA, 2007; Volume 435, pp. 371–383. [Google Scholar]
- Nyhan, M.J.; O’Sullivan, G.C.; McKenna, S.L. Role of the VHL (von Hippel–Lindau) gene in renal cancer: A multifunctional tumour suppressor. Biochem. Soc. Trans. 2008, 36, 472–478. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Von Hippel-Lindau Disease. Annu. Rev. Pathol. 2007, 2, 145–173. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, 35. [Google Scholar] [CrossRef]
- Yin, H.; Zheng, L.; Liu, W.; Zhang, D.; Li, W.; Yuan, L. Rootletin prevents Cep68 from VHL-mediated proteasomal degradation to maintain centrosome cohesion. Biochim. et Biophys. Acta (BBA) Bioenerg. 2017, 1864, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Saitoh, K.; Hirai, S.-I.; Iwai, K.; Takaki, Y.; Baba, M.; Minato, N.; Ohno, S.; Shuin, T. The von Hippel-Lindau Tumor Suppressor Protein Mediates Ubiquitination of Activated Atypical Protein Kinase C. J. Biol. Chem. 2001, 276, 43611–43617. [Google Scholar] [CrossRef]
- Lai, Y.; Qiao, M.; Song, M.; Weintraub, S.T.; Shiio, Y. Quantitative Proteomics Identifies the Myb-Binding Protein p160 as a Novel Target of the von Hippel-Lindau Tumor Suppressor. PLoS ONE 2011, 6, e16975. [Google Scholar] [CrossRef]
- Na, X.; Duan, H.O.; Messing, E.M.; Schoen, S.R.; Ryan, C.K.; Di Sant’Agnese, P.; Golemis, E.A.; Wu, G. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel–Lindau protein. EMBO J. 2003, 22, 4249–4259. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.-D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef]
- Liu, X.; Simon, J.M.; Xie, H.; Hu, L.; Wang, J.; Zurlo, G.; Fan, C.; Ptacek, T.S.; Herring, L.; Tan, X.; et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol. Cell 2020, 77, 1294–1306. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C.; et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2019, 10, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-H.; Wu, Z.-X.; Xie, L.-Q.; Li, C.-X.; Mao, Y.-Q.; Duan, Y.-T.; Han, B.; Han, S.-F.; Yu, Y.; Lu, H.-J.; et al. VHL deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating ALDH2. Nat. Commun. 2017, 8, 15337. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.-S.; Youn, H.-D. The Positive Regulation of p53 by the Tumor Suppressor VHL. Cell Cycle 2006, 5, 2054–2056. [Google Scholar] [CrossRef]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef]
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G. pVHL Acts as an Adaptor to Promote the Inhibitory Phosphorylation of the NF-κB Agonist Card9 by CK2. Mol. Cell 2007, 28, 15–27. [Google Scholar] [CrossRef][Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Hausinger, R.P. Fe(II)/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. [Google Scholar] [CrossRef]
- McNeill, L.A.; Flashman, E.; Buck, M.R.G.; Hewitson, K.S.; Clifton, I.J.; Jeschke, G.; Claridge, T.D.W.; Ehrismann, D.; Oldham, N.J.; Schofield, C.J. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol. BioSyst. 2005, 1, 321–324. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 617–626. [Google Scholar] [CrossRef]
- Ehrismann, D.; Flashman, E.; Genn, D.N.; Mathioudakis, N.; Hewitson, K.S.; Ratcliffe, P.J.; Schofield, C.J. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 2006, 401, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef]
- Takeda, K.; Ho, V.C.; Takeda, H.; Duan, L.-J.; Nagy, A.; Fong, G.-H. Placental but Not Heart Defects Are Associated with Elevated Hypoxia-Inducible Factor α Levels in Mice Lacking Prolyl Hydroxylase Domain Protein 2. Mol. Cell. Biol. 2006, 26, 8336–8346. [Google Scholar] [CrossRef] [PubMed]
- Marxsen, J.H.; Stengel, P.; Doege, K.; Heikkinen, P.; Jokilehto, T.; Wagner, T.; Jelkmann, W.; Jaakkola, P.M.; Metzen, E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-α-prolyl-4-hydroxylases. Biochem. J. 2004, 381, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. A Conserved Family of Prolyl-4-Hydroxylases That Modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef]
- Villar, D.; Vara-Vega, A.; Landázuri, M.O.; Del Peso, L. Identification of a region on hypoxia-inducible-factor prolyl 4-hydroxylases that determines their specificity for the oxygen degradation domains. Biochem. J. 2007, 408, 231–240. [Google Scholar] [CrossRef][Green Version]
- Rodríguez, J.; Pilkington, R.; Munoz, A.G.; Nguyen, L.K.; Rauch, N.; Kennedy, S.; Monsefi, N.; Herrero, A.; Taylor, C.T.; Von Kriegsheim, A. Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell Rep. 2016, 14, 2745–2760. [Google Scholar] [CrossRef]
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-M.; Kang, M.; Jang, Y.J.; Yang, S.-J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef]
- Heir, P.; Srikumar, T.; Bikopoulos, G.; Bunda, S.; Poon, B.P.; Lee, J.E.; Raught, B.; Ohh, M. Oxygen-dependent Regulation of Erythropoietin Receptor Turnover and Signaling. J. Biol. Chem. 2016, 291, 7357–7372. [Google Scholar] [CrossRef]
- Zurlo, G.; Liu, X.; Takada, M.; Fan, C.; Simon, J.; Ptacek, T.S.; Rodriguez, J.; Von Kriegsheim, A.; Liu, J.; Locasale, J.W.; et al. Prolyl hydroxylase substrate adenylosuccinate lyase is an oncogenic driver in triple negative breast cancer. Nat. Commun. 2019, 10, 5177. [Google Scholar] [CrossRef]
- Zheng, X.; Zhai, B.; Koivunen, P.; Shin, S.J.; Lu, G.; Liu, J.; Geisen, C.; Chakraborty, A.A.; Moslehi, J.J.; Smalley, D.M.; et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014, 28, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, C.; Chen, X.; Takada, M.; Fan, C.; Zheng, X.; Wen, H.; Liu, Y.; Pestell, R.G.; Aird, K.M.; et al. EglN2 associates with the NRF 1- PGC 1α complex and controls mitochondrial function in breast cancer. EMBO J. 2015, 34, 2953–2970. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.; McNeill, L.A.; Riordan, M.V.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Schofield, C.J.; Tian, Y.-M.; et al. Hypoxia-inducible Factor (HIF) Asparagine Hydroxylase Is Identical to Factor Inhibiting HIF (FIH) and Is Related to the Cupin Structural Family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef]
- Bracken, C.P.; Fedele, A.O.; Linke, S.; Balrak, W.; Lisy, K.; Whitelaw, M.L.; Peet, D.J. Cell-specific Regulation of Hypoxia-inducible Factor (HIF)-1α and HIF-2α Stabilization and Transactivation in a Graded Oxygen Environment. J. Biol. Chem. 2006, 281, 22575–22585. [Google Scholar] [CrossRef] [PubMed]
- Schödel, J.; Bohr, D.; Klanke, B.; Schley, G.; Schlötzer-Schrehardt, U.; Warnecke, C.; Kurtz, A.; Amann, K.; Eckardt, K.-U.; Willam, C. Factor inhibiting HIF limits the expression of hypoxia-inducible genes in podocytes and distal tubular cells. Kidney Int. 2010, 78, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Clissold, P.M.; Ponting, C.P. JmjC: Cupin metalloenzyme-like domains in jumonji, hairless and phospholipase A2β. Trends Biochem. Sci. 2001, 26, 7–9. [Google Scholar] [CrossRef]
- Tian, Y.-M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential Sensitivity of Hypoxia Inducible Factor Hydroxylation Sites to Hypoxia and Hydroxylase Inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [Google Scholar] [CrossRef]
- Elkins, J.M.; Hewitson, K.S.; McNeill, L.A.; Seibel, J.F.; Schlemminger, I.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Structure of Factor-inhibiting Hypoxia-inducible Factor (HIF) Reveals Mechanism of Oxidative Modification of HIF-1α. J. Biol. Chem. 2002, 278, 1802–1806. [Google Scholar] [CrossRef]
- Cockman, M.E.; Webb, J.D.; Ratcliffe, P.J. FIH-Dependent Asparaginyl Hydroxylation of Ankyrin Repeat Domain-Containing Proteins. Ann. N. Y. Acad. Sci. 2009, 1177, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Kuchnio, A.; Bruning, U.; Carmeliet, P. Emerging novel functions of the oxygen-sensing prolyl hydroxylase domain enzymes. Trends Biochem. Sci. 2013, 38, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ge, W.; Chowdhury, R.; Claridge, T.D.W.; Kramer, H.; Schmierer, B.; McDonough, M.A.; Gong, L.; Kessler, B.M.; Ratcliffe, P.J.; et al. Asparagine and Aspartate Hydroxylation of the Cytoskeletal Ankyrin Family Is Catalyzed by Factor-inhibiting Hypoxia-inducible Factor. J. Biol. Chem. 2010, 286, 7648–7660. [Google Scholar] [CrossRef]
- Ge, W.; Wolf, A.; Feng, T.; Ho, C.-H.; Sekirnik, R.; Zayer, A.; Granatino, N.; Cockman, M.E.; Loenarz, C.; Loik, N.D.; et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat. Chem. Biol. 2012, 8, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Fu, Z.; Linke, S.; Chicher, J.; Gorman, J.J.; Visk, D.W.; Haddad, G.G.; Poellinger, L.; Peet, D.J.; Powell, F.L.; et al. The Asparaginyl Hydroxylase Factor Inhibiting HIF-1α Is an Essential Regulator of Metabolism. Cell Metab. 2010, 11, 364–378. [Google Scholar] [CrossRef]
- Liao, C.; Zhang, Y.; Fan, C.; Herring, L.E.; Liu, J.; Locasale, J.W.; Takada, M.; Zhou, J.; Zurlo, G.; Hu, L.; et al. Identification of BBOX1 as a Therapeutic Target in Triple-Negative Breast Cancer. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nat. Cell Biol. 2010, 466, 1129–1133. [Google Scholar] [CrossRef]
- Gu, T.-P.; Guo, F.; Yang, H.; Wu, H.-P.; Xu, G.-F.; Liu, W.; Xie, Z.-G.; Shi, L.; He, X.; Jin, S.-G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nat. Cell Biol. 2011, 477, 606–610. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Paschka, P.; Späth, D.; Habdank, M.; Köhne, C.-H.; Germing, U.; Von Lilienfeld-Toal, M.; Held, G.; Horst, H.-A.; Haase, D.; et al. TET2 Mutations in Acute Myeloid Leukemia (AML): Results From a Comprehensive Genetic and Clinical Analysis of the AML Study Group. J. Clin. Oncol. 2012, 30, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Lorsbach, R.; Moore, J.; Mathew, S.; Raimondi, S.; Mukatira, S.; Downing, J. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t (10; 11)(q22; q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Äijö, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lähdesmäki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nat. Cell Biol. 2013, 497, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y. Regulation of TET Protein Stability by Calpains. Cell Rep. 2014, 6, 278–284. [Google Scholar] [CrossRef]
- Shi, F.-T.; Kim, H.; Lu, W.; He, Q.; Liu, D.; Goodell, M.A.; Wan, M.; Songyang, Z. Ten-Eleven Translocation 1 (Tet1) Is Regulated by O-Linked N-Acetylglucosamine Transferase (Ogt) for Target Gene Repression in Mouse Embryonic Stem Cells. J. Biol. Chem. 2013, 288, 20776–20784. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nat. Cell Biol. 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-Hydroxymethylcytosine Is an Epigenetic Hallmark of Melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-Antagonism Regulates Breast Cancer Stemness and Metastasis via TET-Family-Dependent Chromatin Remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.; Yu, G.; Rong, F.; Zhang, D.; Xu, C.; Du, J.; Li, Z.; Ouyang, G.; Xiao, W. TET is targeted for proteasomal degradation by the PHD-pVHL pathway to reduce DNA hydroxymethylation. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Chen, T.; Hao, Y.-J.; Zhang, Y.; Li, M.-M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L. m6A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 2015, 16, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.M.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nat. Cell Biol. 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N 6 -Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ren, D.; Du, Z.; Wang, H.; Zhang, H.; Jin, Y. m 6 A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem. Biophys. Res. Commun. 2018, 502, 456–464. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.-F.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.-G.; et al. m 6 A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nat. Cell Biol. 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Hopkinson, R.J.; Schofield, C.J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 2012, 16, 525–534. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Shi, Z.; Bai, X.; Xia, Y.; Zheng, Y.; Xu, D.; Chen, F.; You, Y.; Fang, J.; et al. KDM3A Senses Oxygen Availability to Regulate PGC-1α-Mediated Mitochondrial Biogenesis. Mol. Cell 2019, 76, 885–895. [Google Scholar] [CrossRef]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R.; et al. Jmjd6 Catalyses Lysyl-Hydroxylation of U2AF65, a Protein Associated with RNA Splicing. Science 2009, 325, 90–93. [Google Scholar] [CrossRef]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-Associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Markolovic, S.; Wilkins, S.E.; Schofield, C.J. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 2015, 290, 20712–20722. [Google Scholar] [CrossRef]
- Vogler, M.; Zieseniss, A.; Hesse, A.R.; Levent, E.; Tiburcy, M.; Heinze, E.; Burzlaff, N.; Schley, G.; Eckardt, K.U.; Willam, C.; et al. Pre- and post-conditional inhibition of prolyl-4-hydroxylase domain enzymes protects the heart from an ischemic insult. Pflügers Archiv Eur. J. Physiol. 2015, 467, 2141–2149. [Google Scholar] [CrossRef]
- Fraisl, P.; Aragonés, J.; Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 2009, 8, 139–152. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The Expression and Distribution of the Hypoxia-Inducible Factors HIF-1α and HIF-2α in Normal Human Tissues, Cancers, and Tumor-Associated Macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef]
- Fang, J.; Xia, C.; Cao, Z.; Zheng, J.Z.; Reed, E.; Jiang, B.-H. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005, 19, 342–353. [Google Scholar] [CrossRef]
- Fang, J.; Zhou, Q.; Liu, L.-Z.; Xia, C.; Hu, X.; Shi, X.; Jiang, B.-H. Apigenin inhibits tumor angiogenesis through decreasing HIF-1α and VEGF expression. Carcinogenesis 2007, 28, 858–864. [Google Scholar] [CrossRef]
- Zhong, X.-S.; Zheng, J.Z.; Reed, E.; Jiang, B.-H. SU5416 inhibited VEGF and HIF-1α expression through the PI3K/AKT/p70S6K1 signaling pathway. Biochem. Biophys. Res. Commun. 2004, 324, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Tinnefeld, H.; Metzner, A.; Jücker, M.; Butzal, M.; Bruweleit, M.; Fischer, U.; Draab, E.; Schuch, G.; O’-Farrel, A.M.; et al. Downregulation of VEGF-A, STAT5 and AKT in acute myeloid leukemia blasts of patients treated with SU5416. Leuk. Lymphoma 2006, 47, 2601–2609. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.-H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. CCAAT/Enhancer-binding Protein-Homologous Protein Sensitizes to SU5416 by Modulating p21 and PI3K/Akt Signal Pathway in FRO Anaplastic Thyroid Carcinoma Cells. Horm. Metab. Res. 2012, 45, 9–14. [Google Scholar] [CrossRef]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef]
- Dat, N.T.; Jin, X.; Lee, K.; Hong, Y.-S.; Kim, Y.H.; Lee, J.J. Hypoxia-Inducible Factor-1 Inhibitory Benzofurans and Chalcone-Derived Diels−Alder Adducts fromMorusSpecies. J. Nat. Prod. 2009, 72, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-L.; Liu, Y.-N.; Huang, Y.-T.; Pan, S.-L.; Huang, D.-Y.; Guh, J.-H.; Lee, F.-Y.; Kuo, S.-C.; Teng, C.-M. YC-1 inhibits HIF-1 expression in prostate cancer cells: Contribution of Akt/NF-κB signaling to HIF-1α accumulation during hypoxia. Oncogene 2007, 26, 3941–3951. [Google Scholar] [CrossRef]
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin inhibits breast cancer growth by repressing HIF-1α-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, O.; Borau, P.G.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Grande, E. Targeting HIF-2 α in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit. Rev. Oncol. 2017, 111, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Motto, I.; Bordogna, A.; Soshilov, A.A.; Denison, M.S.; Bonati, L. New Aryl Hydrocarbon Receptor Homology Model Targeted To Improve Docking Reliability. J. Chem. Inf. Model. 2011, 51, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Li, Q.; Ma, H.-W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nat. Cell Biol. 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, I.C.S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nat. Cell Biol. 2016, 539, 107–111. [Google Scholar] [CrossRef]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, L.; Chen, J.; Sun, Y.; Lin, H.; Jin, R.-A.; Tang, M.; Liang, X.; Cai, X. Increasing AR by HIF-2α inhibitor (PT-2385) overcomes the side-effects of sorafenib by suppressing hepatocellular carcinoma invasion via alteration of pSTAT3, pAKT and pERK signals. Cell Death Dis. 2017, 8, e3095. [Google Scholar] [CrossRef]
- Rogers, J.L.; Bayeh, L.; Scheuermann, T.H.; Longgood, J.; Key, J.; Naidoo, J.; Melito, L.; Shokri, C.; Frantz, D.E.; Bruick, R.K.; et al. Development of Inhibitors of the PAS-B Domain of the HIF-2α Transcription Factor. J. Med. Chem. 2013, 56, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Metelo, A.M.; Noonan, H.; Iliopoulos, O. HIF2a inhibitors for the treatment of VHL disease. Oncotarget 2015, 6, 23036–23037. [Google Scholar] [CrossRef]
- Harrison, H.; Rogerson, L.; Gregson, H.J.; Brennan, K.R.; Clarke, R.B.; Landberg, G. Contrasting Hypoxic Effects on Breast Cancer Stem Cell Hierarchy Is Dependent on ER- Status. Cancer Res. 2012, 73, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Belvedere, S.; Witter, D.J.; Yan, J.; Secrist, J.P.; Richon, V.; Miller, T.A. Aminosuberoyl hydroxamic acids (ASHAs): A potent new class of HDAC inhibitors. Bioorganic Med. Chem. Lett. 2007, 17, 3969–3971. [Google Scholar] [CrossRef]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients with CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R.; McDonough, M.A.; King, O.N.F.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 2011, 40, 4364–4397. [Google Scholar] [CrossRef]
- Sakurai, M.; Rose, N.R.; Schultz, L.; Quinn, A.M.; Jadhav, A.; Ng, S.S.; Oppermann, U.; Schofield, C.J.; Simeonov, A. A miniaturized screen for inhibitors of Jumonji histone demethylases. Mol. BioSyst. 2010, 6, 357–364. [Google Scholar] [CrossRef]
- Hamada, S.; Suzuki, T.; Mino, K.; Koseki, K.; Oehme, F.; Flamme, I.; Ozasa, H.; Itoh, Y.; Ogasawara, D.; Komaarashi, H.; et al. Design, Synthesis, Enzyme-Inhibitory Activity, and Effect on Human Cancer Cells of a Novel Series of Jumonji Domain-Containing Protein 2 Histone Demethylase Inhibitors. J. Med. Chem. 2010, 53, 5629–5638. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, J.; Liu, Y.; Xia, J.; Li, Y.; Wang, Z.P.; Wei, W. PROTACs: A Novel Strategy for Cancer Therapy. In Seminars in Cancer Biology; Academic Press: Waltham, MA, USA, 2020. [Google Scholar]
- Rodriguez-Gonzalez, A.; Cyrus, K.; Salcius, M.; Kim, K.; Crews, C.; Deshaies, R.; Sakamoto, K. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201–7211. [Google Scholar] [CrossRef] [PubMed]
- Kounde, C.S.; Shchepinova, M.M.; Saunders, C.N.; Muelbaier, M.; Rackham, M.D.; Harling, J.D.; Tate, E.W. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem. Commun. 2020, 56, 5532–5535. [Google Scholar] [CrossRef]
- Naro, Y.; Darrah, K.; Deiters, A. Optical Control of Small Molecule-Induced Protein Degradation. J. Am. Chem. Soc. 2020, 142, 2193–2197. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Cancer Type | Mechanism/Outcomes | Reference |
|---|---|---|---|---|
| Apigenin | HIF-1 | Ovarian cancer | Targets PI3K/AKT signaling and downregulates HIF-1 and VEGF expression | [165,166] |
| SU5416 | HIF-1 | Acute myeloid leukemia, ovarian cancer, anaplastic thyroid carcinoma | Inhibits ability of HIF-1 to bind DNA; decreases VEGF and HIF-1α expression; downregulates VEGF, PI3K, AKT, and p70S6K1 | [167,168,169] |
| PX-478 | HIF-1 | Prostate, breast, colon, and pancreatic cancer | Modulates HIF-1α deubiquitylation, inhibits HIF-1α transcription and translation | [170] |
| BAY 87-2243 | HIF-1, HIF-2 | Non-small cell lung cancer | Prevents accumulation of HIF-1α and HIF-2α | [171] |
| Moracin O and moracin P | HIF-1 | Hepatocellular carcinoma | Inhibits HIF-1 activation | [172] |
| YC-1 | HIF-1 | Prostate cancer | Activates soluble guanylyl cyclase, induces HIF-1 degradation, targets PI3K/Akt/mTOR and NF-κB signaling | [173] |
| Cardamonin | HIF-1 | Triple-negative breast cancer | Inhibits the mTOR/p70S6K pathway and HIF-1α expression | [174] |
| PT-2399 | HIF-2 | Clear cell renal cell carcinoma | Inhibits HIF-2α-ARNT dimerization | [175,177,178] |
| PT-2385 | HIF-2 | Clear cell renal cell carcinoma, lung cancer cell xenograft, hepatocellular carcinoma | Reduces the levels of tumor-induced circulating VEGFA, represses HIF-2α-related Stat-3/Akt/Erk signaling | [175,177,178,180,181] |
| TC-S 7009 | HIF-2 | Hepatoma | Prevents DNA binding, heterodimerization, and the transcription activation of HIF-2α by inducing allosteric conformational changes in PAS-B β-sheets | [177,182] |
| Compound-76 | HIF-2 | Breast cancer | Inhibits HIF-2α translation by promoting interaction of IRP1 and HIF-2α mRNA, represses chemoresistance and stem-ness | [183,184] |
| Vorinostat/SAHA | JMJD2 | Cutaneous T cell lymphoma | Inhibits JMJD2 | [185] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Shi, R.; Zhang, Q. Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression. Int. J. Mol. Sci. 2020, 21, 8162. https://doi.org/10.3390/ijms21218162
Yang G, Shi R, Zhang Q. Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression. International Journal of Molecular Sciences. 2020; 21(21):8162. https://doi.org/10.3390/ijms21218162
Chicago/Turabian StyleYang, Guang, Rachel Shi, and Qing Zhang. 2020. "Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression" International Journal of Molecular Sciences 21, no. 21: 8162. https://doi.org/10.3390/ijms21218162
APA StyleYang, G., Shi, R., & Zhang, Q. (2020). Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression. International Journal of Molecular Sciences, 21(21), 8162. https://doi.org/10.3390/ijms21218162