STVNa Attenuates Isoproterenol-Induced Cardiac Hypertrophy Response through the HDAC4 and Prdx2/ROS/Trx1 Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
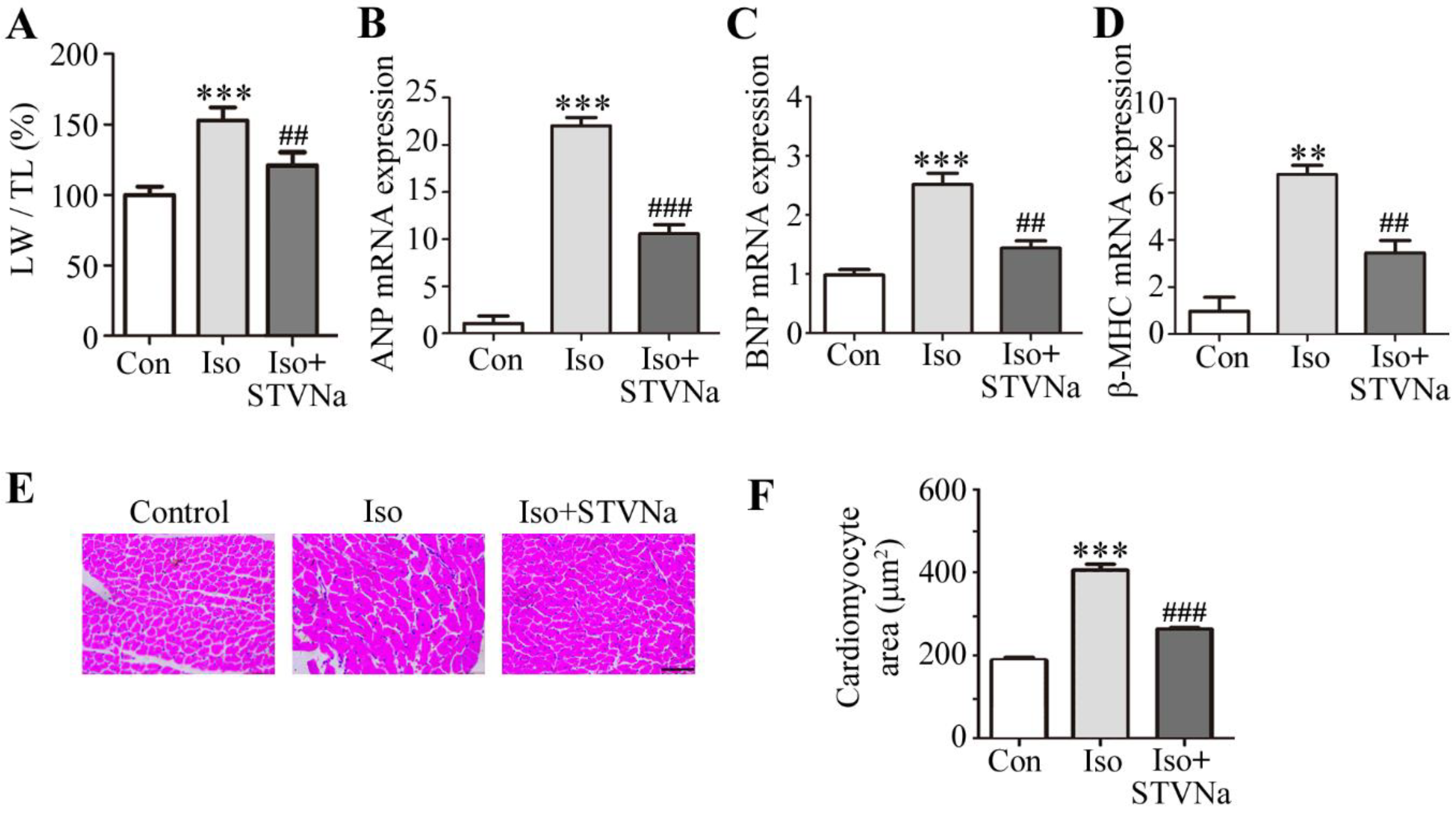
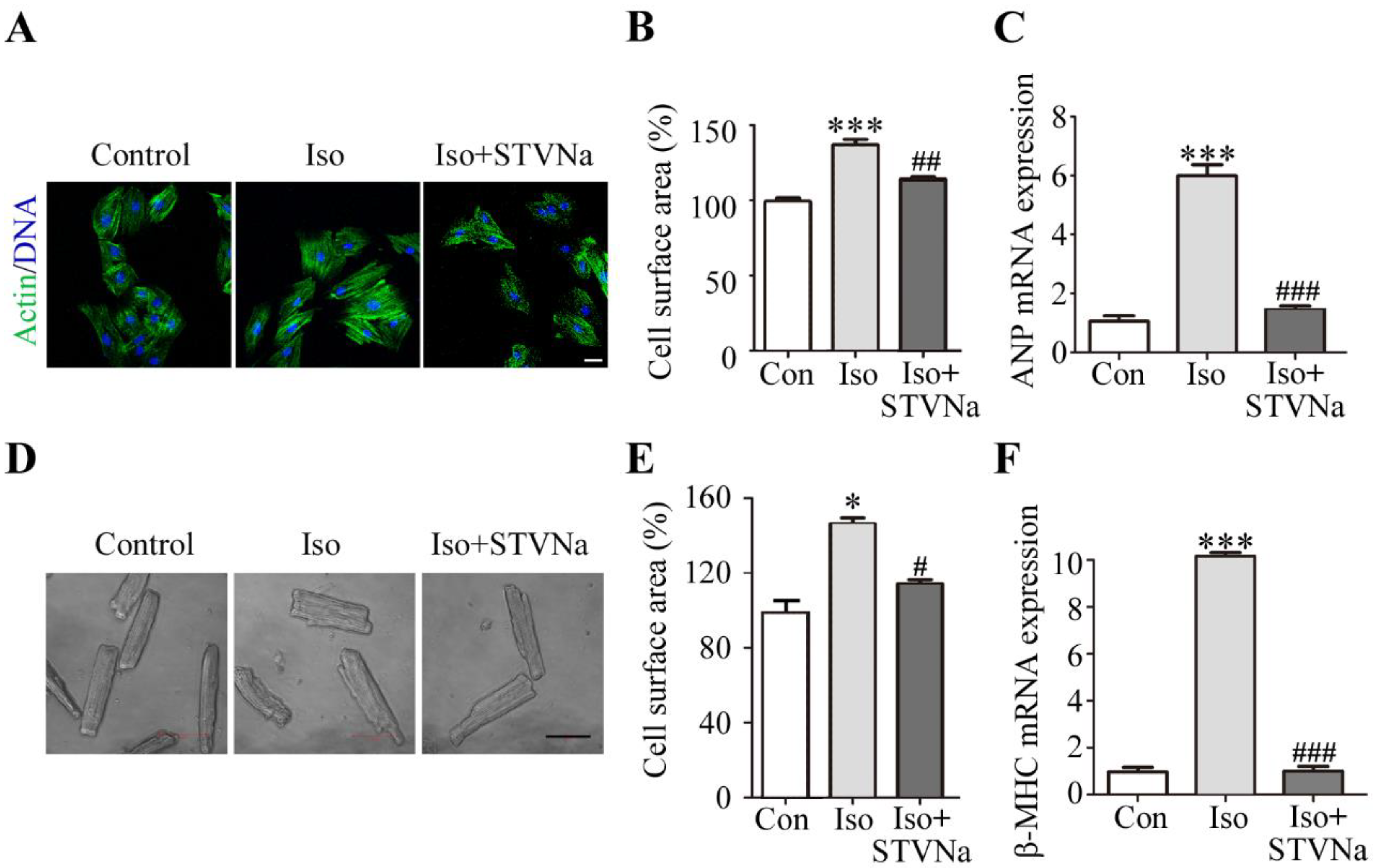
2.1. STVNa Reversed Cardiomyocyte Hypertrophy Induced by Iso
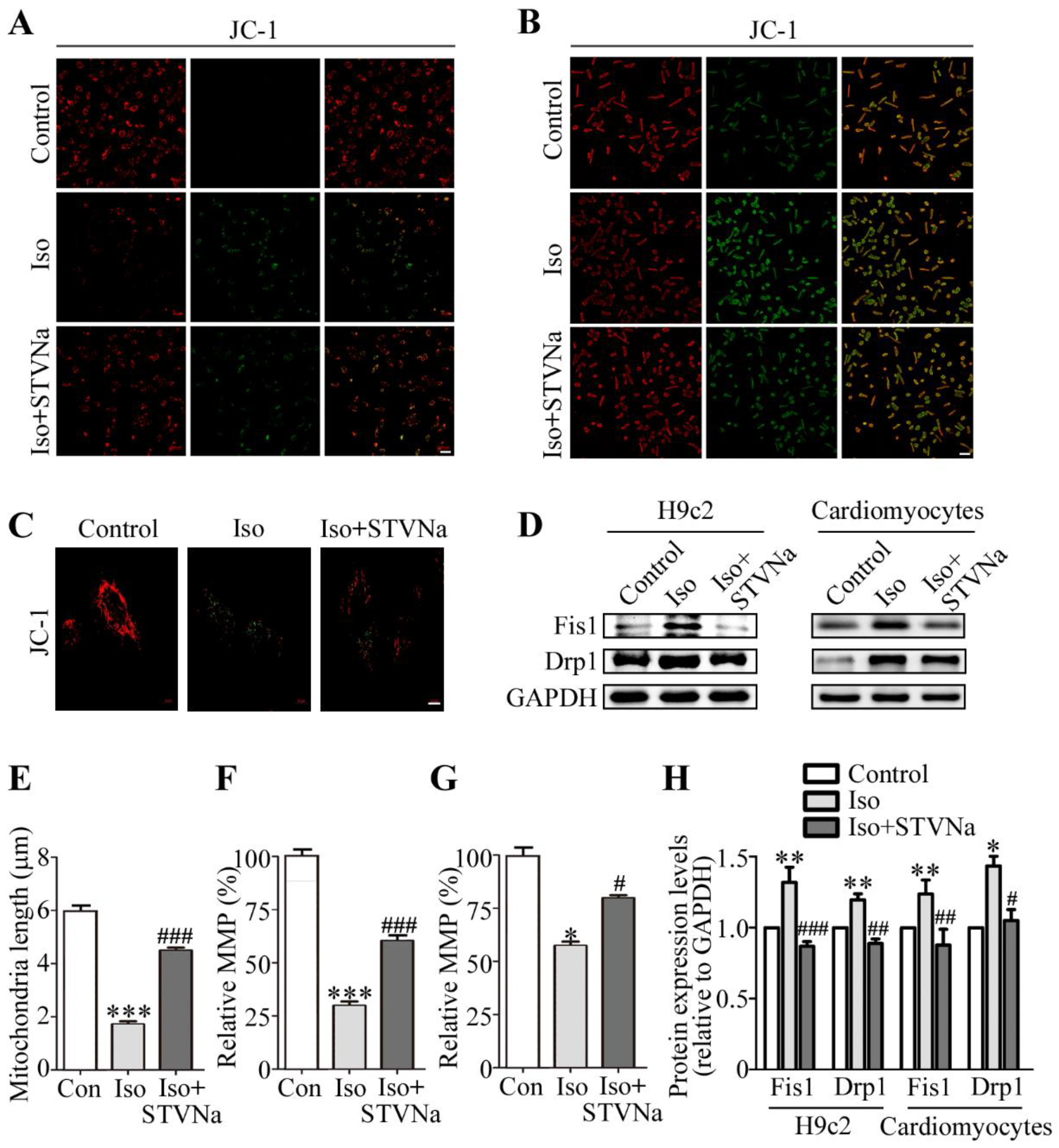
2.2. Effect of STVNa on Mitochondrial Integrity After Iso Induction
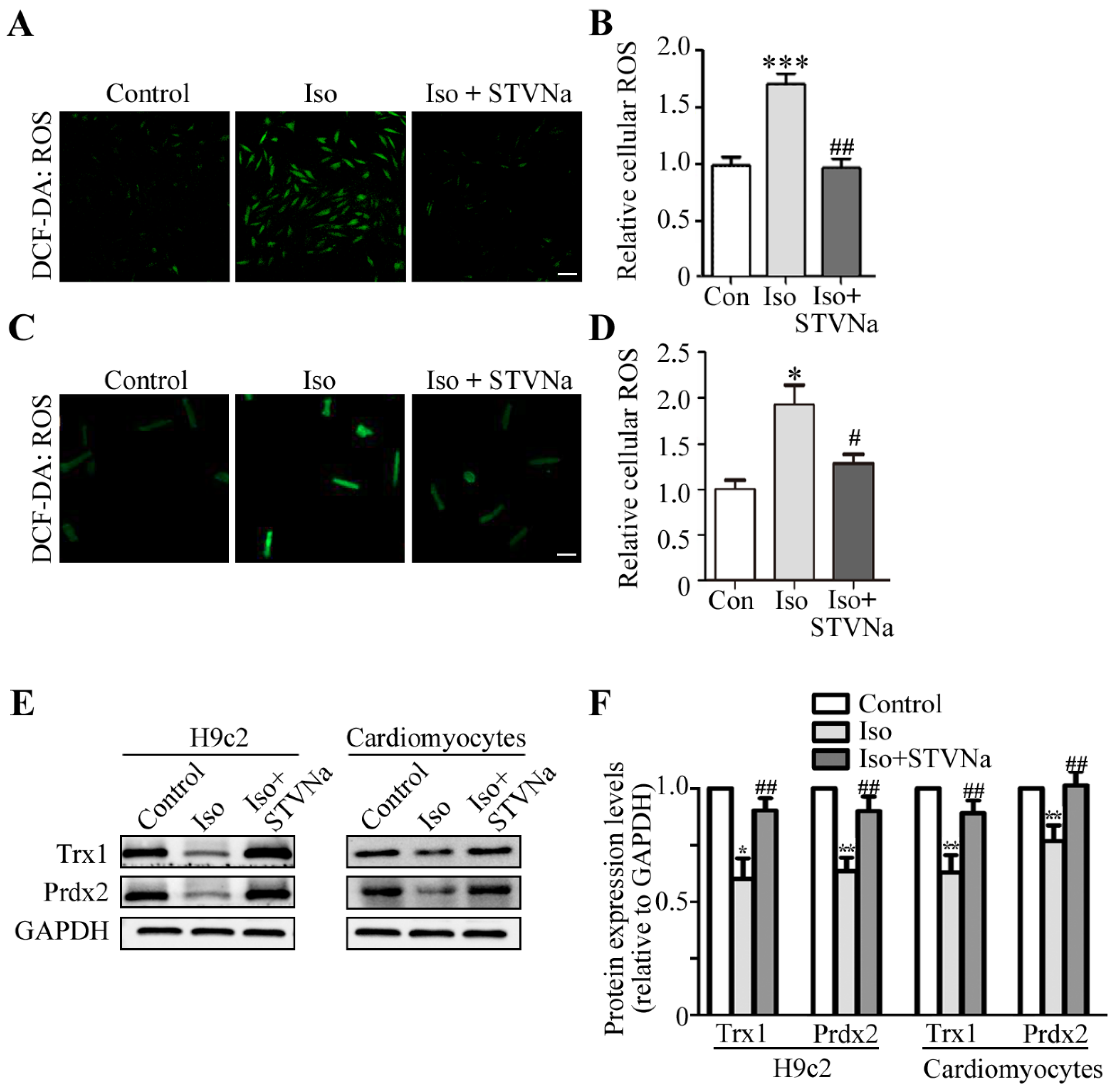
2.3. STVNa Inhibited ROS Burst and Increased Anti-Oxidant Trx1 and Prdx2 Expression
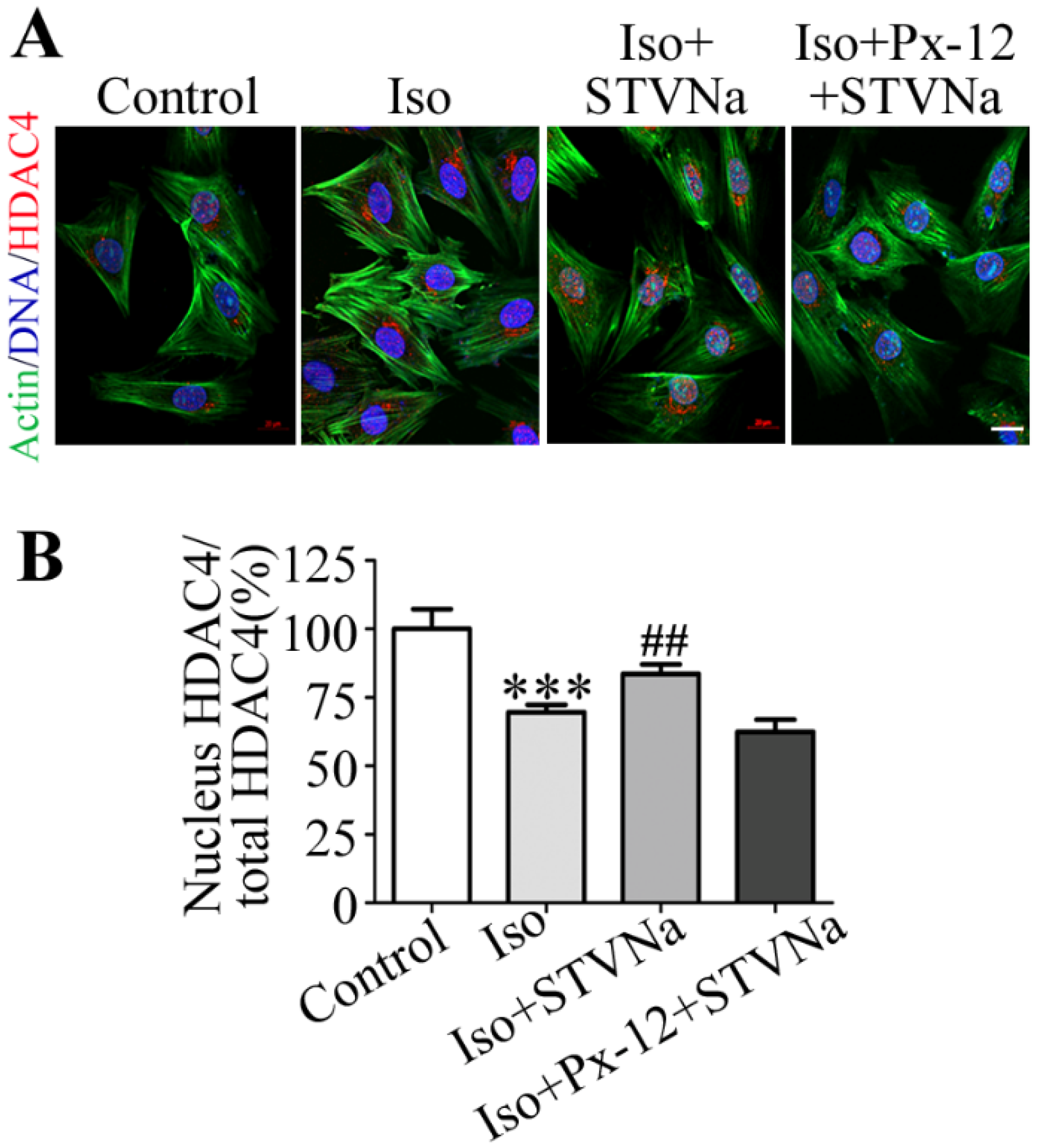
2.4. STVNa Inhibits Iso-Induced Cardiac Hypertrophy by Regulating Nuclear Translocation of HDAC4
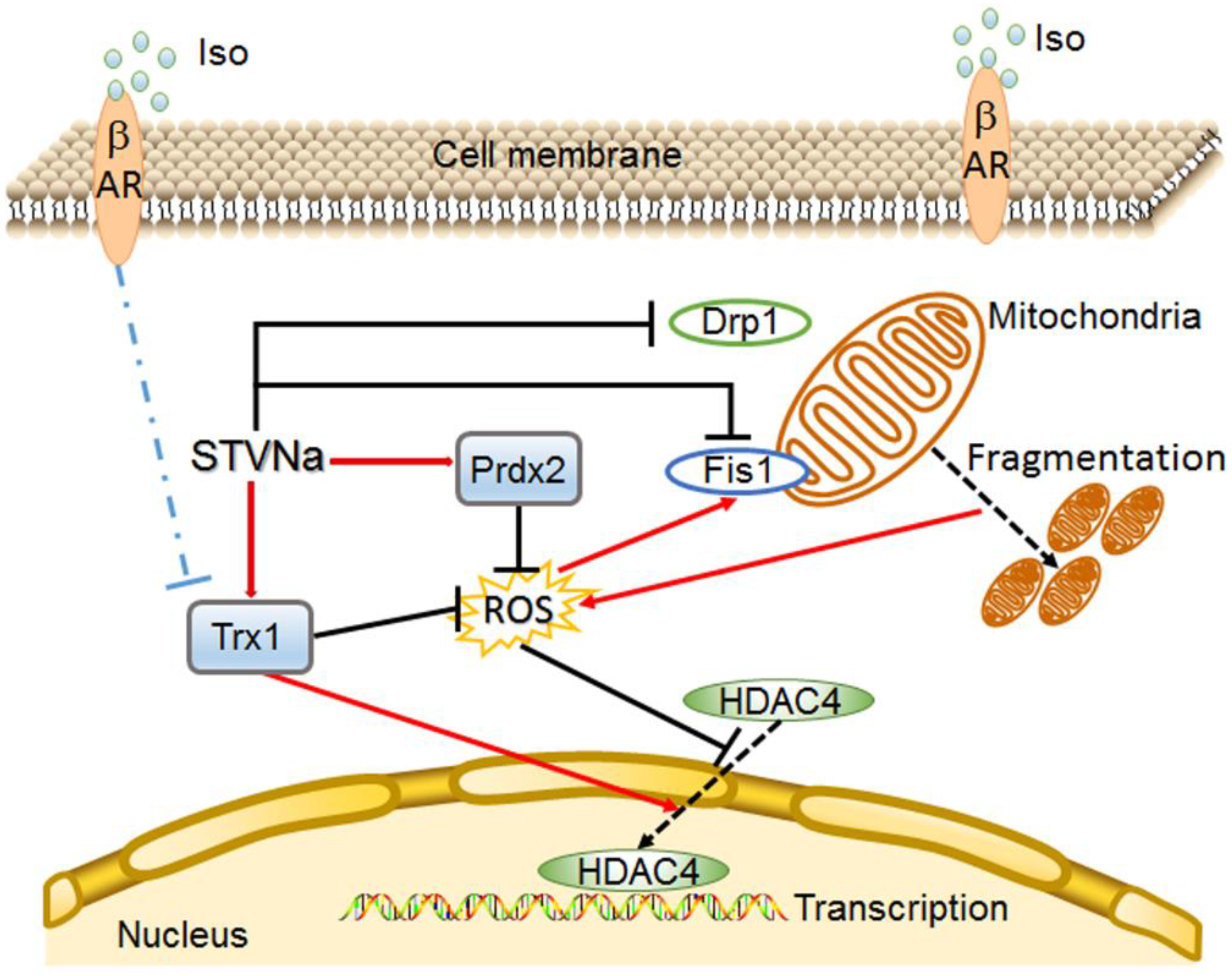
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Experiments and Primary Cardiomyocyte Isolation
4.3. H9c2 Culture and Interventions
4.4. Measurement of Cell Surface Area
4.5. ROS Measurement
4.6. Measurement of Mitochondrial Membrane Potential
4.7. Real-Time quantitative PCR
4.8. Western Blot Analysis
4.9. Immunofluorescence (IF) Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Santambrogio, G.M.; Maloberti, A.; Vallerio, P.; Peritore, A.; Spanò, F.; Occhi, L.; Musca, F.; Belli, O.; De Chiara, B.; Casadei, F.; et al. Could two-dimensional radial strain be considered as a novel tool to identify pre-clinical hypertrophic cardiomyopathy mutation carriers? Int. J. Cardiovasc. Imaging 2019, 35, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Rohini, A.; Agrawal, N.; Koyani, C.N.; Singh, R. Molecular targets and regulators of cardiac hypertrophy. Pharm. Res. 2010, 61, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Youle, R.J. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol. Cell 2008, 29, 409–410. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W.N. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W.N. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in AdultMouse Hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef]
- Xue, R.; Sun, L.; Yu, X.; Li, D.; Zang, W. Vagal nerve stimulation improves mitochondrial dynamics via an 350 M3receptor/CaMKKbeta/AMPK pathway in isoproterenol-induced myocardial ischaemia. J. Cell Mol. 2017, 21, 58–71. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Xue, R.; Zhao, M.; Wu, Q.; Yang, S.; Cui, Y.; Yu, X.; Liu, J.; Zang, W. Regulation of mitochondrial cristae remodelling by acetylcholine alleviates palmitate-induced cardiomyocyte hypertrophy. Free Radic. Bio. Med. 2019, 145, 103–117. [Google Scholar] [CrossRef]
- Xiong, S.; Sun, H.; Cao, L.; Zhu, M.; Liu, T.; Wu, Z.; Bian, J. Stimulation of Na+ /K+ -ATPase with an Antibody against Its 4th Extracellular Region Attenuates Angiotensin II-Induced H9c2 Cardiomyocyte Hypertrophy via an AMPK/SIRT3/PPARγSignaling Pathway. Oxid. Med. Cell Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Jong, C.J.; Yeung, J.; Tseung, E.; Karmazyn, M. Leptin-induced cardiomyocyte hypertrophy is associated with enhanced mitochondrial fission. Mol. Cell Biochem. 2019, 454, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.Y.; Zhang, Z.L.; Cheng, X.; Li, X.D.; Chuai, M.; Lee, K.K.; Kurihara, H.; Yang, X. Excess ROS induced by AAPH causes myocardial hypertrophy in the developing chick embryo. Int. J. Cardiol. 2014, 176, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Date, M.; Morita, T.; Yamashita, N.; Nishida, K.; Yamaguchi, O.; Higuchi, Y.; Hirotani, S.; Matsumura, Y.; Hori, M.; Tada, M.; et al. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricularhypertrophy in in vivo murine pressure-overload model. J. Am. Coll. Cardiol. 2002, 39, 907–912. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Long, Z.; Wang, C.; Wang, L.; Sun, P.; Li, P.; Wang, T. Hydrogen (H(2)) Inhibits Isoproterenol-Induced Cardiac Hypertrophy via Antioxidative Pathways. Front. Pharm. 2016, 7, 392. [Google Scholar] [CrossRef] [PubMed]
- Zschauer, T.; Matsushima, S.; Altschmied, J.; Shao, D.; Sadoshima, J.; Haendeler, J. Interacting with thioredoxin-1--disease or no disease? Antioxid. Redox Sign. 2013, 18, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Ding, Y.; Ryan, S.J.; Kannan, R.; Hinton, D.R. Regulation of thioredoxin by ceramide in retinal pigment epithelial cells. Exp. Eye Res. 2009, 88, 410–417. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rong Jin, Y.G.S.Z. Trx1/TrxR1 system regulates post-selected DP thymocytes survival by modulating ASK1-JNK/p38 MAPK activities. Immunol. Cell Biol. 2015, 8, 744–752. [Google Scholar]
- Luan, R.; Liu, S.; Yin, T.; Lau, W.B.; Wang, Q.; Guo, W.; Wang, H.; Tao, L. High glucose sensitizes adult cardiomyocytes to ischaemia/reperfusion injury through nitrative thioredoxin inactivation. Cardiovasc. Res. 2009, 83, 294–302. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yang, G.; Hong, C.; Liu, J.; Holle, E.; Yu, X.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Investig. 2003, 112, 1395–1406. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. Iubmb. Life 2001, 52, 35–41. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid. Redox Sign. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Hecht, H.J.; Flohé, L. Peroxiredoxins. Biol. Chem. 2002, 383, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Salzano, S.; Checconi, P.; Hanschmann, E.; Lillig, C.H.; Bowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylatedperoxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef]
- Kang, S.W.; Chang, T.; Lee, T.; Kim, E.S.; Yu, D.; Rhee, S.G. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J. Biol. Chem. 2004, 279, 2535–2543. [Google Scholar] [CrossRef]
- Persson, T.; Lattanzio, F.; Calvo-Garrido, J.; Rimondini, R.; Rubio-Rodrigo, M.; Sundström, E.; Maioli, S.; Sandebring-Matton, A.; Cedazo-Mínguez, Á. Apolipoprotein E4 Elicits Lysosomal Cathepsin D Release, Decreased Thioredoxin-1 Levels, and Apoptosis. J. Alzheimer’s Dis. 2017, 56, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, Z.; Wang, J.; Milne, R.W.; Xu, D.; Davey, A.K.; Evans, A.M. Isosteviol reduces plasma glucose levels in the intravenous glucose tolerancetest in Zucker diabetic fatty rats. DiabetesObes. Metab. 2007, 9, 597–599. [Google Scholar]
- Xu, D.; Xia, J.; Liu, H.; Chen, H. Acute toxicological study for isosteviol. Chin. J. Pharmacol. Toxicol. 1994, 8, 33. [Google Scholar]
- Zhang, H.; Zhong, K.; Lu, M.; Mei, Y.; Tan, E.; Sun, X.; Tan, W. Neuroprotective effects of isosteviol sodium through increasing CYLD by the downregulation of miRNA-181b. Brain Res. Bull. 2018, 140, 392–401. [Google Scholar] [CrossRef]
- Shi, X.; Chen, R.; Zhang, Y.; Yun, J.; Brand-Arzamendi, K.; Liu, X.; Wen, X.Y. Zebrafish heart failure models: Opportunities and challenges. Amino Acids 2018, 50, 787–798. [Google Scholar] [CrossRef]
- Li, H.; Xu, C.; Li, Q.; Gao, X.; Sugano, E.; Tomita, H.; Yang, L.; Shi, S. Thioredoxin 2 Offers Protection against Mitochondrial Oxidative Stress in H9c2 Cells and against Myocardial Hypertrophy Induced by Hyperglycemia. Int. J. Mol. Sci. 2017, 18, 1958. [Google Scholar] [CrossRef]
- Su, H.; Pistolozzi, M.; Shi, X.; Sun, X.; Tan, W. Alterations in NO/ROS ratio and expression of Trx1 and Prdx2 inisoproterenol-induced cardiac hypertrophy. Acta. Bioch. Bioph. Sin. 2017, 49, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Chen, Y.; Wu, H.; Xu, D.; Tan, W. Attenuation of ischemia/reperfusion-induced inhibition of the rapid component of delayed rectifier potassium current by Isosteviol through scavenging reactive oxygen species. Biochim. Et Biophys. Acta Biomembr. 2017, 1859, 2447–2453. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Hong, Y.; Kemp, B.K.; Barrientos, A.A.; Erusalimsky, J.D. Evaluation of fluorescent dyes for the detection of mitochondrial membranepotential changes in cultured cardiomyocytes. Cardiovasc. Res. 2000, 46, 126–138. [Google Scholar] [CrossRef]
- Xiong, W.; Hua, J.; Liu, Z.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; Xu, D. PTEN induced putative kinase 1 (PINK1) alleviates angiotensin II-induced cardiac injury by ameliorating mitochondrial dysfunction. Int. J. Cardiol. 2018, 266, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, S.; Nair, R.R. Mitoprotective antioxidant EUK-134 stimulates fatty acid oxidation and preventshypertrophy in H9C2 cells. Mol. Cell Biochem. 2016, 420, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Sabri, A.; Hughie, H.H.; Lucchesi, P.A. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygenspecies in cardiac myocytes. Antioxid Redox Sign. 2003, 5, 731–740. [Google Scholar] [CrossRef]
- Zhao, W.; Fan, G.; Zhang, Z.; Bandyopadhyay, A.; Zhou, X.; Kranias, E.G. Protection of peroxiredoxin II on oxidative stress-induced cardiomyocyte deathand apoptosis. Basic Res. Cardiol. 2009, 104, 377–389. [Google Scholar] [CrossRef]
- Wu, Y.; Hou, F.; Wang, X.; Kong, Q.; Han, X.; Bai, B. Aberrant Expression of Histone Deacetylases 4 in Cognitive Disorders: Molecular Mechanisms and a Potential Target. Front. Mol. Neurosci. 2016, 9, 114. [Google Scholar] [CrossRef]
- Stanley, B.A.; Sivakumaran, V.; Shi, S.; McDonald, I.; Lloyd, D.; Watson, W.H.; Aon, M.A.; Paolocci, N. Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J. Biol. Chem. 2011, 286, 33669–33677. [Google Scholar] [CrossRef]
- Fan, Z.; Lv, N.; Luo, X.; Tan, W. Isosteviol prevents the prolongation of action potential in hypertrophiedcardiomyoctyes by regulating transient outward potassium and L-type calciumchannels. Biochim. Et Biophys. Acta. Biomembr. 2017, 1859, 1872–1879. [Google Scholar] [CrossRef]
- Hoffmann, P.; Richards, D.; Heinroth-Hoffmann, I.; Mathias, P.; Wey, H.; Toraason, M. Arachidonic acid disrupts calcium dynamics in neonatal rat cardiac myocytes. Cardiovasc. Res. 1995, 30, 889–898. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Y.; Xie, Y.; Shi, X.; Huang, L.; Tan, W. Protective role of STVNa in myocardial ischemia reperfusion injury by inhibiting mitochondrial fission. Oncotarget 2018, 9, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, D.; Wang, Y.; Liu, S.; Qin, J.; Wang, J.; Ran, J.; Zhang, Y.; Huang, Q.; Liu, X.; et al. Discovery of Centrosomal Protein 70 as an Important Player in the Development and Progression of Breast Cancer. Am. J. Pathol. 2017, 187, 679–688. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Su, H.; Liu, B.; Mei, Y.; Ke, Q.; Sun, X.; Tan, W. STVNa Attenuates Isoproterenol-Induced Cardiac Hypertrophy Response through the HDAC4 and Prdx2/ROS/Trx1 Pathways. Int. J. Mol. Sci. 2020, 21, 682. https://doi.org/10.3390/ijms21020682
Liu F, Su H, Liu B, Mei Y, Ke Q, Sun X, Tan W. STVNa Attenuates Isoproterenol-Induced Cardiac Hypertrophy Response through the HDAC4 and Prdx2/ROS/Trx1 Pathways. International Journal of Molecular Sciences. 2020; 21(2):682. https://doi.org/10.3390/ijms21020682
Chicago/Turabian StyleLiu, Fei, Hao Su, Bo Liu, Ying Mei, Qingjin Ke, Xiaoou Sun, and Wen Tan. 2020. "STVNa Attenuates Isoproterenol-Induced Cardiac Hypertrophy Response through the HDAC4 and Prdx2/ROS/Trx1 Pathways" International Journal of Molecular Sciences 21, no. 2: 682. https://doi.org/10.3390/ijms21020682
APA StyleLiu, F., Su, H., Liu, B., Mei, Y., Ke, Q., Sun, X., & Tan, W. (2020). STVNa Attenuates Isoproterenol-Induced Cardiac Hypertrophy Response through the HDAC4 and Prdx2/ROS/Trx1 Pathways. International Journal of Molecular Sciences, 21(2), 682. https://doi.org/10.3390/ijms21020682