Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L.

 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Results
2.1. De Novo Reconstruction of Transcriptomes
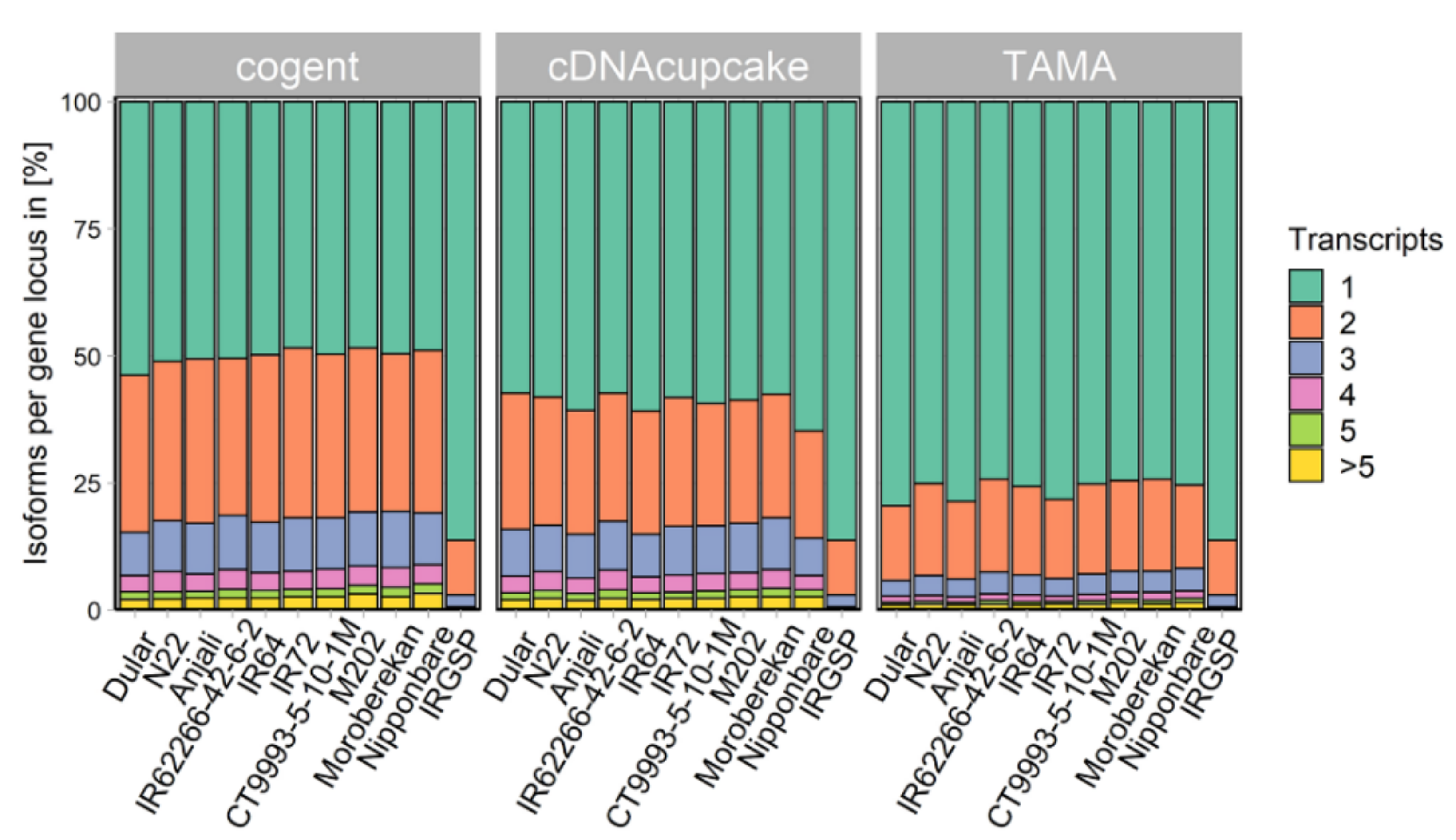
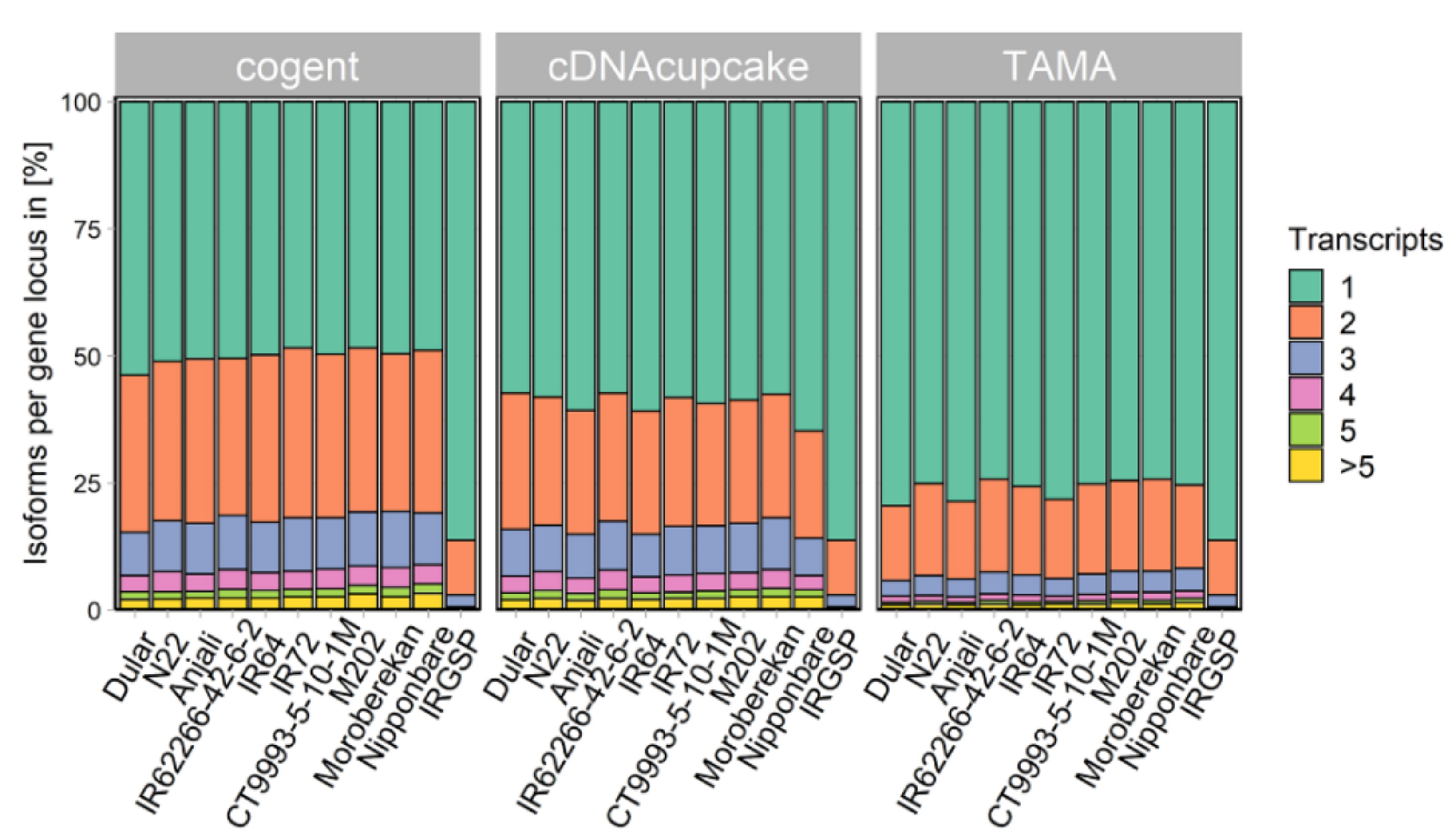
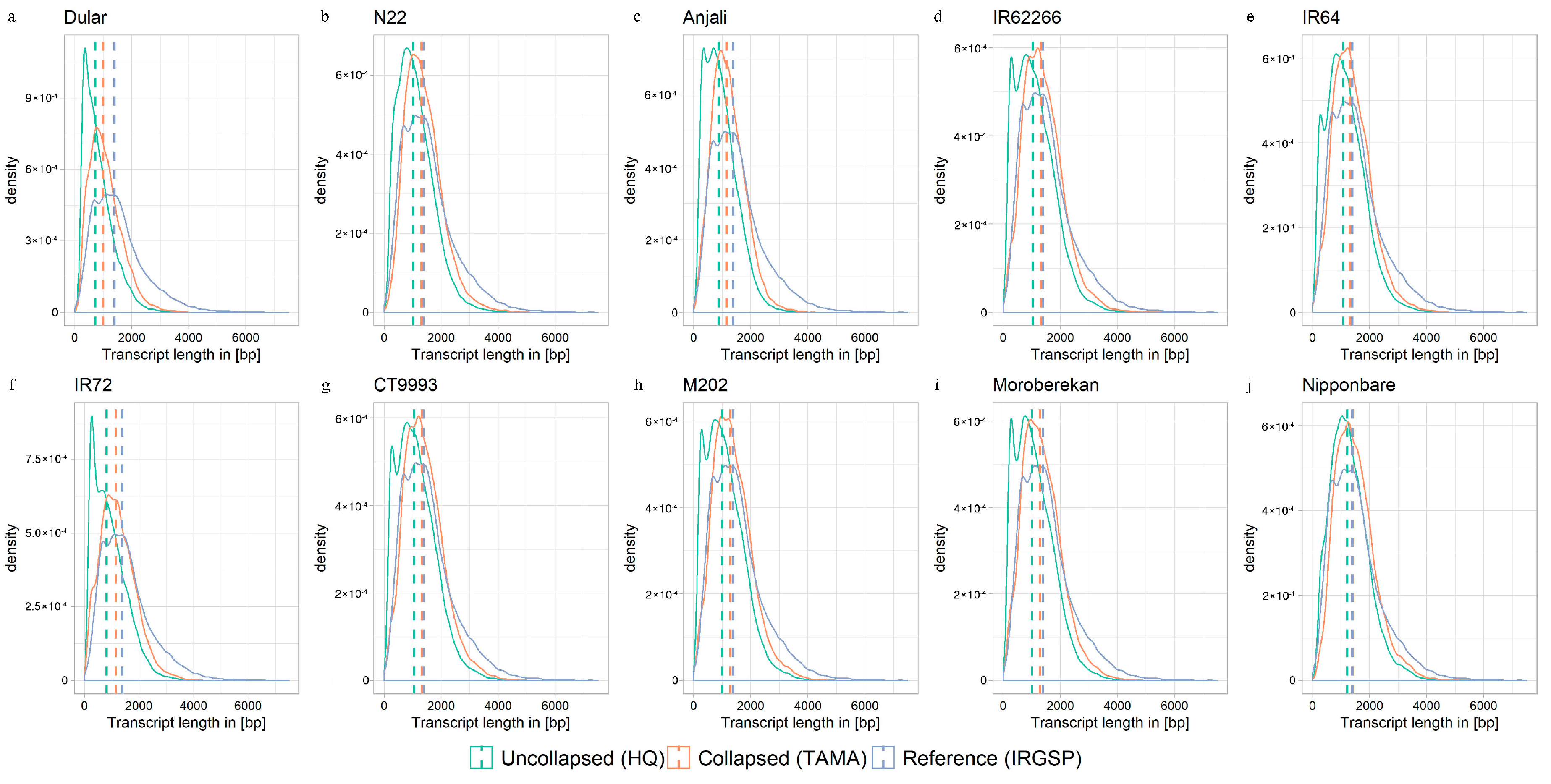
2.2. Collapsing Redundant Isoforms
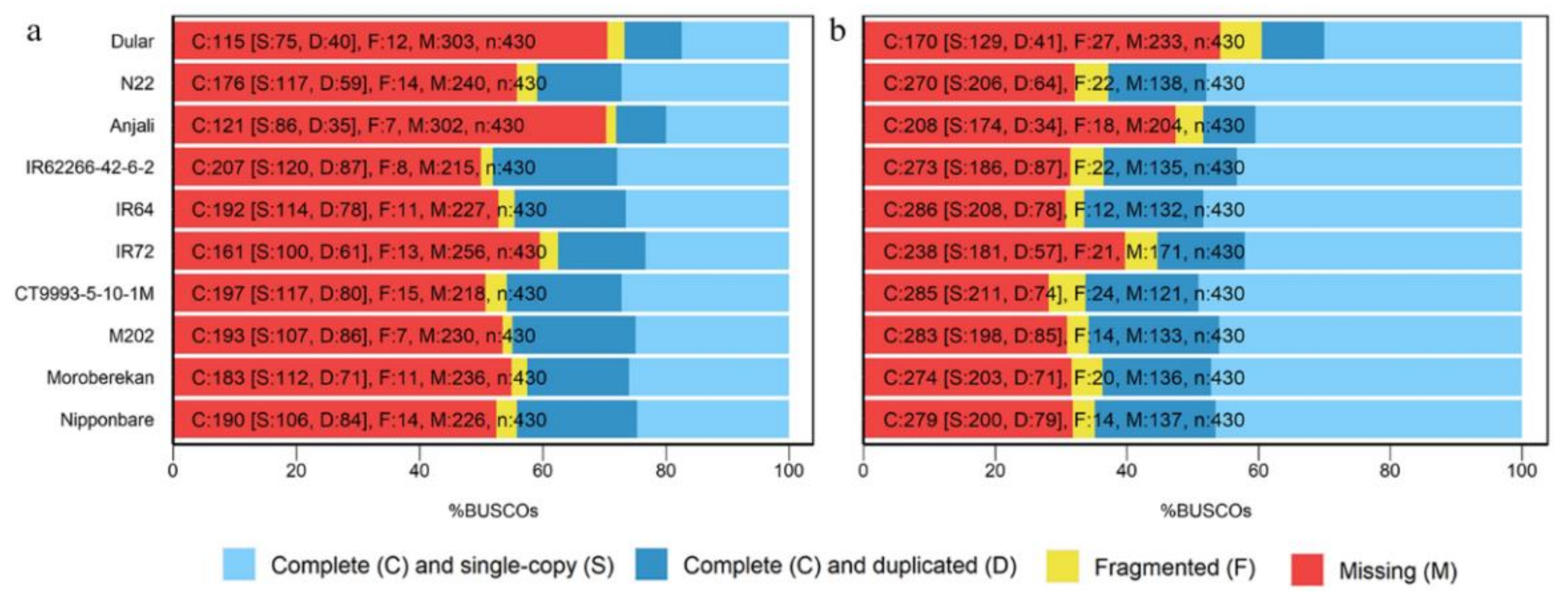
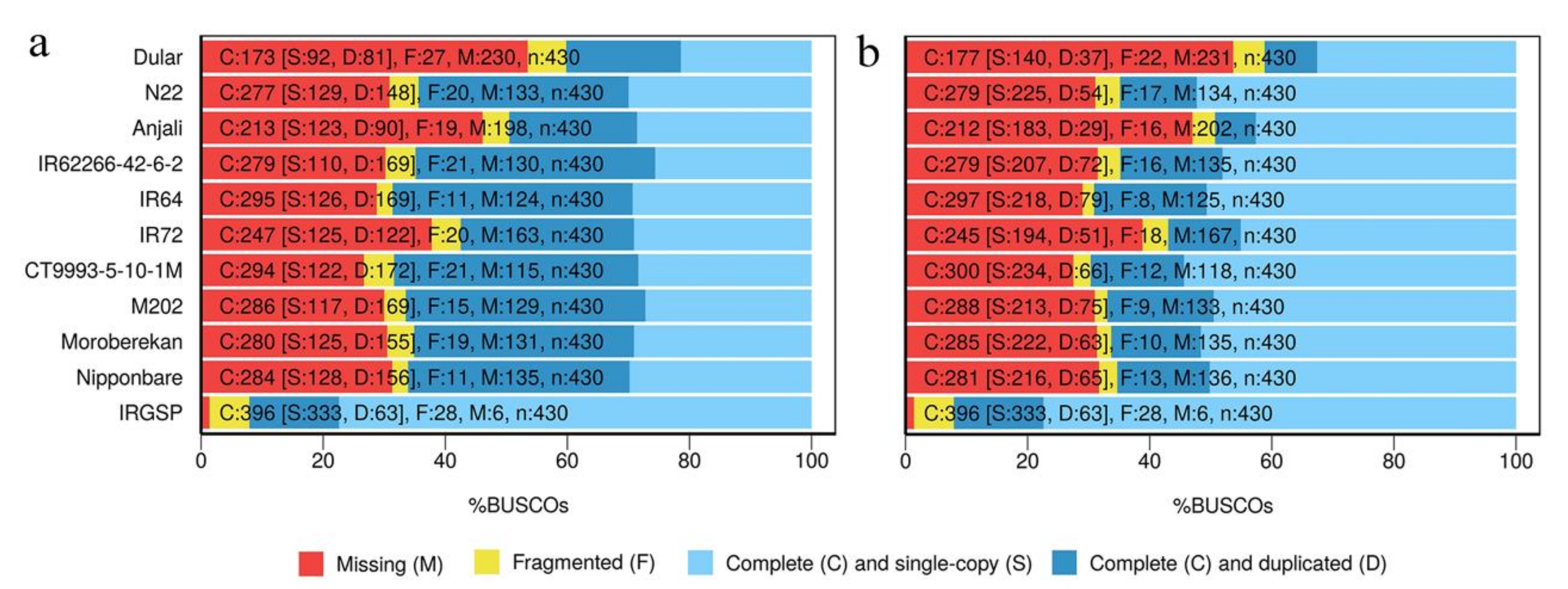
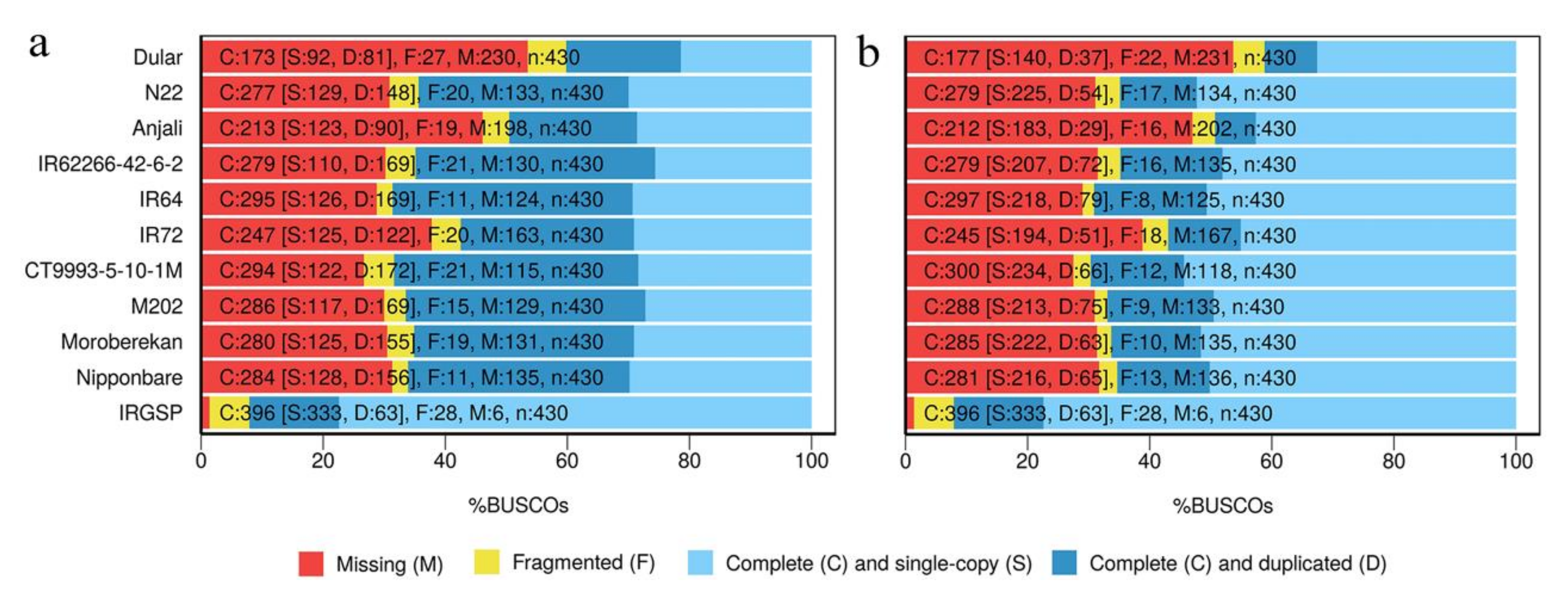
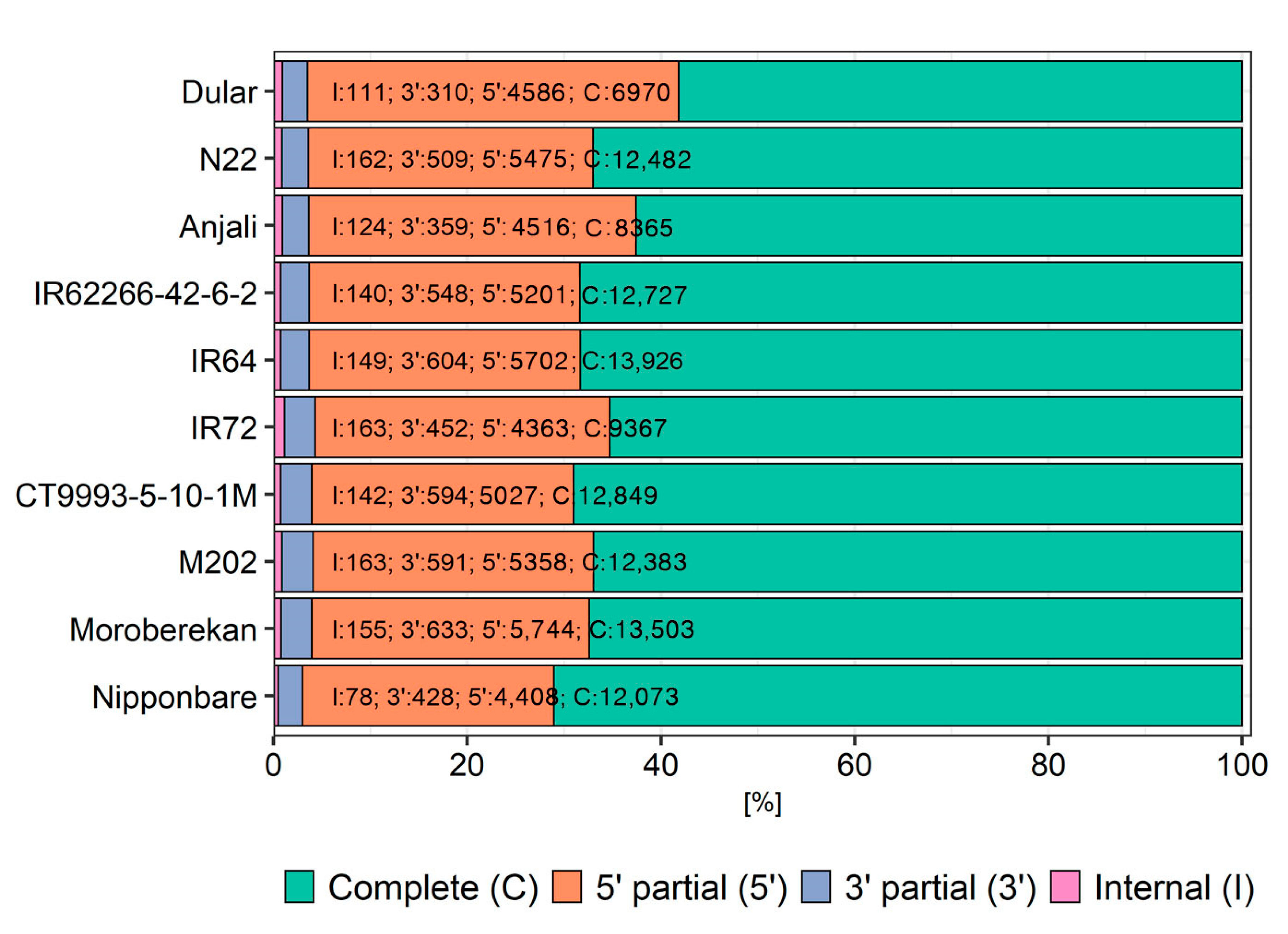
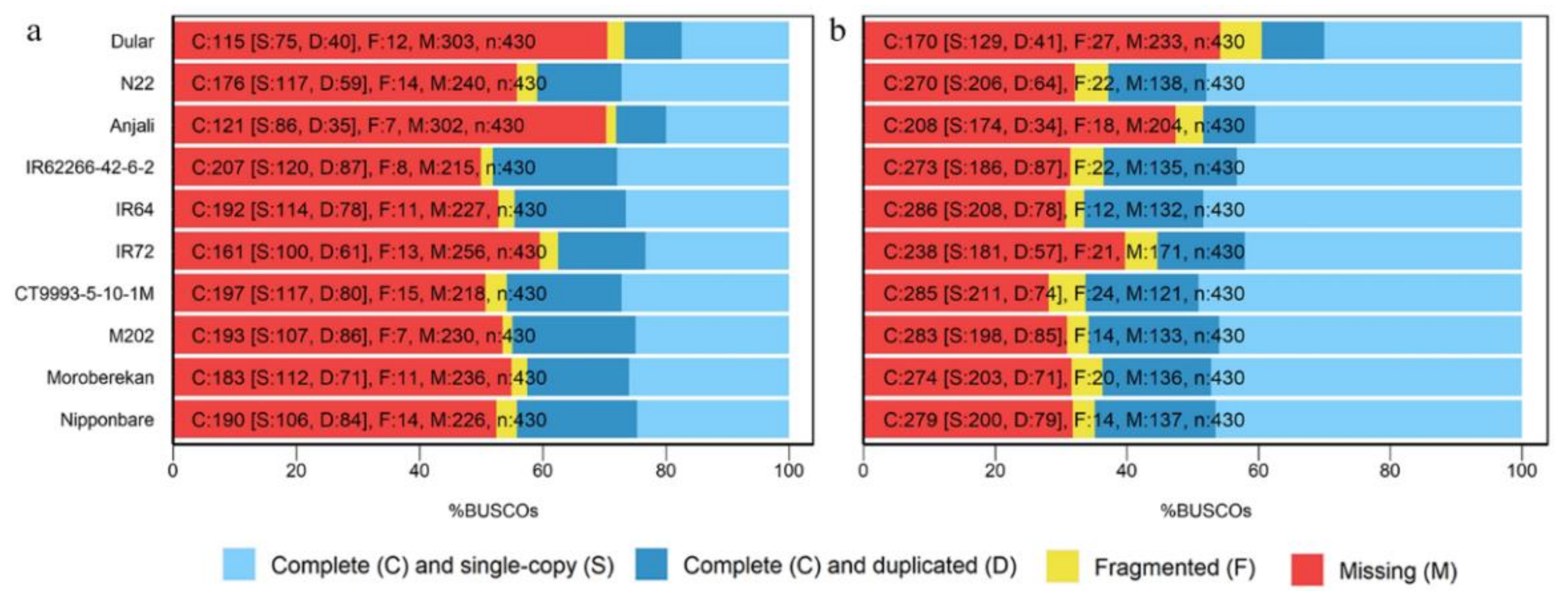
2.3. Evaluation of Reconstructed Transcriptomes
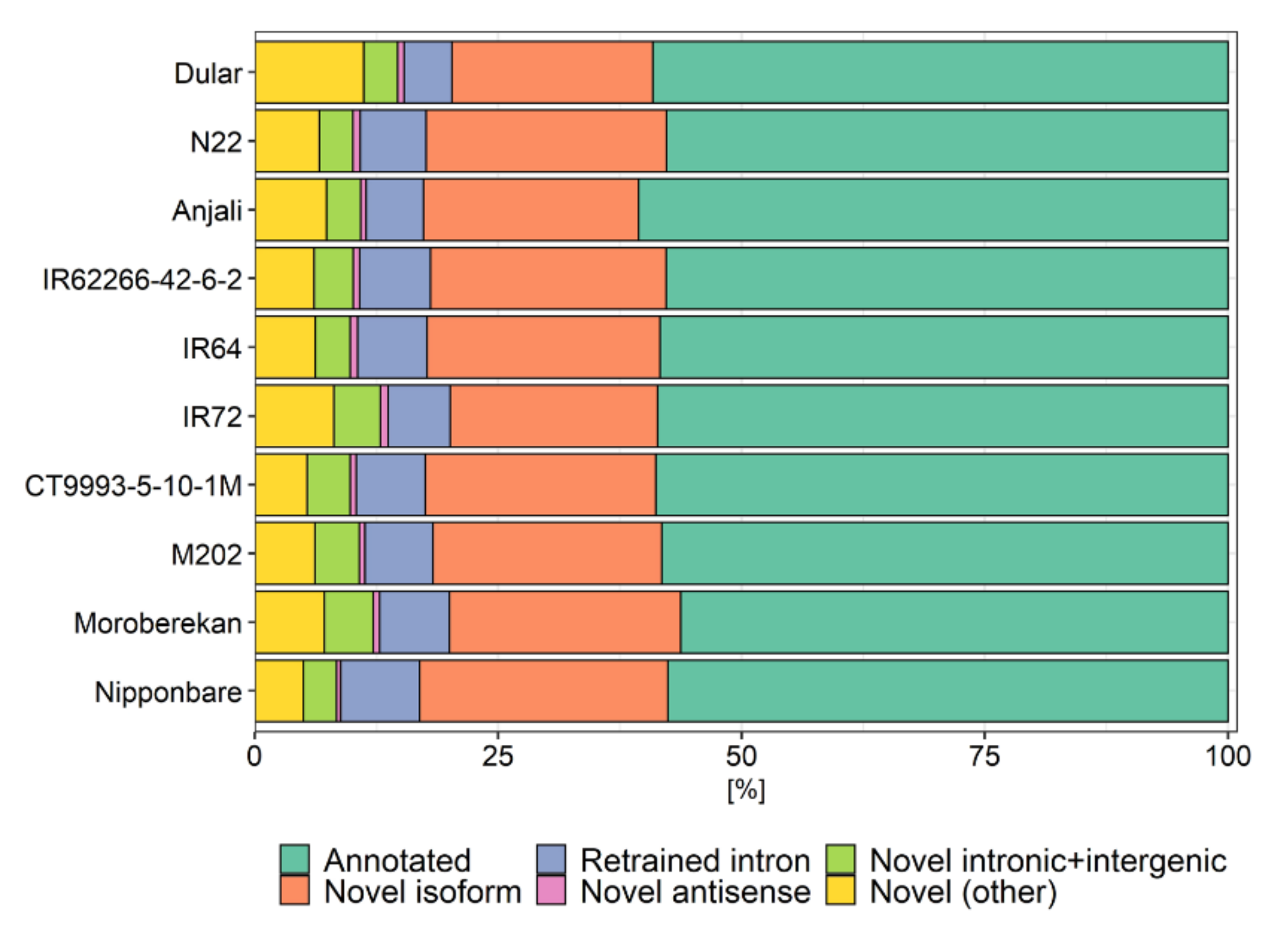
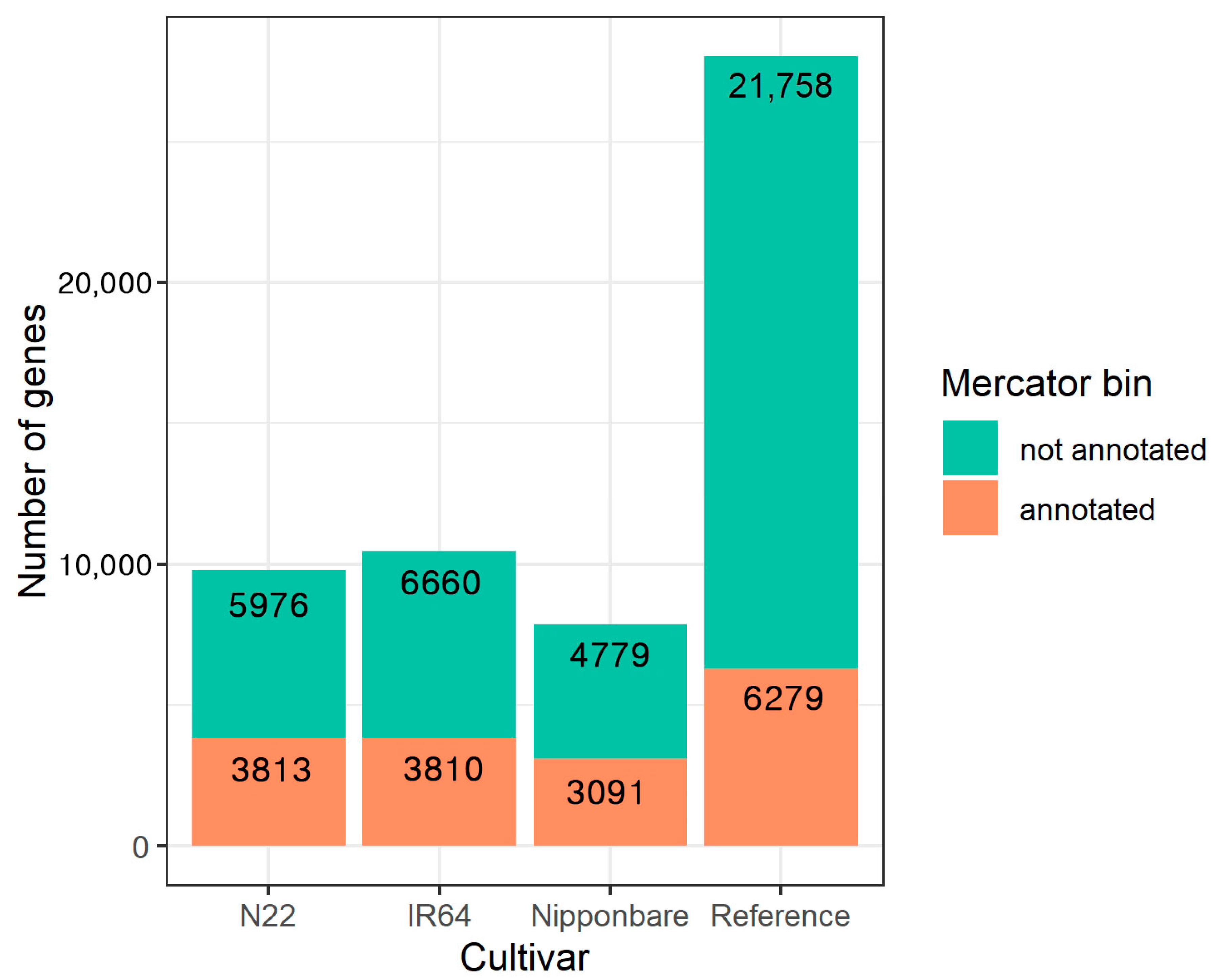
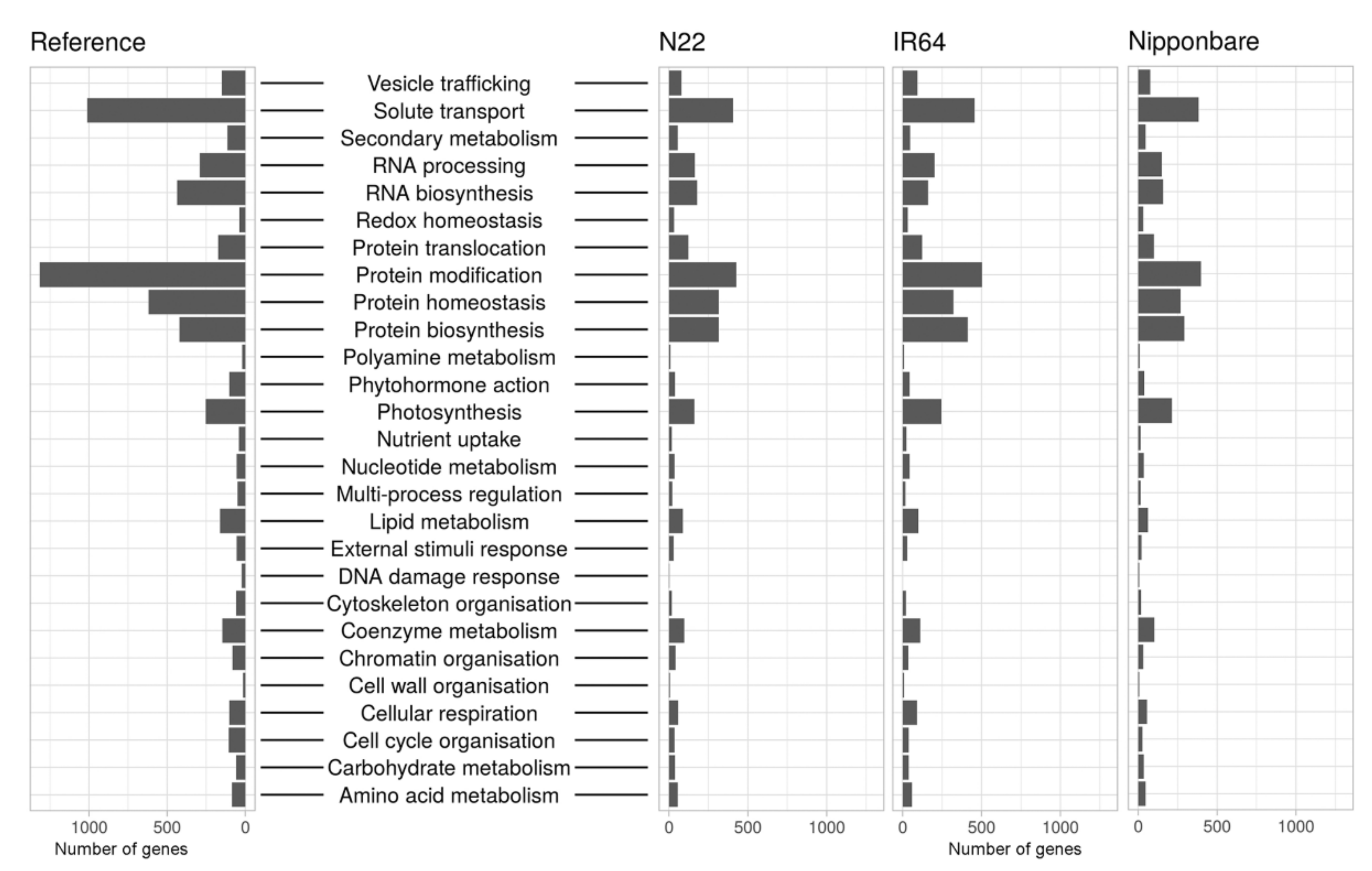
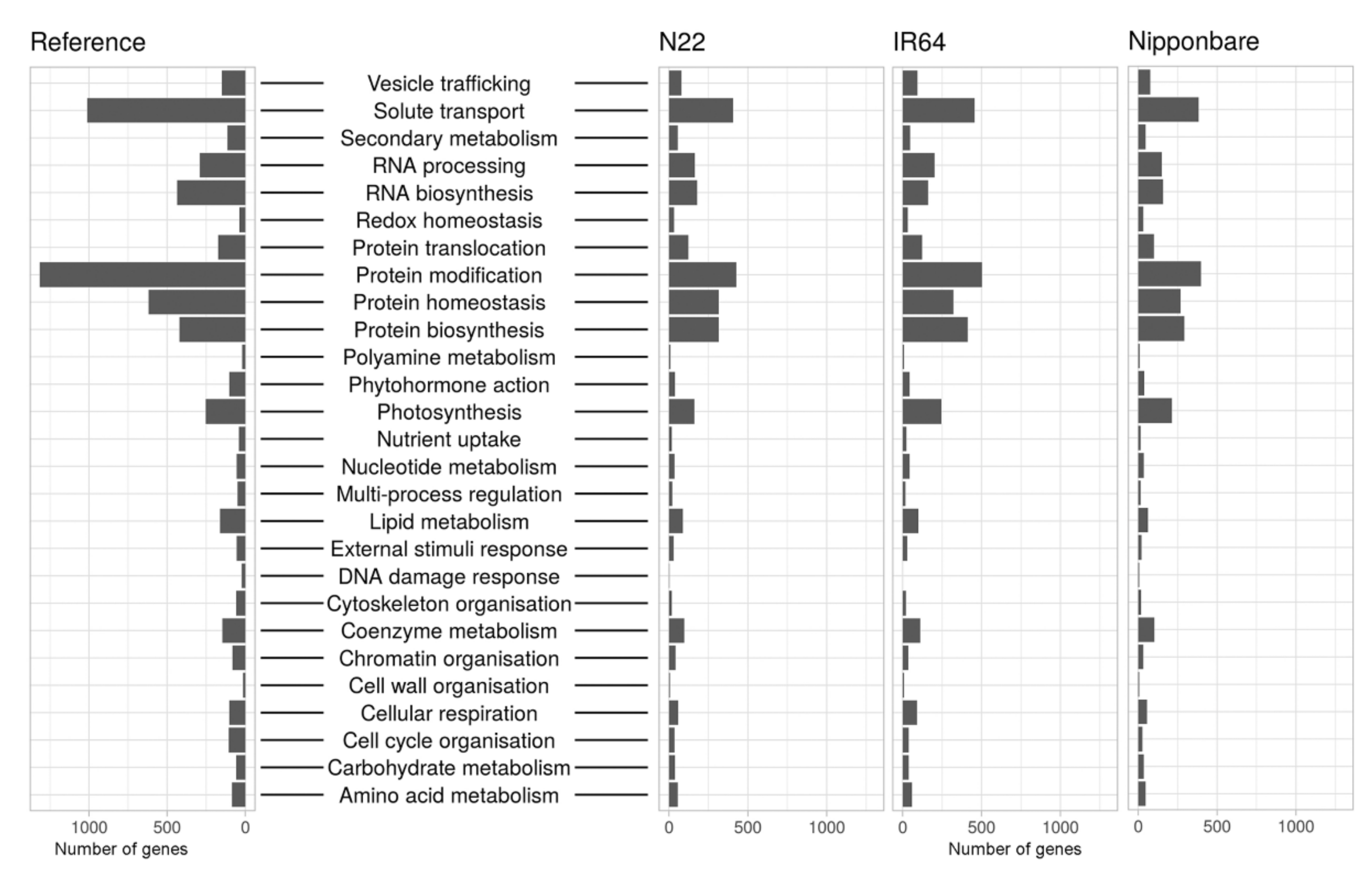
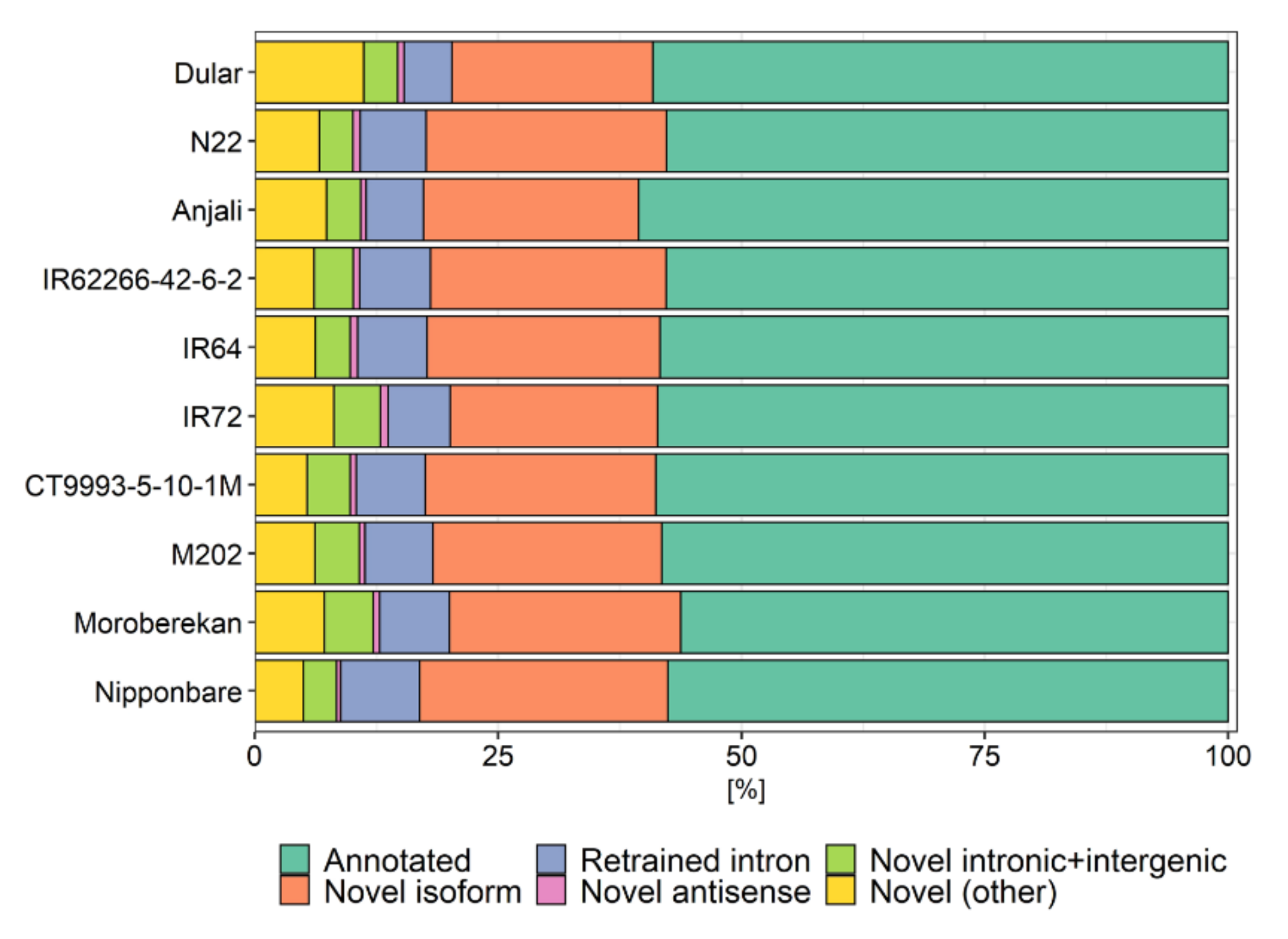
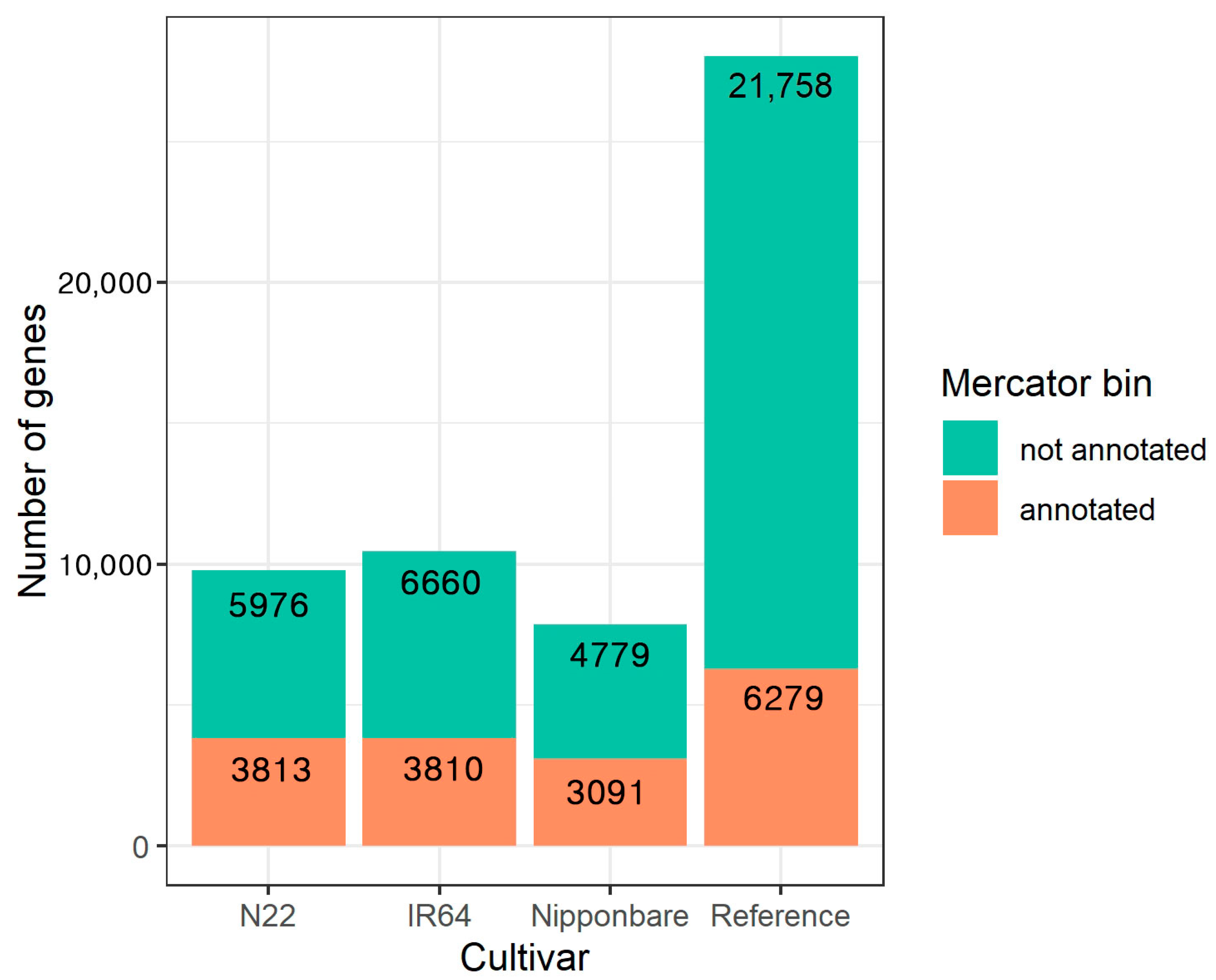
2.4. Functional Annotation
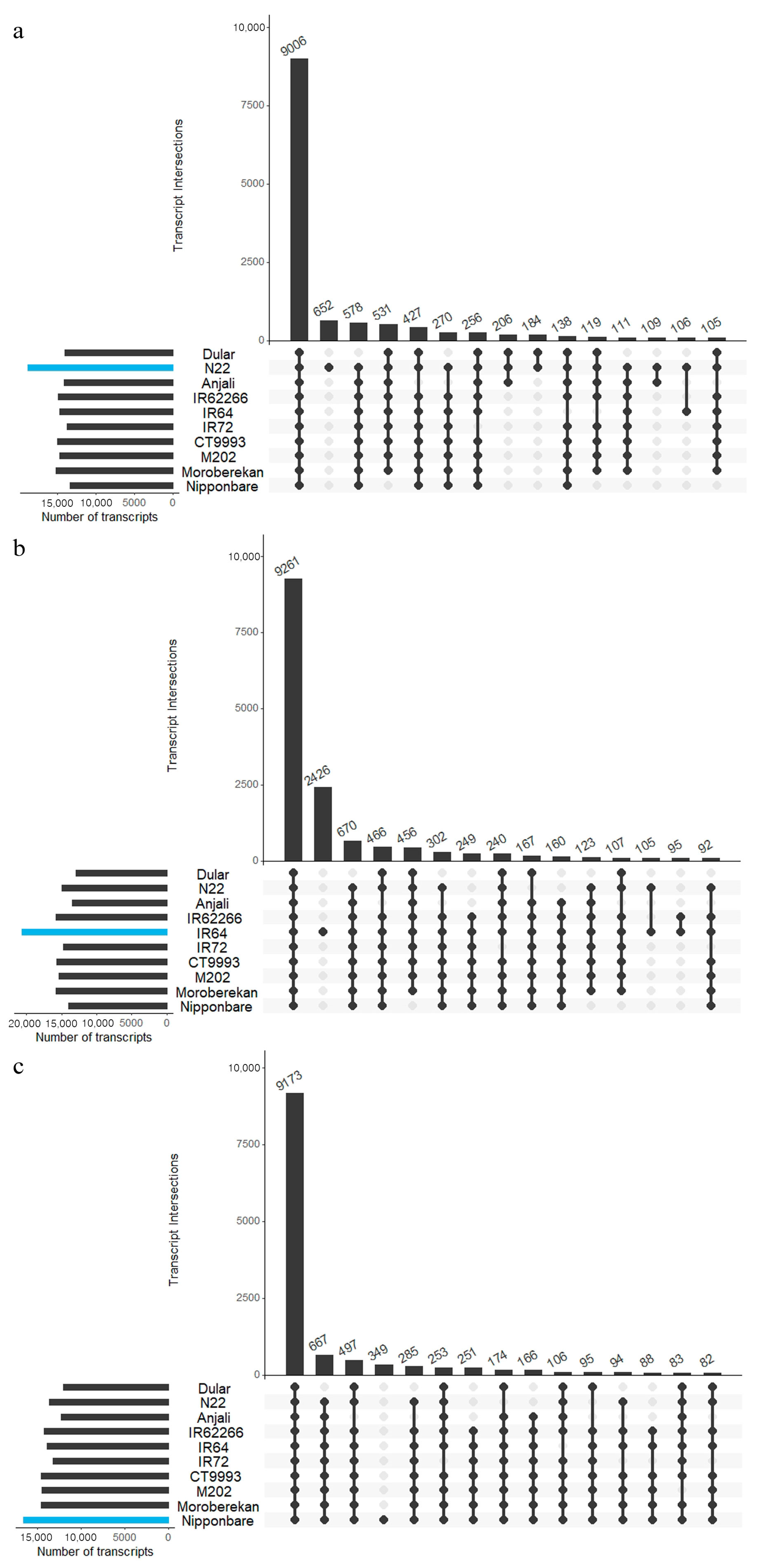
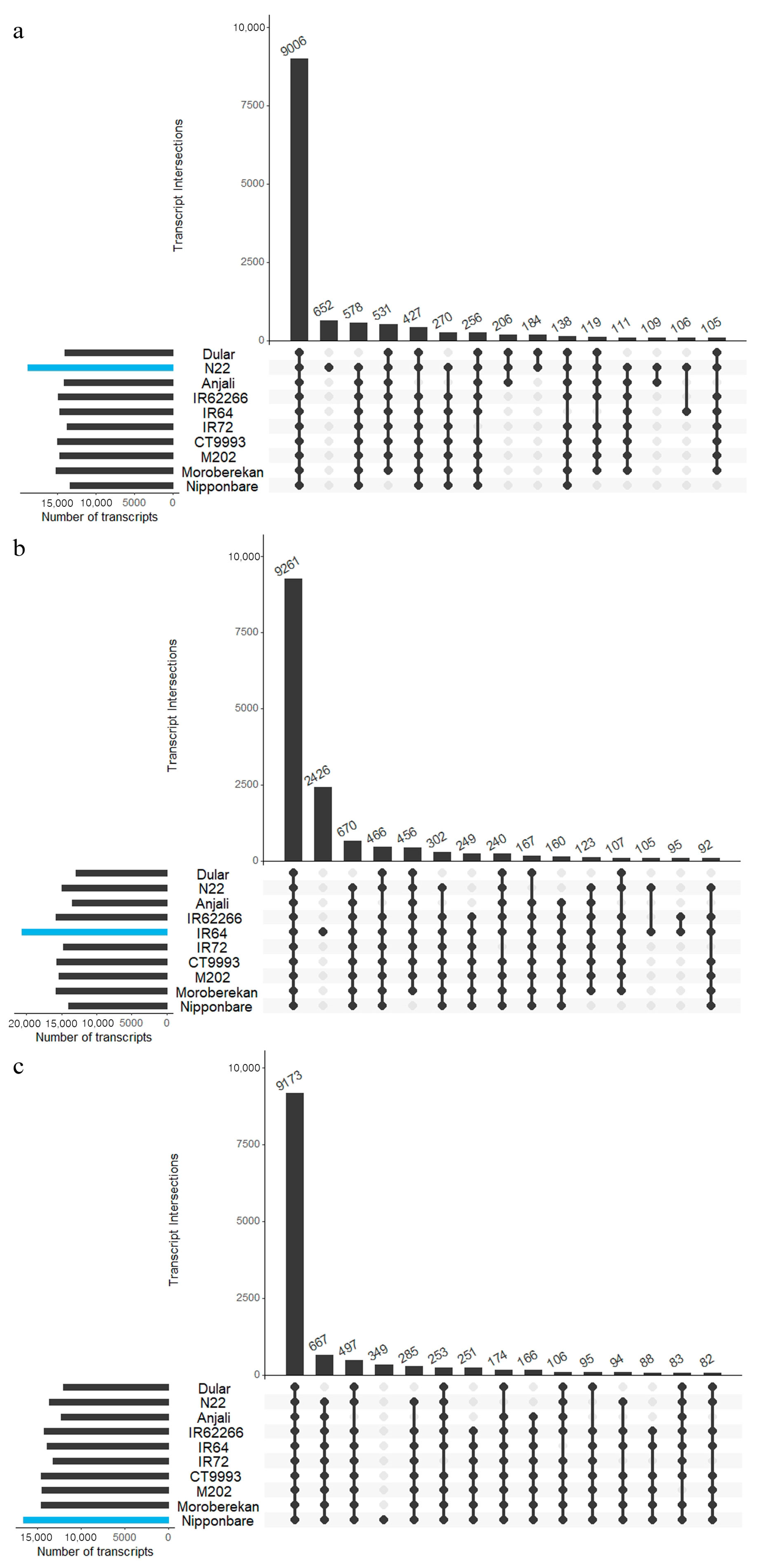
2.5. Common and Specific Transcripts among Cultivars
2.6. Differential Gene Expression Analysis for Aus Specific Transcripts
3. Discussion
3.1. Sequencing Performance
3.2. Collapsing Redundant Transcripts and Transcriptome Quality Assessment
3.3. Common Transcripts and Differential Gene Expression Analysis
4. Materials and Methods
4.1. Plant Material
4.2. RNA Extraction and Sequencing
4.3. De novo Transcriptome Reconstruction
- ccs $in.subreads.bam $out.bam --noPolish --minPasses=1
- lima $in.xml primer.fasta $out.demux.ccs.bam --isoseq --no-pbi --dump-clips
- isoseq3 cluster $in.demux.ccs.bam $out.unpolished.bam
- isoseq3 polish $in.unpolished.bam $out.polished.bam
4.4. Genome References
4.5. InDel Analysis
4.6. Collapsing Redundant Isoforms
4.7. BUSCO Analysis
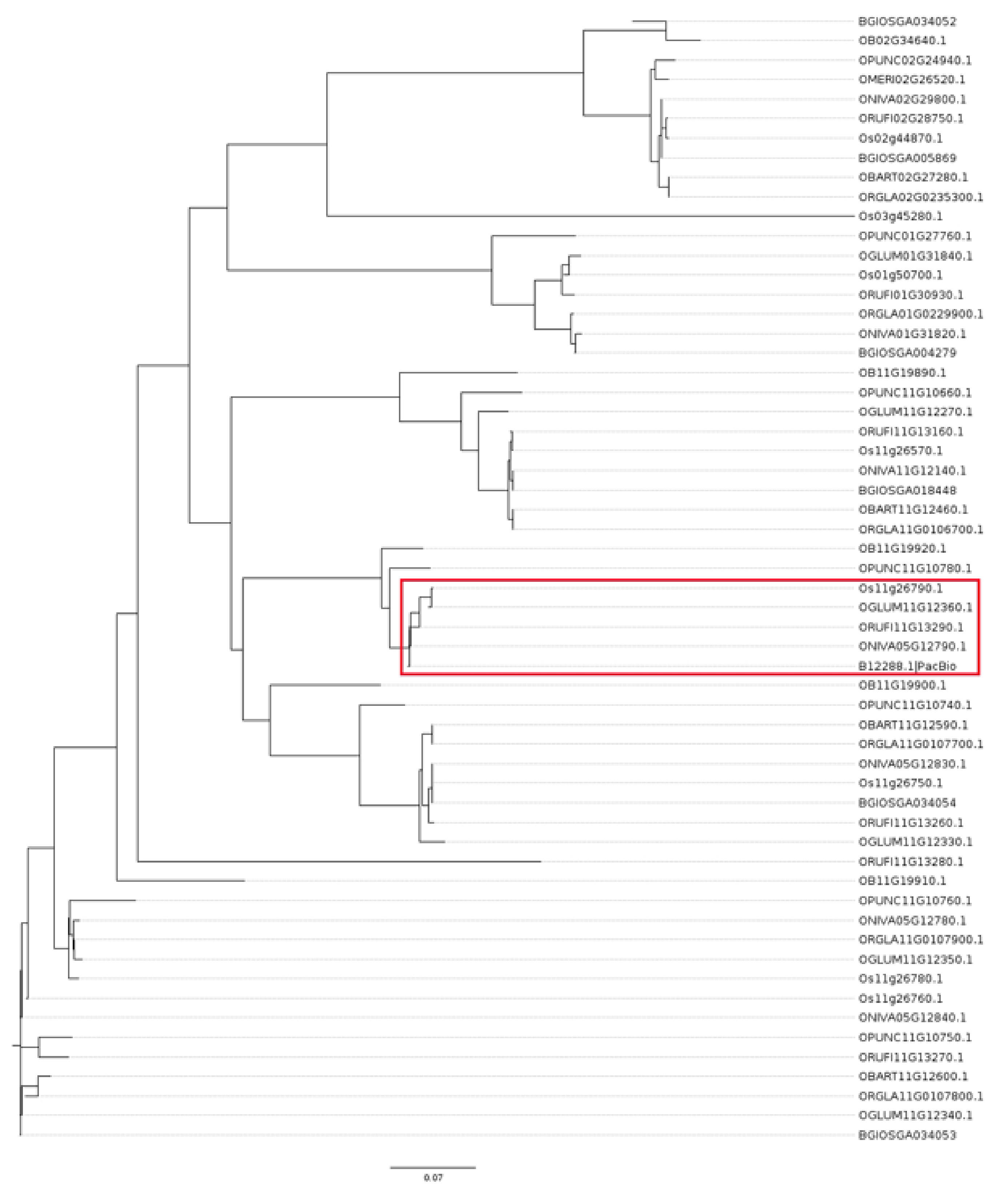
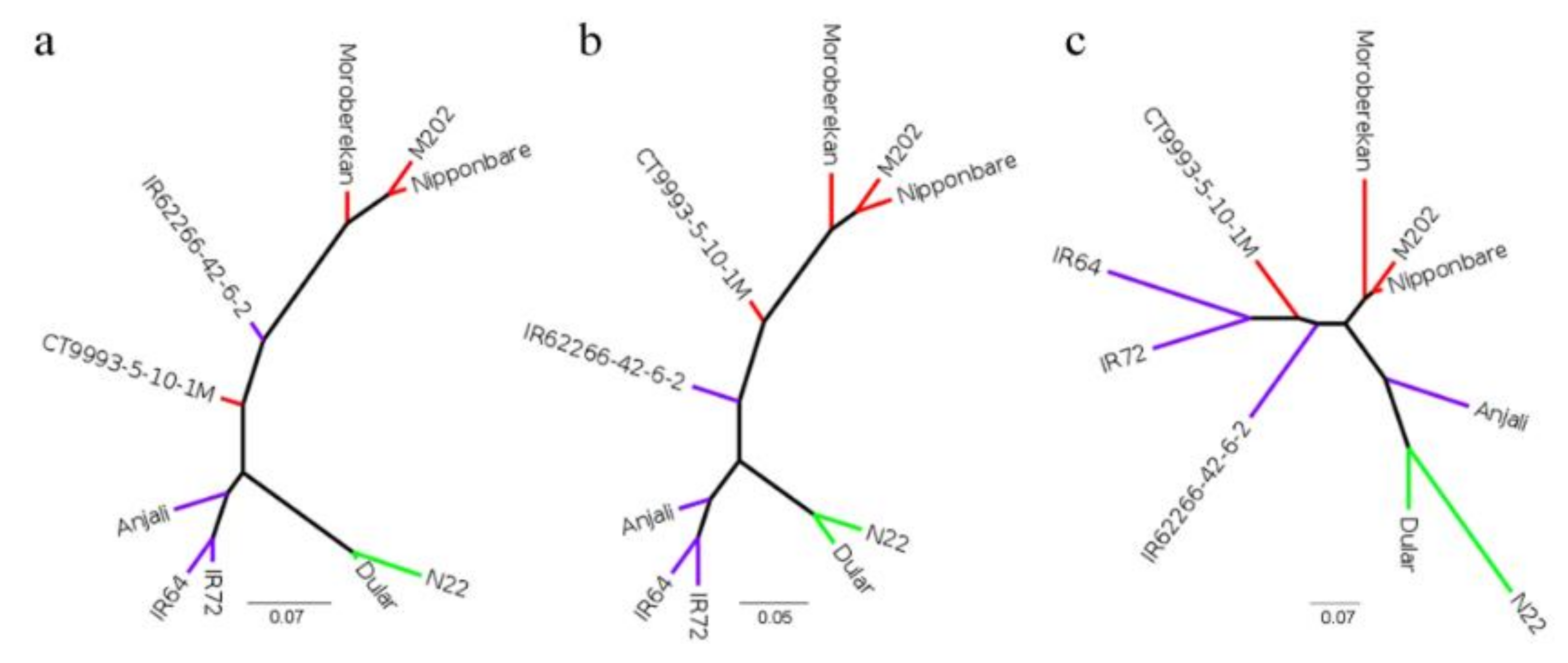
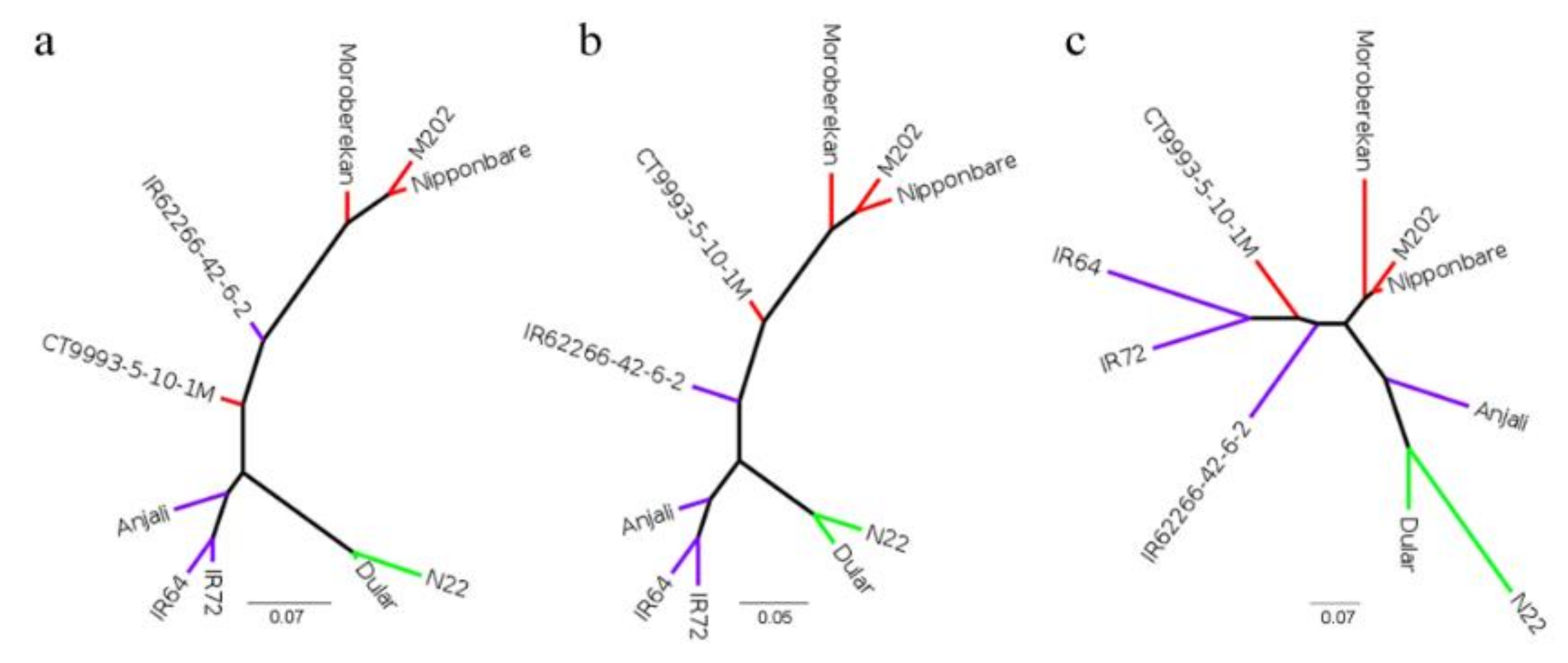
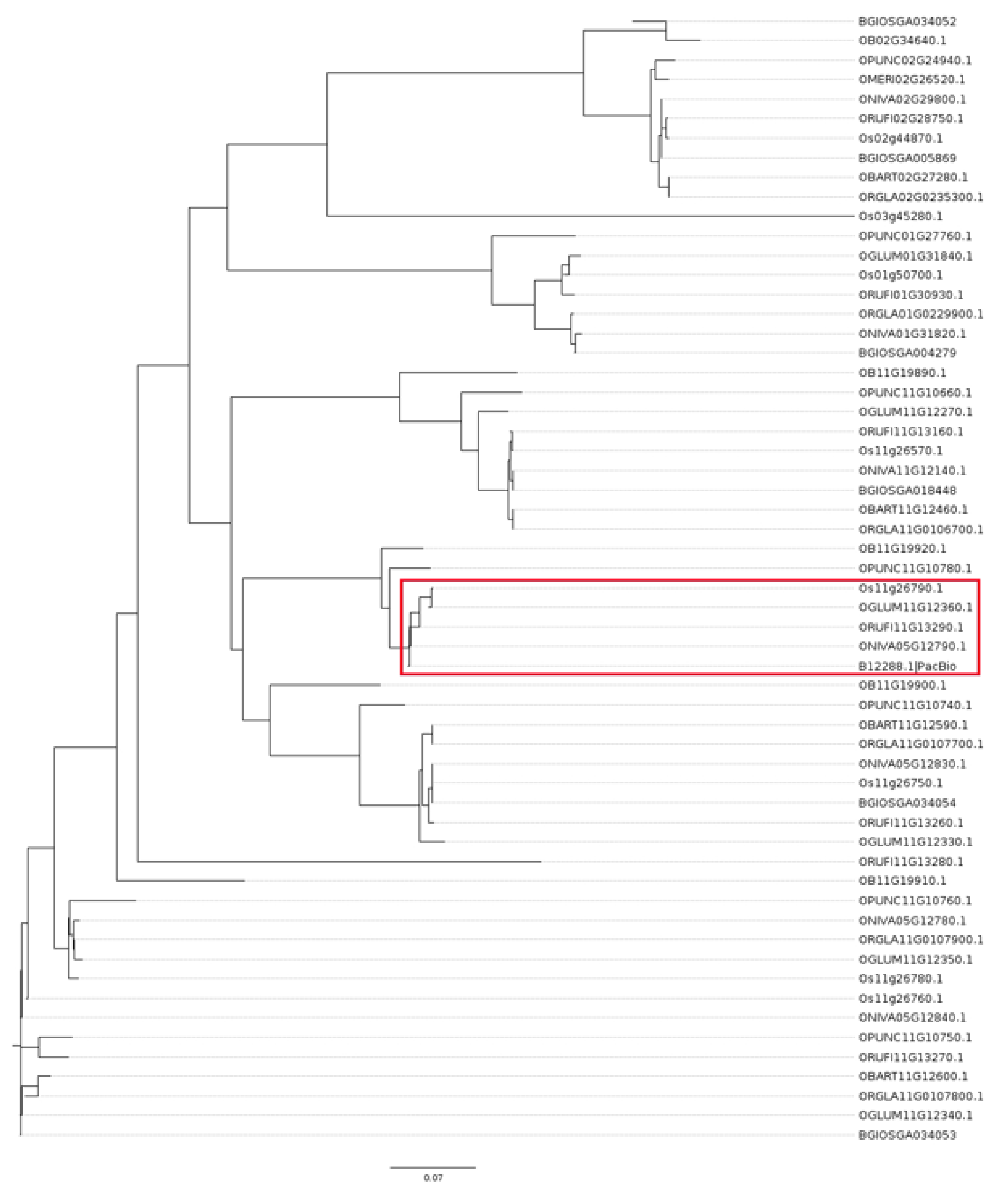
4.8. Phylogenetic Analysis
4.9. Comparison of Reconstructed Transcriptomes
4.10. Functional Annotation
4.11. Determination of Common Overlap
4.12. Differential Gene Expression Analysis
4.13. Graphical Visualization
4.14. Availability of Data and Material
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| bp | Basepairs |
| BUSCO | Benchmarking Universal Single-Copy Orthologs |
| FLNC | Full-Length Non-Chimeric |
| GB | Gigabases |
| HNT | High Night Temperature |
| HQ | High Quality |
| InDel | Insertion/Deletion |
| IRGSP | International Rice Genome Sequencing Project |
| IsoSeq | Isoform Sequencing |
| LQ | Low Quality |
| ORF | Open Reading Frame |
| RNA-Seq | RNA Sequencing |
| SMRT | Single-Molecule, Real-Time |
| SNP | Single Nucleotide Polymorphism |
Appendix A

References
- Lamaoui, M.; Jemo, M.; Datla, R.; Bekkaoui, F. Heat and drought stresses in crops and approaches for their mitigation. Front. Chem. 2018, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.B.; Huang, Y.; Huang, M.; Yao, Y.; Bassu, S.; Ciais, P.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizumi, T.; Ramankutty, N. Changes in yield variability of major crops for 1981–2010 explained by climate change. Environ. Res. Lett. 2016, 11, 034003. [Google Scholar] [CrossRef]
- Dawson, T.P.; Perryman, A.H.; Osborne, T.M. Modelling impacts of climate change on global food security. Clim. Chang. 2016, 134, 429–440. [Google Scholar] [CrossRef]
- Peng, S.; Huang, J.; Sheehy, J.E.; Laza, R.C.; Visperas, R.M.; Zhong, X.; Centeno, G.S.; Khush, G.S.; Cassman, K.G. Rice yields decline with higher night temperature from global warming. Proc. Natl. Acad. Sci. USA 2004, 101, 9971–9975. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L.; et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef]
- FAO. Food Systems for Better Nutrition; FAO: Rome, Italy, 2013. [Google Scholar]
- The Rice Genome Project. The 3000 rice genomes project. GigaScience 2014, 3, 7. [Google Scholar]
- Mahesh, H.B.; Shirke, M.D.; Singh, S.; Rajamani, A.; Hittalmani, S.; Wang, G.L.; Gowda, M. Indica rice genome assembly, annotation and mining of blast disease resistance genes. BMC Genom. 2016, 17, 242. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.L.; Sun, S.; Kudrna, D.; Copetti, D.; Li, W.; Mu, T.; Jiao, W.B.; Xing, F.; Lee, S.; et al. Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Sci. Data 2016, 3, 160076. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Kanamori, H.; Arai-Kichise, Y.; Shibata-Hatta, M.; Ebana, K.; Oono, Y.; Kurita, K.; Fujisawa, H.; Katagiri, S.; Mukai, Y.; et al. Construction of pseudomolecule sequences of the aus rice cultivar Kasalath for comparative genomics of asian cultivated rice. DNA Res. 2014, 21, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, K.L.; Childs, K.L.; Bohnert, R.; Davidson, R.M.; Zhao, K.; Ulat, V.J.; Zeller, G.; Clark, R.M.; Hoen, D.R.; Bureau, T.E.; et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. USA 2009, 106, 12273–12278. [Google Scholar] [CrossRef] [Green Version]
- Garris, A.J.; McCouch, S.R.; Kresovich, S. Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 2003, 165, 759–769. [Google Scholar] [PubMed]
- Gamuyao, R.; Chin, J.H.; Pariasca-Tanaka, J.; Pesaresi, P.; Catausan, S.; Dalid, C.; Slamet-Loedin, I.; Tecson-Mendoza, E.M.; Wissuwa, M.; Heuer, S. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 2012, 488, 535. [Google Scholar] [CrossRef]
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Nagai, K.; Furukawa, S.; Song, X.J.; Kawano, R.; Sakakibara, H.; Wu, J.; Matsumoto, T.; Yoshimura, A.; Kitano, H.; et al. The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 2009, 460, 1026–1030. [Google Scholar] [CrossRef]
- Baltazar, M.D.; Ignacio, J.C.I.; Thomson, M.J.; Ismail, A.M.; Mendioro, M.S.; Septiningsih, E.M. QTL mapping for tolerance of anaerobic germination from IR64 and the aus landrace Nanhi using SNP genotyping. Euphytica 2014, 197, 251–260. [Google Scholar] [CrossRef]
- Baltazar, M.D.; Ignacio, J.C.I.; Thomson, M.J.; Ismail, A.M.; Mendioro, M.S.; Septiningsih, E.M. QTL mapping for tolerance to anaerobic germination in rice from IR64 and the aus landrace Kharsu 80A. Breed. Sci. 2019, 69, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Bernier, J.; Kumar, A.; Venuprasad, R.; Spaner, D.; Verulkar, S.; Mandal, N.P.; Sinha, P.K.; Peeraju, P.; Dongre, P.R.; Mahto, R.N.; et al. Characterization of the effect of a QTL for drought resistance in rice, qtl12.1, over a range of environments in the Philippines and eastern India. Euphytica 2009, 166, 207–217. [Google Scholar] [CrossRef]
- Slabaugh, E.; Desai, J.S.; Sartor, R.C.; Lawas, L.M.F.; Jagadish, S.V.K.; Doherty, C.J. Analysis of differential gene expression and alternative splicing is significantly influenced by choice of reference genome. RNA 2019, 25, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Feng, S.; Xu, M.; Liu, F.; Cui, C.; Zhou, B. Reconstruction of the full-length transcriptome atlas using PacBio Iso-Seq provides insight into the alternative splicing in Gossypium australe. BMC Plant Biol. 2019, 19, 365. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, D.S.; Nishimwe, A.V.; Schnable, J.C. IsoSeq transcriptome assembly of C3 panicoid grasses provides tools to study evolutionary change in the Panicoideae. Plant Direct 2020, 4, e00203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [Green Version]
- Alam, R.; Hummel, M.; Yeung, E.; Locke, A.M.; Ignacio, J.C.I.; Baltazar, M.D.; Jia, Z.; Ismail, A.M.; Septiningsih, E.M.; Bailey-Serres, J. Flood resilience loci SUBMERGENCE 1 and ANAEROBIC GERMINATION 1 interact in seedlings established underwater. Plant Direct 2020, 4, e00240. [Google Scholar] [CrossRef] [PubMed]
- Schaarschmidt, S.; Lawas, L.M.F.; Glaubitz, U.; Li, X.; Erban, A.; Kopka, J.; Jagadish, S.V.K.; Hincha, D.K.; Zuther, E. Season affects yield and metabolic profiles of rice (Oryza sativa) under high night temperature stress in the field. IJMS 2020, 21, 3187. [Google Scholar] [CrossRef] [PubMed]
- Lawas, L.M.F.; Shi, W.; Yoshimoto, M.; Hasegawa, T.; Hincha, D.K.; Zuther, E.; Jagadish, S.V.K. Combined drought and heat stress impact during flowering and grain filling in contrasting rice cultivars grown under field conditions. Field Crops Res. 2018, 229, 66–77. [Google Scholar] [CrossRef]
- Li, X.; Lawas, L.M.; Malo, R.; Glaubitz, U.; Erban, A.; Mauleon, R.; Heuer, S.; Zuther, E.; Kopka, J.; Hincha, D.K.; et al. Metabolic and transcriptomic signatures of rice floral organs reveal sugar starvation as a factor in reproductive failure under heat and drought stress. Plant Cell Environ. 2015, 38, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, U.; Li, X.; Köhl, K.I.; van Dongen, J.T.; Hincha, D.K.; Zuther, E. Differential physiological responses of different rice (Oryza sativa) cultivars to elevated night temperature during vegetative growth. Funct. Plant Biol. 2014, 41, 437. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration, The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A refined protein classification and annotation framework applicable to multi-omics data analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef]
- Verma, G.; Dhar, Y.V.; Srivastava, D.; Kidwai, M.; Chauhan, P.S.; Bag, S.K.; Asif, M.H.; Chakrabarty, D. Genome-wide analysis of rice dehydrin gene family: Its evolutionary conservedness and expression pattern in response to PEG induced dehydration stress. PLoS ONE 2017, 12, e0176399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graether, S.P.; Boddington, K.F. Disorder and function: A review of the dehydrin protein family. Front. Plant Sci. 2014, 5, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundertmark, M.; Hincha, D.K. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Gao, Y.; Chen, W.; Wang, W.; Gong, L.; Liu, X. Spatiotemporal distribution of phenolamides and the genetics of natural variation of hydroxycinnamoyl spermidine in rice. Mol. Plant 2015, 8, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Workman, R.E.; Myrka, A.M.; Wong, G.W.; Tseng, E.; Welch, K.C., Jr.; Timp, W. Single-molecule, full-length transcript sequencing provides insight into the extreme metabolism of the ruby-throated hummingbird Archilochus colubris. GigaScience 2018, 7. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef]
- Xie, L.; Teng, K.; Tan, P.; Chao, Y.; Li, Y.; Guo, W.; Han, L. PacBio single-molecule long-read sequencing shed new light on the transcripts and splice isoforms of the perennial ryegrass. Mol. Genet. Genom. 2020, 295, 475–489. [Google Scholar] [CrossRef]
- Kuo, R.I.; Cheng, Y.; Smith, J.; Archibald, A.L.; Burt, D.W. Illuminating the dark side of the human transcriptome with TAMA Iso-Seq analysis. bioRxiv 2019, 780015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Regulski, M.; Tseng, E.; Olson, A.; Goodwin, S.; McCombie, W.R.; Ware, D. A comparative transcriptional landscape of maize and sorghum obtained by single-molecule sequencing. Genome Res. 2018, 28, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Kuo, R. TAMA: Transcriptome Annotation by Modular Algorithms. Available online: https://github.com/GenomeRIK/tama (accessed on 20 February 2019).
- Tung, L.H.; Shao, M.; Kingsford, C. Quantifying the benefit offered by transcript assembly on single-molecule long reads. bioRxiv 2019, 632703. [Google Scholar] [CrossRef] [Green Version]
- Olsen, A.N.; Mundy, J.; Skriver, K. Peptomics, identification of novel cationic arabidopsis peptides with conserved sequence motifs. Silico Biol. 2002, 2, 441–451. [Google Scholar]
- Mundy, J.; Chua, N.H. Abscisic acid and water-stress induce the expression of a novel rice gene. EMBO J. 1988, 7, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Koubaa, S.; Bremer, A.; Hincha, D.K.; Brini, F. Structural properties and enzyme stabilization function of the intrinsically disordered LEA_4 protein TdLEA3 from wheat. Sci. Rep. 2019, 9, 3720. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, D.; Kalmar, E.; Torok, Z.; Tompa, P. Chaperone activity of ERD10 and ERD14, two disordered stress-related plant proteins. Plant Physiol. 2008, 147, 381. [Google Scholar] [CrossRef]
- Sowemimo, O.T.; Knox-Brown, P.; Borcherds, W.; Rindfleisch, T.; Thalhammer, A.; Daughdrill, G.W. Conserved glycines control disorder and function in the cold-regulated protein, COR15A. Biomolecules 2019, 9, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Trick, H.N. Rapid method for high-quality RNA isolation from seed endosperm containing high levels of starch. BioTechniques 2005, 38, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Do, P.T.; Degenkolbe, T.; Erban, A.; Heyer, A.G.; Kopka, J.; Kohl, K.I.; Hincha, D.K.; Zuther, E. Dissecting rice polyamine metabolism under controlled long-term drought stress. PLoS ONE 2013, 8, e60325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, E. cDNA Cupcake. Available online: https://github.com/Magdoll/cDNA_Cupcake (accessed on 29 November 2019).
- Tseng, E. Cogent. Available online: https://github.com/Magdoll/Cogent (accessed on 29 November 2019).
- Tseng, E. Cogent Tutorial. Available online: https://github.com/Magdoll/Cogent/wiki/Tutorial%3A-Using-Cogent-to-collapse-redundant-transcripts-in-absence-of-genome (accessed on 29 November 2019).
- Tseng, E. cDNA cupcake Wiki. Available online: https://github.com/Magdoll/cDNA_Cupcake/wiki (accessed on 29 November 2019).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Figshare. Available online: www.figshare.com (accessed on 30 October 2020).
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 1471. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 29 November 2019).
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare [version 1; peer review: 3 approved]. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- The UniProt Consortium, UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515.
- Eddy, S. Hidden Markov Models. Available online: http://hmmer.org/ (accessed on 19 November 2019).
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Usadel, B. Mercator4 Webtool. Available online: https://plabipd.de/portal/mercator4 (accessed on 30 March 2020).
- EnsemblPlants, Oryza Wildspecies. Available online: https://plants.ensembl.org/index.html (accessed on 2 April 2019).
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kassambara, A. Ggpubr R Package: Ggplot2-Based Publication Ready Plots. Available online: https://github.com/kassambara/ggpubr (accessed on 30 April 2019).
- Auguie, B. GridExtra: Miscellaneous Functions for “Grid” Graphics. R Package Version 2.3. Available online: http://CRAN.R-project.org/package=gridExtra (accessed on 30 April 2019).
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 2074. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cultivar | ssp. | ID No. | Organ | Set-up | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL | LE | PA | FS | DS | SH | RO | SO | PP | AN | CC | F | NH | |||
| Dular | aus | IRGC 636 | X | X | X | X | |||||||||
| N22 | IRTP 3911 | X | X | X | X | ||||||||||
| Anjali | indica | IRTP 23206 | X | X | X | X | |||||||||
| IR62266-42-6-2 | IRGC 117597 | X | X | X | X | X | X | X | |||||||
| IR64 | IRTP 12158 | X | X | X | X | X | X | X | X | ||||||
| IR72 | IRTP 14747 | X | X | X | X | X | X | X | |||||||
| CT9993-5-10-1M | japonica | IRIS 71-1229921 | X | X | X | X | X | X | X | ||||||
| M202 | IRGC 77142 | X | X | X | X | X | X | X | |||||||
| Moroberekan | IRGC 12048 | X | X | X | X | X | X | X | X | ||||||
| Nipponbare | IRGC 12731 | X | X | X | X | X | |||||||||
| Cultivar | ssp. | PB | GB | FLNC | HQ | LQ | HQ after Filtering |
|---|---|---|---|---|---|---|---|
| Dular | aus | 2 | 18.46 | 460,340 | 42,252 | 1960 | 41,396 |
| N22 | 3 | 24.17 | 736,747 | 54,572 | 1807 | 52,333 | |
| Anjali | indica | 2 | 15.49 | 481,094 | 40,208 | 1732 | 39,438 |
| IR62266-42-6-2 | 2 | 22.48 | 649,085 | 50,569 | 1659 | 50,510 | |
| IR64 | 2 | 21.97 | 622,881 | 49,633 | 1279 | 49,327 | |
| IR72 | 2 | 20.31 | 554,872 | 44,176 | 2170 | 44,049 | |
| CT9993-5-10-1M | japonica | 2 | 20.81 | 620,595 | 48,537 | 1465 | 48,401 |
| M202 | 2 | 24.07 | 656,740 | 48,836 | 1501 | 48,676 | |
| Moroberekan | 2 | 24.51 | 675,251 | 54,684 | 1721 | 54,594 | |
| Nipponbare | 3 | 15.65 | 544,792 | 37,951 | 1233 | 37,535 |
| Cultivar | ssp. | Reference | # Tr. | Reference-Based | Reference-Free | |||
|---|---|---|---|---|---|---|---|---|
| TAMA | cDNA Cupcake | Unmapped | Cogent | Unmapped | ||||
| Dular | aus | n22 | 41,396 | 13,995 | 18,239 | 313 | 13,107 | 7340 |
| N22 | 52,333 | 18,787 | 23,954 | 149 | 19,026 | 6603 | ||
| Anjali | indica | S498 | 39,438 | 14,371 | 18,170 | 178 | 13,237 | 6476 |
| IR62266-42-6-2 | 50,510 | 18,926 | 23,803 | 220 | 18,773 | 6913 | ||
| IR64 | 49,327 | 19,064 | 23,435 | 1911 | 17,874 | 7979 | ||
| IR72 | 44,049 | 15,954 | 20,646 | 143 | 15,251 | 7426 | ||
| CT9993-5-10-1M | japonica | Nipponbare | 48,401 | 18,789 | 23,415 | 223 | 18,359 | 6611 |
| M202 | 48,676 | 18,925 | 23,670 | 240 | 18,091 | 6695 | ||
| Moroberekan | 54,594 | 20,604 | 26,009 | 268 | 20,378 | 7358 | ||
| Nipponbare | 37,535 | 16,584 | 19,674 | 42 | 14,345 | 5441 | ||
| Cultivar | ssp. | # GL | # TR | # TR/GL | Total # bp | Min [bp] | Max [bp] | Median [bp] | GC [%] |
|---|---|---|---|---|---|---|---|---|---|
| Dular | aus | 10,511 | 14,255 | 1.4 | 15,447,641 | 56 | 4551 | 986 | 50.87 |
| N22 | 13,343 | 18,913 | 1.4 | 26,290,969 | 62 | 5911 | 1295 | 52.26 | |
| Anjali | indica | 10,616 | 14,499 | 1.4 | 17,717,403 | 75 | 4216 | 1156 | 51.99 |
| IR62266-42-6-2 | 13,227 | 19,093 | 1.4 | 26,791,848 | 51 | 7190 | 1314 | 51.37 | |
| IR64 | 15,011 | 20,672 | 1.4 | 28,663,408 | 56 | 6919 | 1299 | 52.76 | |
| IR72 | 11,647 | 16,081 | 1.4 | 19,678,018 | 53 | 5475 | 1149 | 51.16 | |
| CT9993-5-10-1M | japonica | 13,354 | 18,963 | 1.4 | 26,757,988 | 55 | 5752 | 1318 | 51.97 |
| M202 | 13,143 | 19,105 | 1.5 | 26,258,012 | 59 | 6644 | 1287 | 51.74 | |
| Moroberekan | 14,324 | 20,803 | 1.5 | 28,446,682 | 57 | 7072 | 1278 | 51.80 | |
| Nipponbare | 11,366 | 16,622 | 1.5 | 24,760,098 | 75 | 6035 | 1394 | 52.60 | |
| IRGSP | japonica | 38,866 | 45,660 | 1.2 | 69,184,066 | 30 | 16,029 | 1385 | 51.24 |
| Cultivar | ssp. | Mercator | Blastx | Blastp | PFAM | GO | No Annotation | Homologs WS |
|---|---|---|---|---|---|---|---|---|
| Dular | aus | 61.60 | 65.17 | 59.57 | 59.81 | 37.98 | 27.60 | 90.54 |
| N22 | 68.40 | 72.05 | 68.43 | 70.01 | 45.52 | 19.24 | 91.33 | |
| Anjali | indica | 65.77 | 69.46 | 65.43 | 66.90 | 43.06 | 22.03 | 89.82 |
| IR62266-46-6-2 | 68.08 | 71.53 | 67.16 | 68.64 | 44.85 | 20.19 | 91.19 | |
| IR64 | 67.78 | 71.27 | 67.37 | 69.55 | 45.31 | 20.23 | 82.03 | |
| IR72 | 63.55 | 67.20 | 62.26 | 63.78 | 41.22 | 24.96 | 88.54 | |
| CT9993-5-10-1M | japonica | 68.57 | 71.80 | 67.58 | 69.24 | 45.01 | 19.62 | 92.43 |
| M202 | 67.78 | 71.08 | 66.69 | 67.97 | 44.44 | 20.68 | 90.71 | |
| Moroberekan | 65.72 | 69.03 | 64.66 | 66.85 | 43.42 | 22.37 | 91.68 | |
| Nipponbare | 71.25 | 74.35 | 70.26 | 72.16 | 47.59 | 16.81 | 91.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaarschmidt, S.; Fischer, A.; Lawas, L.M.F.; Alam, R.; Septiningsih, E.M.; Bailey-Serres, J.; Jagadish, S.V.K.; Huettel, B.; Hincha, D.K.; Zuther, E. Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L. Int. J. Mol. Sci. 2020, 21, 8148. https://doi.org/10.3390/ijms21218148
Schaarschmidt S, Fischer A, Lawas LMF, Alam R, Septiningsih EM, Bailey-Serres J, Jagadish SVK, Huettel B, Hincha DK, Zuther E. Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L. International Journal of Molecular Sciences. 2020; 21(21):8148. https://doi.org/10.3390/ijms21218148
Chicago/Turabian StyleSchaarschmidt, Stephanie, Axel Fischer, Lovely Mae F. Lawas, Rejbana Alam, Endang M. Septiningsih, Julia Bailey-Serres, S. V. Krishna Jagadish, Bruno Huettel, Dirk K. Hincha, and Ellen Zuther. 2020. "Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L." International Journal of Molecular Sciences 21, no. 21: 8148. https://doi.org/10.3390/ijms21218148
APA StyleSchaarschmidt, S., Fischer, A., Lawas, L. M. F., Alam, R., Septiningsih, E. M., Bailey-Serres, J., Jagadish, S. V. K., Huettel, B., Hincha, D. K., & Zuther, E. (2020). Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L. International Journal of Molecular Sciences, 21(21), 8148. https://doi.org/10.3390/ijms21218148