Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites

 ,
,
Abstract
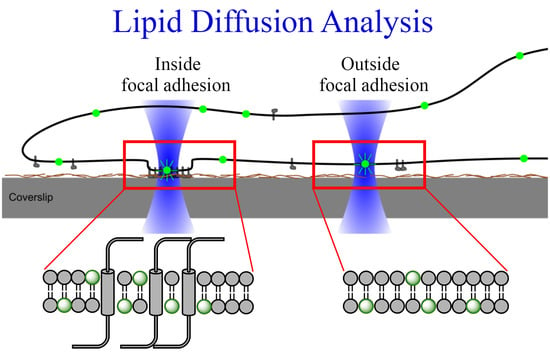
1. Introduction
2. Results
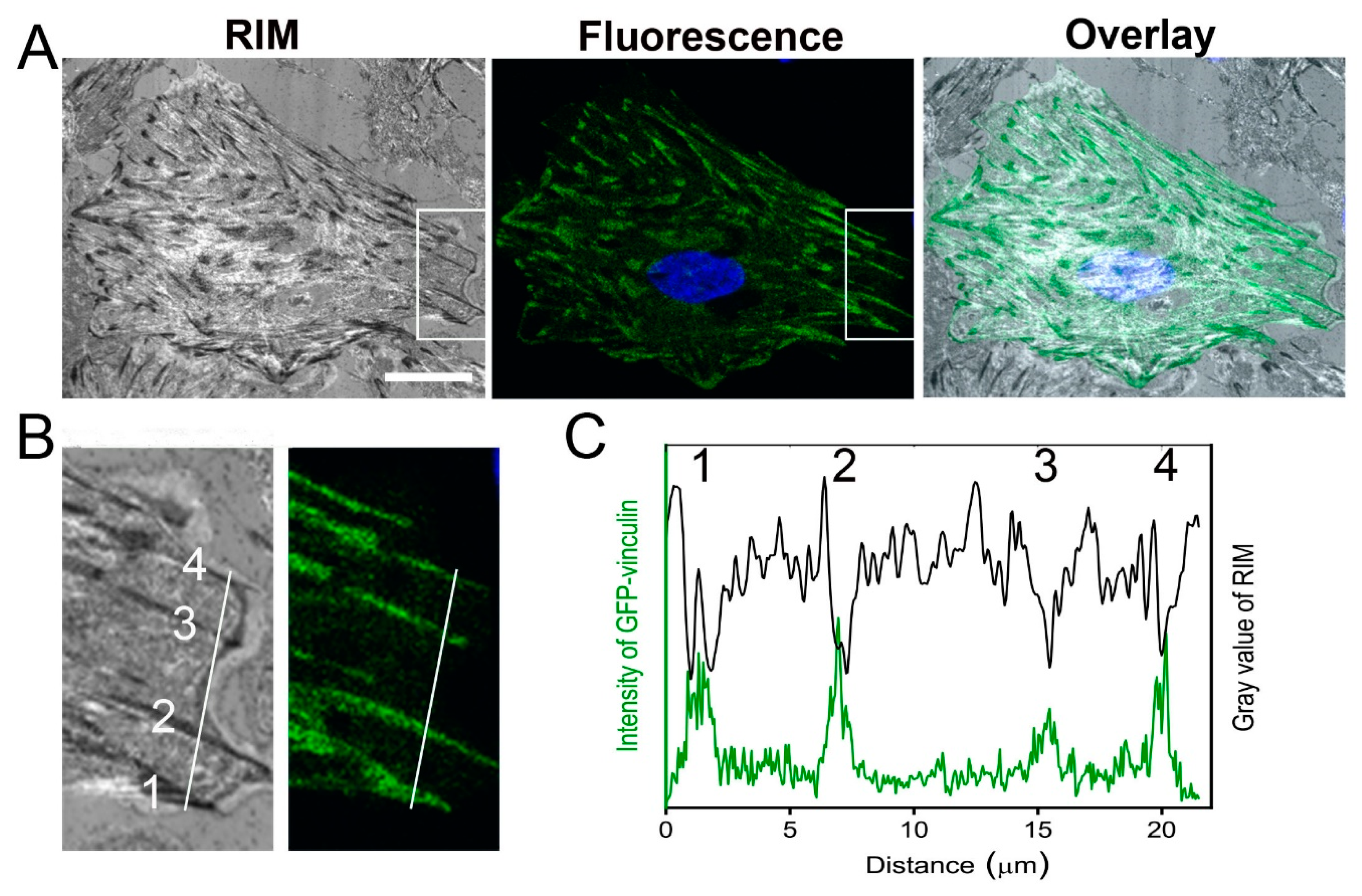
2.1. Localization of Focal Adhesion Sites of Cardiac Myofibroblasts
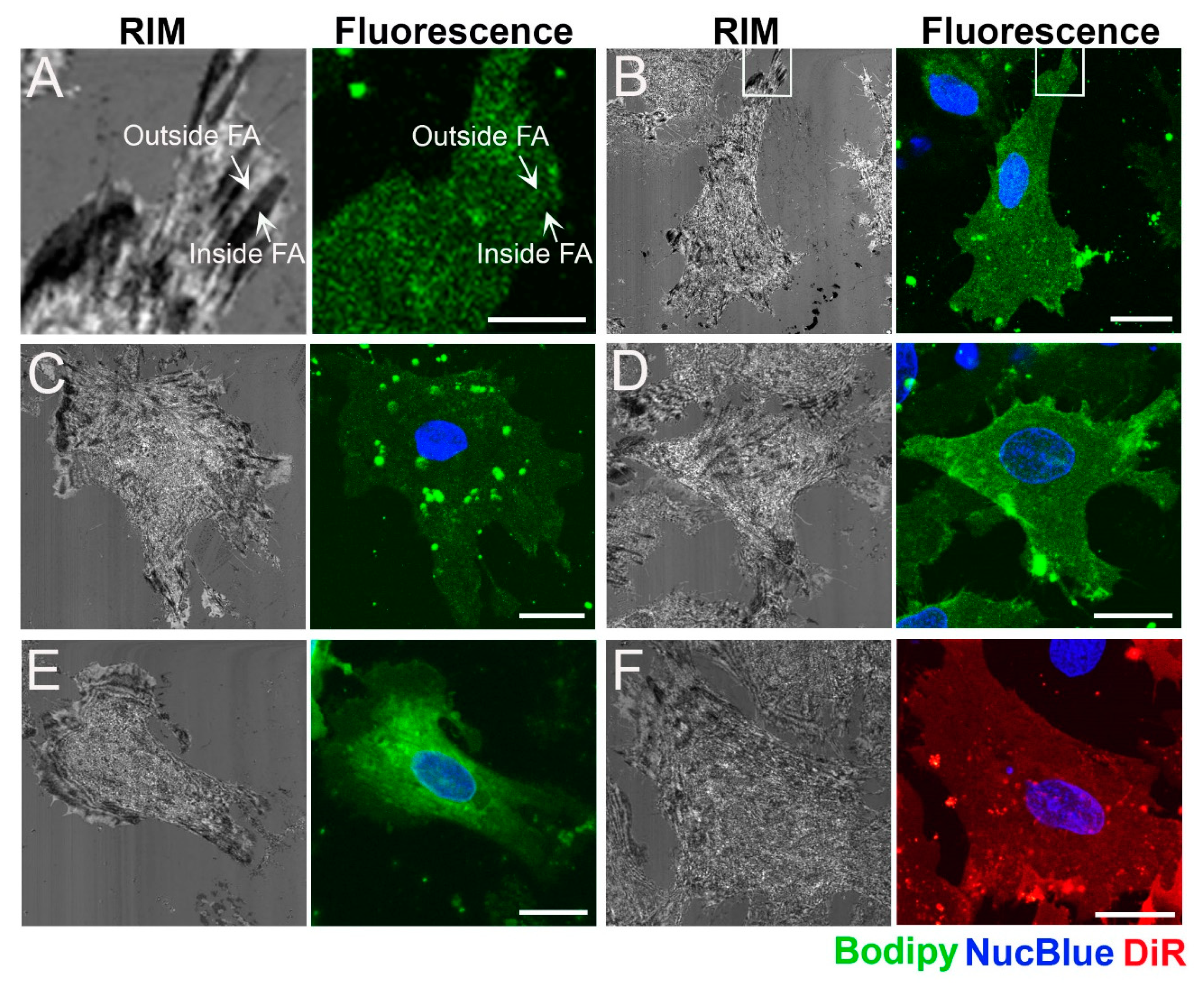
2.2. Distribution of Fluorescent Membrane Components in the Plasma Membrane of Cardiac Myofibroblasts
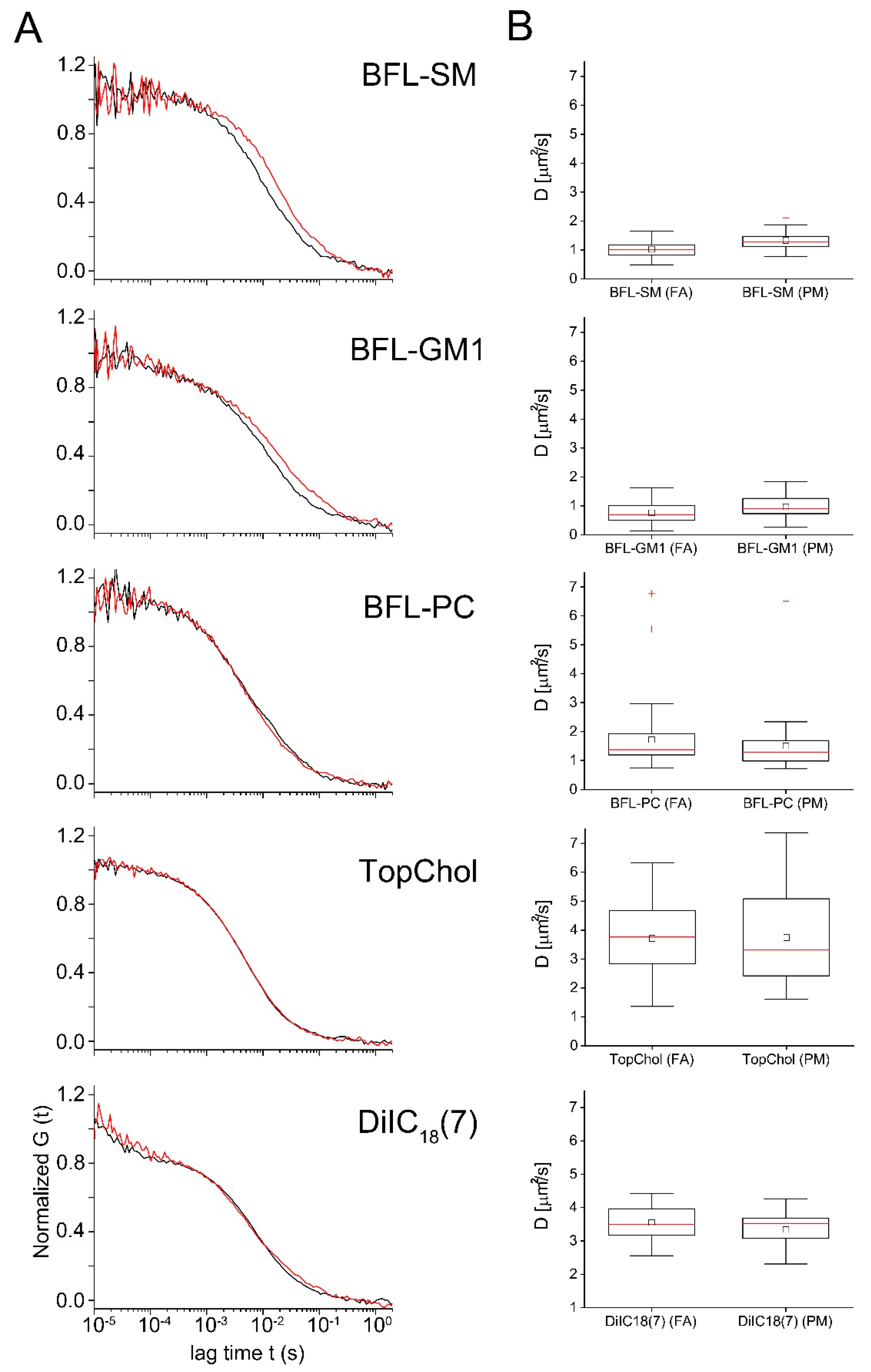
2.3. Diffusion of Fluorescent Lipids in the Plasma Membrane of Cardiac Myofibroblasts
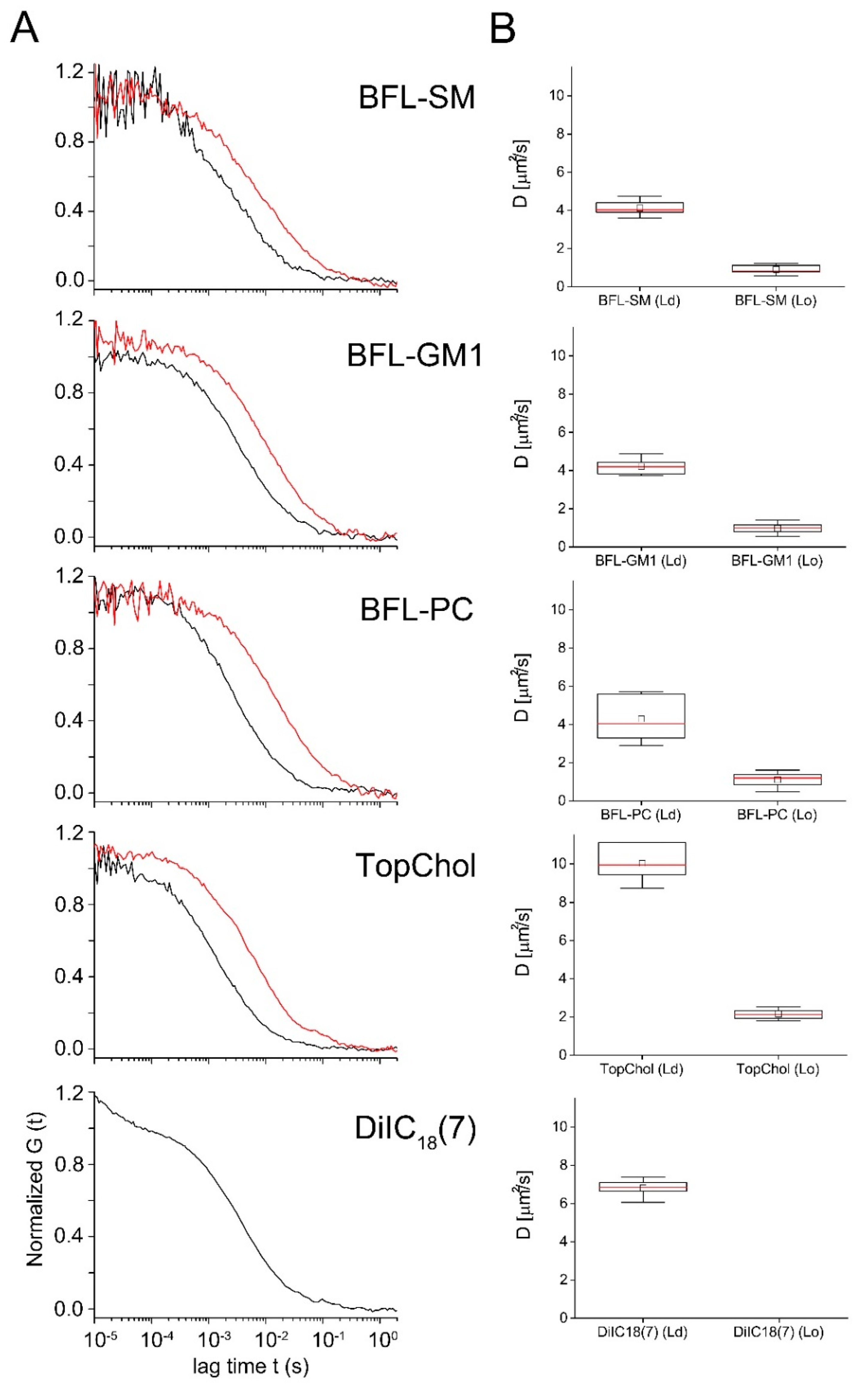
2.4. Influence of Lipid Order on the Lipid Diffusion in Giant Unilamellar Vesicles (GUVs)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Identification of Myocytes and Fibroblasts by Immunostaining and Phase Contrast Microscopy
4.3. Transfection of Myofibroblasts with GFP-Vinculin
4.4. Lipids
4.5. Cell Staining Using Fusogenic Liposomes
4.6. Giant Unilamellar Vesicles (GUVs)
4.7. Supported Lipid Bilayer
4.8. Confocal Microscopy and Fluorescence Correlation Spectroscopy (FCS)
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abercrombie, M.; Dunn, G.A. Adhesions of fibroblasts to substratum during contact inhibition observed by interference reflection microscopy. Exp. Cell Res. 1975, 92, 57–62. [Google Scholar] [CrossRef]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, 5033. [Google Scholar] [CrossRef]
- Lawson, C.; Schlaepfer, D.D. Integrin adhesions: Who’s on first? What’s on second? Connections between FAK and talin. Cell Adh. Migr. 2012, 6, 302–306. [Google Scholar] [CrossRef][Green Version]
- Wehrle-Haller, B. Structure and function of focal adhesions. Curr. Opin. Cell Biol. 2012, 24, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Cohen, M.; Addadi, L.; Geiger, B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 2004, 32, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, A.; Sonnenberg, A. Function and interactions of integrins. Cell Tissue Res. 2001, 305, 285–298. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef]
- Gahmberg, C.G.; Fagerholm, S.C.; Nurmi, S.M.; Chavakis, T.; Marchesan, S.; Gronholm, M. Regulation of integrin activity and signalling. Biochim. Biophys. Acta 2009, 1790, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell. Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.J. A lipid matrix model of membrane raft structure. Prog. Lipid. Res. 2010, 49, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, T.K. Lipid-protein interplay and lateral organization in biomembranes. Chem. Phys. Lipids 2015, 189, 48–55. [Google Scholar] [CrossRef]
- Lu, S.M.; Fairn, G.D. Mesoscale organization of domains in the plasma membrane—Beyond the lipid raft. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 192–207. [Google Scholar] [CrossRef]
- Saltel, F.; Mortier, E.; Hytonen, V.P.; Jacquier, M.C.; Zimmermann, P.; Vogel, V.; Liu, W.; Wehrle-Haller, B. New PI(4,5)P2- and membrane proximal integrin-binding motifs in the talin head control beta3-integrin clustering. J. Cell Biol. 2009, 187, 715–731. [Google Scholar] [CrossRef]
- Kalli, A.C.; Rog, T.; Vattulainen, I.; Campbell, I.D.; Sansom, M.S.P. The Integrin Receptor in Biologically Relevant Bilayers: Insights from Molecular Dynamics Simulations. J. Membr. Biol. 2017, 250, 337–351. [Google Scholar] [CrossRef]
- Schmidt, T.; Suk, J.E.; Ye, F.; Situ, A.J.; Mazumder, P.; Ginsberg, M.H.; Ulmer, T.S. Annular anionic lipids stabilize the integrin alphaIIbbeta3 transmembrane complex. J. Biol. Chem. 2015, 290, 8283–8293. [Google Scholar] [CrossRef]
- Veatch, S.L.; Keller, S.L. Seeing spots: Complex phase behavior in simple membranes. Biochim. Biophys. Acta 2005, 1746, 172–185. [Google Scholar] [CrossRef]
- Marsh, D. Cholesterol-induced fluid membrane domains: A compendium of lipid-raft ternary phase diagrams. Biochim. Biophys. Acta 2009, 1788, 2114–2123. [Google Scholar] [CrossRef]
- Krause, M.R.; Regen, S.L. The structural role of cholesterol in cell membranes: From condensed bilayers to lipid rafts. Acc. Chem. Res. 2014, 47, 3512–3521. [Google Scholar] [CrossRef]
- Wiseman, P.W.; Brown, C.M.; Webb, D.J.; Hebert, B.; Johnson, N.L.; Squier, J.A.; Ellisman, M.H.; Horwitz, A.F. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J. Cell Sci. 2004, 117, 5521–5534. [Google Scholar] [CrossRef]
- Cavalcanti-Adam, E.A.; Volberg, T.; Micoulet, A.; Kessler, H.; Geiger, B.; Spatz, J.P. Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 2007, 92, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Selhuber-Unkel, C.; Erdmann, T.; Lopez-Garcia, M.; Kessler, H.; Schwarz, U.S.; Spatz, J.P. Cell adhesion strength is controlled by intermolecular spacing of adhesion receptors. Biophys. J. 2010, 98, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.C.; Fujiwara, T.K.; Chen, L.; Suzuki, K.G.; Ishikawa, Y.; Nemoto, Y.L.; Miwa, Y.; Kalay, Z.; Chadda, R.; Naruse, K.; et al. Archipelago architecture of the focal adhesion: Membrane molecules freely enter and exit from the focal adhesion zone. Cytoskeleton 2012, 69, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Wirth, V.F.; List, F.; Diez, G.; Goldmann, W.H. Vinculin’s C-terminal region facilitates phospholipid membrane insertion. Biochem. Biophys. Res. Commun. 2010, 398, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Ramachandran, S.; Case, L.B.; Tolbert, C.E.; Tandon, A.; Pershad, M.; Dokholyan, N.V.; Waterman, C.M.; Campbell, S.L. A Structural Model for Vinculin Insertion into PIP2-Containing Membranes and the Effect of Insertion on Vinculin Activation and Localization. Structure 2017, 25, 264–275. [Google Scholar] [CrossRef]
- Dwivedi, M.; Winter, R. Binding of Vinculin to Lipid Membranes in Its Inhibited and Activated States. Biophys. J. 2016, 111, 1444–1453. [Google Scholar] [CrossRef][Green Version]
- Martel, V.; Racaud-Sultan, C.; Dupe, S.; Marie, C.; Paulhe, F.; Galmiche, A.; Block, M.R.; Albiges-Rizo, C. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 2001, 276, 21217–21227. [Google Scholar] [CrossRef]
- Ye, X.; McLean, M.A.; Sligar, S.G. Phosphatidylinositol 4,5-Bisphosphate Modulates the Affinity of Talin-1 for Phospholipid Bilayers and Activates Its Autoinhibited Form. Biochemistry 2016, 55, 5038–5048. [Google Scholar] [CrossRef]
- Gaus, K.; Le Lay, S.; Balasubramanian, N.; Schwartz, M.A. Integrin-mediated adhesion regulates membrane order. J. Cell. Biol. 2006, 174, 725–734. [Google Scholar] [CrossRef]
- Fuentes, D.E.; Butler, P.J. Coordinated Mechanosensitivity of Membrane Rafts and Focal Adhesions. Cell Mol. Bioeng. 2012, 5, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Moroney, G.J.; Butler, P.J. beta1-Integrin-Mediated Adhesion Is Lipid-Bilayer Dependent. Biophys. J. 2017, 113, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Gaul, V.; Lopez, S.G.; Lentz, B.R.; Moran, N.; Forster, R.J.; Keyes, T.E. The lateral diffusion and fibrinogen induced clustering of platelet integrin alphaIIbbeta3 reconstituted into physiologically mimetic GUVs. Integr. Biol. 2015, 7, 402–411. [Google Scholar] [CrossRef]
- Lietha, D.; Izard, T. Roles of membrane domains in integrin-mediated cell adhesion. Int. J. Mol. Sci. 2020, 21, 5531. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef]
- Dugina, V.; Fontao, L.; Chaponnier, C.; Vasiliev, J.; Gabbiani, G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 2001, 114, 3285–3296. [Google Scholar]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef]
- Wang, T.-Y.; Silvius, J.R. Sphingomyelin partitioning into ordered domains in cholesterol-free and cholesterol-containing lipid bilayers. Biophys. J. 2003, 84, 367–378. [Google Scholar] [CrossRef]
- Veatch, S.L.; Keller, S.L. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys. Rev. Lett. 2005, 94, 148101. [Google Scholar] [CrossRef]
- Heberle, F.A.; Feigenson, G.W. Phase separation in lipid membranes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004630. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, T.; Hunt, G.; Farkas, E.R.; Webb, W.W.; Feigenson, G.W. Fluorescence probe partitioning between Lo/Ld phases in lipid membranes. Biochim. Biophys. Acta 2007, 1768, 2182–2194. [Google Scholar] [CrossRef]
- Sengupta, P.; Hammond, A.; Holowka, D.; Baird, B. Structural determinants for partitioning of lipids and proteins between coexisting fluid phases in giant plasma membrane vesicles. Biochim. Biophys. Acta 2008, 1778, 20–32. [Google Scholar] [CrossRef]
- Geiger, B.; Tokuyasu, K.T.; Dutton, A.H.; Singer, S.J. Vinculin, an intracellular protein localized at specialized sites where microfilament bundles terminate at cell membranes. Proc. Natl. Acad. Sci. USA 1980, 77, 4127–4131. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Enomoto-Iwamoto, M.; Yoshida, T.; Sakakura, T. Vinculin, talin, integrin α6β1 and laminin can serve as components of attachment complex mediating contraction force transmission from cardiomyocytes to extracellular matrix. Cell Mot. Cytoskel. 1999, 42, 1–11. [Google Scholar] [CrossRef]
- Cesa, C.M.; Kirchgeßner, N.; Mayer, D.; Schwarz, U.S.; Hoffmann, B.; Merkel, R. Micropatterned silicone elastomer substrates for high resolution analysis of cellular force patterns. Rev. Sci. Instrum. 2007, 7, 034301. [Google Scholar] [CrossRef]
- Kleusch, C.; Hersch, N.; Hoffmann, B.; Merkel, R.; Csiszar, A. Fluorescent lipids: Functional parts of fusogenic liposomes and tools for cell membrane labeling and visualization. Molecules 2012, 17, 1055–1073. [Google Scholar] [CrossRef]
- van Echten-Deckert, G.; Herget, T. Sphingolipid metabolism in neural cells. Biochim. Biophys. Acta 2006, 1758, 1978–1994. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Sleight, R.G.; Pagano, R.E. Transport of a fluorescent phosphatidylcholine analog from the plasma memrane to the Golgi apparatus. J. Cell Biol. 1984, 99, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Lin, S.X.; Karylowski, O.J.; Wustner, D.; McGraw, T.E.; Maxfield, F.R. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J. Biol. Chem. 2002, 277, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.F.; Morris, R. Transmembrane asymmetry and lateral domains in biological membranes. Traffic 2004, 5, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Patla, I.; Volberg, T.; Elad, N.; Hirschfeld-Warneken, V.; Grashoff, C.; Fässler, R.; Spatz, J.P.; Geiger, B.; Medalia, O. Dissecting the molecular architecture of integrin adhesion sites by cryo-electron tomography. Nat. Cell Biol. 2010, 12, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Medalia, O.; Geiger, B. Frontiers of microscopy-based research into cell–matrix adhesions. Curr. Opin. Cell Biol. 2010, 22, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Veatch, S.L.; Keller, S.L. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 2003, 85, 3074–3083. [Google Scholar] [CrossRef]
- Barenholz, Y. Sphingomyelin and cholesterol: From membrane biophysics and rafts to potential medical applications. Sub-Cell. Biochem. 2004, 37, 167–215. [Google Scholar] [CrossRef]
- Solanko, L.M.; Honigmann, A.; Midtiby, H.S.; Lund, F.W.; Brewer, J.R.; Dekaris, V.; Bittman, R.; Eggeling, C.; Wustner, D. Membrane orientation and lateral diffusion of BODIPY-cholesterol as a function of probe structure. Biophys. J. 2013, 105, 2082–2092. [Google Scholar] [CrossRef]
- Honigmann, A.; Mueller, V.; Ta, H.; Schoenle, A.; Sezgin, E.; Hell, S.W.; Eggeling, C. Scanning STED-FCS reveals spatiotemporal heterogeneity of lipid interaction in the plasma membrane of living cells. Nat. Commun. 2014, 5, 5412. [Google Scholar] [CrossRef]
- Zamir, E.; Katz, B.Z.; Aota, S.; Yamada, K.M.; Geiger, B.; Kam, Z. Molecular diversity of cell-matrix adhesions. J. Cell Sci. 1999, 112, 1655–1669. [Google Scholar]
- Dimitrov, D.S.; Angelova, M.I. Lipid swelling and liposome formation mediated by electric fields. Bioelectrochem. Bioenerg. 1988, 19, 323–336. [Google Scholar] [CrossRef]
- Schwille, P.; Korlach, J.; Webb, W.W. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry 1999, 36, 176–182. [Google Scholar] [CrossRef]
- Schwille, P. Fluorescence Correlation Spectroscopy and Its Potential for Intracellular Applications. Cell Biochem. Biophys. 2001, 34, 383–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D (μm2/s) | sdD (µm2/s) | Signi. D | NPart | sdPart | Signi. NPart | Nmeas | |||
|---|---|---|---|---|---|---|---|---|---|
| T-Test | Rank Sum Test | T-Test | Rank Sum Test | ||||||
| BFL-SM (FA) | 1.0 | 0.3 | *** | *** | 23 | 17 | n.s. | n.s. | 54 |
| BFL-SM (PM) | 1.3 | 0.3 | 21 | 17 | 54 | ||||
| BFL-GM1 (FA) | 0.8 | 0.3 | *** | * | 38 | 30 | n.s. | n.s. | 37 |
| BFL-GM1 (PM) | 1.0 | 0.4 | 41 | 54 | 37 | ||||
| BFL-PC (FA) | 1.5 | 0.9 | n.s. | n.s. | 21 | 14 | n.s. | n.s. | 45 |
| BFL-PC (PM) | 1.7 | 1.1 | 27 | 18 | 45 | ||||
| TopChol (FA) | 3.7 | 1.4 | n.s. | n.s. | 29 | 15 | n.s. | n.s. | 35 |
| TopChol (PM) | 3.7 | 1.6 | 32 | 18 | 35 | ||||
| DiIC18(7) (FA) | 3.1 | 0.4 | n.s. | n.s. | 18 | 15 | n.s. | n.s. | 17 |
| DiIC18(7) (PM) | 3.7 | 0.7 | 25 | 20 | 16 | ||||
| D (μm2/s) | sdD (µm2/s) | Signi. D | NPart | sdPart | Signi. NPart | Nmeas | |||
|---|---|---|---|---|---|---|---|---|---|
| T-test | Rank Sum Test | T-test | Rank Sum Test | ||||||
| BFL-SM (Lo) | 0.9 | 0.2 | *** | *** | 72 | 59 | n.s. | n.s. | 10 |
| BFL-SM (Ld) | 4.1 | 0.4 | 146 | 134 | 8 | ||||
| BFL-GM1 (Lo) | 1.0 | 0.3 | *** | *** | 36 | 26 | n.s. | n.s. | 9 |
| BFL-GM1 (Ld) | 4.2 | 0.4 | 48 | 23 | 10 | ||||
| BFL-PC (Lo) | 1.1 | 0.3 | *** | *** | 12 | 7 | *** | *** | 12 |
| BFL-PC (Ld) | 4.3 | 1.1 | 58 | 38 | 11 | ||||
| TopChol (Lo) | 2.2 | 0.2 | *** | *** | 34 | 7 | *** | ** | 7 |
| TopChol (Ld) | 10 | 0.9 | 62 | 11 | 7 | ||||
| DiIC18(7) (Ld) | 6.8 | 0.4 | - | - | 3 | 1 | - | - | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleusch, C.; Monzel, C.; Sridhar, K.C.; Hoffmann, B.; Csiszár, A.; Merkel, R. Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites. Int. J. Mol. Sci. 2020, 21, 8149. https://doi.org/10.3390/ijms21218149
Kleusch C, Monzel C, Sridhar KC, Hoffmann B, Csiszár A, Merkel R. Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites. International Journal of Molecular Sciences. 2020; 21(21):8149. https://doi.org/10.3390/ijms21218149
Chicago/Turabian StyleKleusch, Christian, Cornelia Monzel, Krishna Chander Sridhar, Bernd Hoffmann, Agnes Csiszár, and Rudolf Merkel. 2020. "Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites" International Journal of Molecular Sciences 21, no. 21: 8149. https://doi.org/10.3390/ijms21218149
APA StyleKleusch, C., Monzel, C., Sridhar, K. C., Hoffmann, B., Csiszár, A., & Merkel, R. (2020). Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites. International Journal of Molecular Sciences, 21(21), 8149. https://doi.org/10.3390/ijms21218149