Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research

 ,
, 
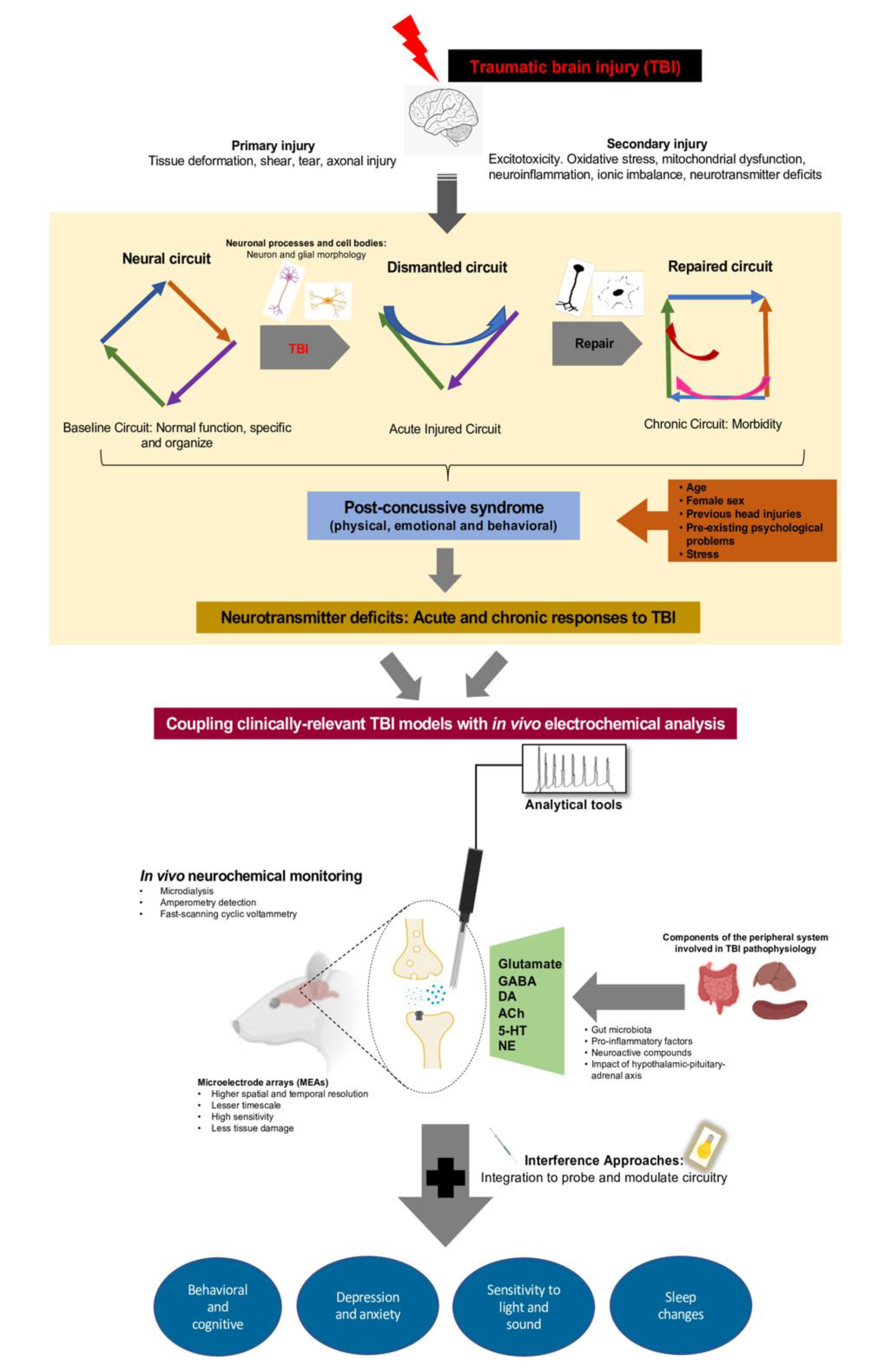{kind=link}
Abstract
1. Introduction: Acute Chronic Deficits of Mild TBI
2. Current Animal Models of TBI
3. Circuit Dysfunction after Diffuse TBI: Adaptive and Maladaptive Responses
4. TBI-Induced Changes in Neurotransmitter Signaling: Acute and Chronic Responses to TBI
4.1. Primary Neurotransmitters
4.1.1. Glutamate
4.1.2. GABA (γ-Aminobutyric Acid)
4.2. Modulatory Neurotransmitters
4.2.1. Dopamine
4.2.2. Norepinephrine
4.2.3. Acetylcholine
4.2.4. Serotonin (5-Hydroxytryptamine, 5-HT)
4.3. Conclusion
5. Biosensors for In Vivo Neurotransmitter Monitoring
6. Brain-Injured Circuitry and Behavioral Morbidities: The Whisker Barrel Circuit
7. Electrochemical Biosensors for In Vivo Monitoring of Neurochemical Signaling
8. Moving into the Future: Recordings in Freely-Moving Animals
9. Peripheral Influences on Neurotransmission and PCSs
9.1. Neuroendocrine and Neuroimmune Interactions
9.2. Sex Hormones
9.3. Gut–Brain Axis
9.4. Other Organs
10. Concluding Remarks
Funding
Conflicts of Interest
References
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The epidemiology and impact of traumatic brain injury: A brief overview. J. Head Trauma Rehabil. 2006, 21, 375–378. [Google Scholar] [CrossRef]
- Peterson, A.B.; Xu, L.; Daugherty, J.; Breiding, M.J. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths, United States, 2014. Centers for Disease Control and Prevention, Department of Health and Human Services. 2019. Available online: https://www.google.com.hk/search?newwindow=1&safe=strict&source=hp&ei=sSocXpaxAY6JoASz0ZnACA&q=Surveillance+report+of+traumatic+brain+injury-related+emergency+department+visits%2C+hospitalizations%2C+and+deaths%2C+United+States%2C+2014.+Centers+for+Disease+Control+and+Prevention%2C+Department+of+Health+and+Human+Services&oq=Surveillance+report+of+traumatic+brain+injury-related+emergency+department+visits%2C+hospitalizations%2C+and+deaths%2C+United+States%2C+2014.+Centers+for+Disease+Control+and+Prevention%2C+Department+of+Health+and+Human+Services&gs_l=psy-ab.3...403.403..1296...0.0..0.0.0.......0....2j1..gws-wiz.cFcNrTBM2Yc&ved=0ahUKEwjWza7TlIDnAhWOBIgKHbNoBogQ4dUDCAU&uact=5 (accessed on 13 January 2020).
- Zetterberg, H.; Blennow, K. Fluid biomarkers for mild traumatic brain injury and related conditions. Nat. Rev. Neurol. 2016, 12, 563–574. [Google Scholar] [CrossRef]
- Sharp, D.J.; Fleminger, S.; Powell, J. Traumatic Brain Injury; Oxford University Press: New York, NY, USA, 2016. [Google Scholar]
- Smith, K. Traumatic brain injury: CT scan does not predict outcome of mild traumatic brain injury. Nat. Rev. Neurol. 2012, 8, 474. [Google Scholar] [CrossRef]
- Povlishock, J.T.; Christman, C.W. The pathobiology of traumatically induced axonal injury in animals and humans: A review of current thoughts. J. Neurotrauma 1995, 12, 555–564. [Google Scholar] [CrossRef]
- Farkas, O.; Povlishock, J.T. Cellular and subcellular change evoked by diffuse traumatic brain injury: A complex web of change extending far beyond focal damage. Prog. Brain Res. 2007, 161, 43–59. [Google Scholar]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood–brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef]
- King, A.I. Fundamentals of impact biomechanics: Part I-biomechanics of the head, neck, and thorax. Annu. Rev. Biomed. Eng. 2000, 2, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Medana, I.M.; Esiri, M.M. Axonal damage: A key predictor of outcome in human CNS diseases. Brain 2003, 126, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.L.; Jenkins, L.W.; Lyeth, B.G. Neurotransmitter-mediated mechanisms of traumatic brain injury: Acetylcholine and excitatory amino acids. J. Neurotrauma 1992, 9, S173–S187. [Google Scholar] [PubMed]
- Giza, C.C.; Hovda, D.A. The neurometabolic cascade of concussion. J. Athl. Train. 2001, 36, 228. [Google Scholar] [CrossRef]
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75, S24–S33. [Google Scholar] [CrossRef]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Primers 2016, 2, 16084. [Google Scholar] [CrossRef]
- Langlois, J.A.; Rutland-Brown, W.; Thomas, K.E. Traumatic brain injury in the United States; emergency department visits, hospitalizations, and deaths: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. 2006. Available online: https://stacks.cdc.gov/view/cdc/5571 (accessed on 13 January 2020).
- Centre Task Force on Mild Traumatic Brain Injury. Methodological issues and research recommendations for mild traumatic brain injury: The WHO collaborating centre task force on mild traumatic brain injury. J. Rehabil. Med. 2004, 43, 113–125. [Google Scholar]
- Donovan, J.; Cancelliere, C.; Cassidy, J.D. Summary of the findings of the International collaboration on mild traumatic brain injury prognosis. Chiropr. Man. Ther. 2014, 22, 38. [Google Scholar] [CrossRef]
- Harmon, K.G.; Drezner, J.; Gammons, M.; Guskiewicz, K.; Halstead, M.; Herring, S.; Kutcher, J.; Pana, A.; Putukian, M.; Roberts, W. American Medical Society for Sports Medicine position statement: Concussion in sport. Clin. J. Sport Med. 2013, 23, 1–18. [Google Scholar] [CrossRef]
- Hall, K.D.; Lifshitz, J. Diffuse traumatic brain injury initially attenuates and later expands activation of the rat somatosensory whisker circuit concomitant with neuroplastic responses. Brain Res. 2010, 1323, 161–173. [Google Scholar] [CrossRef]
- Hou, R.; Moss-Morris, R.; Peveler, R.; Mogg, K.; Bradley, B.P.; Belli, A. When a minor head injury results in enduring symptoms: A prospective investigation of risk factors for postconcussional syndrome after mild traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2012, 83, 217–223. [Google Scholar] [CrossRef]
- King, N.S.; Kirwilliam, S. Permanent post-concussion symptoms after mild head injury. Brain Inj. 2011, 25, 462–470. [Google Scholar] [CrossRef]
- Binder, L.M. Persisting symptoms after mild head injury: A review of the postconcussive syndrome. J. Clin. Exp. Neuropsychol. 1986, 8, 323–346. [Google Scholar] [CrossRef]
- Polinder, S.; Cnossen, M.C.; Real, R.G.; Covic, A.; Gorbunova, A.; Voormolen, D.C.; Master, C.L.; Haagsma, J.; Diaz-Arrastia, R.; van Steinbuechel, N. A multidimensional approach to post-concussion symptoms in mild traumatic brain injury: A focused review. Front. Neurol. 2018, 9, 1113. [Google Scholar] [CrossRef]
- Selassie, A.W.; Zaloshnja, E.; Langlois, J.A.; Miller, T.; Jones, P.; Steiner, C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. J. Head Trauma Rehabil. 2008, 23, 123–131. [Google Scholar] [CrossRef]
- Chauhan, N.B. Chronic neurodegenerative consequences of traumatic brain injury. Restor. Neurol. Neurosci. 2014, 32, 337–365. [Google Scholar] [CrossRef]
- Silverberg, N.D.; Iverson, G.L. Etiology of the post-concussion syndrome: Physiogenesis and psychogenesis revisited. NeuroRehabilitation 2011, 29, 317–329. [Google Scholar] [CrossRef]
- Thomas, T.C.; Colburn, T.A.; Korp, K.; Khodadad, A.; Lifshitz, J. Translational considerations for behavioral impairment and rehabilitation strategies after diffuse traumatic brain injury. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; CRC Press: Oxfordshire, UK; Taylor & Francis: Oxfordshire, UK, 2015. [Google Scholar]
- Patricia, B.; O’Neil, D.A.; LaPorte, M.J.; Cheng, J.P.; Beitchman, J.A.; Thomas, T.C.; Bondi, C.O.; Kline, A.E. Elucidating opportunities and pitfalls in the treatment of experimental traumatic brain injury to optimize and facilitate clinical translation. Neurosci. Biobehav. Rev. 2018, 85, 160–175. [Google Scholar]
- Barkhoudarian, G.; Hovda, D.A.; Giza, C.C. The molecular pathophysiology of concussive brain injury. Clin. Sports Med. 2011, 30, 33–48. [Google Scholar] [CrossRef]
- Bargmann, C.I. Beyond the connectome: How neuromodulators shape neural circuits. Bioessays 2012, 34, 458–465. [Google Scholar] [CrossRef]
- Rowe, R.K.; Harrison, J.L.; Ellis, T.W.; Adelson, P.D.; Lifshitz, J. Midline (central) fluid percussion model of traumatic brain injury in pediatric and adolescent rats. J. Neurosurg. Pediatr. 2018, 1, 1–9. [Google Scholar] [CrossRef]
- Lifshitz, J. Fluid percussion injury model. In Animal Models of Acute Neurological Injuries; Springer: Berlin, Germany, 2009; pp. 369–384. [Google Scholar]
- Spain, A.; Daumas, S.; Lifshitz, J.; Rhodes, J.; Andrews, P.J.; Horsburgh, K.; Fowler, J.H. Mild fluid percussion injury in mice produces evolving selective axonal pathology and cognitive deficits relevant to human brain injury. J. Neurotrauma 2010, 27, 1429–1438. [Google Scholar] [CrossRef]
- Lifshitz, J.; Rowe, R.K.; Griffiths, D.R.; Evilsizor, M.N.; Thomas, T.C.; Adelson, P.D.; McIntosh, T.K. Clinical relevance of midline fluid percussion brain injury: Acute deficits, chronic morbidities and the utility of biomarkers. Brain Inj. 2016, 30, 1293–1301. [Google Scholar] [CrossRef]
- Kilbourne, M.; Kuehn, R.; Tosun, C.; Caridi, J.; Keledjian, K.; Bochicchio, G.; Scalea, T.; Gerzanich, V.; Simard, J.M. Novel model of frontal impact closed head injury in the rat. J. Neurotrauma 2009, 26, 2233–2243. [Google Scholar] [CrossRef]
- Risling, M.; Davidsson, J. Experimental animal models for studies on the mechanisms of blast-induced neurotrauma. Front. Neurol. 2012, 3, 30. [Google Scholar] [CrossRef]
- Cernak, I.; Noble-Haeusslein, L.J. Traumatic brain injury: An overview of pathobiology with emphasis on military populations. J. Cereb. Blood Flow Metab. 2010, 30, 255–266. [Google Scholar] [CrossRef]
- Mattiasson, G.J.; Philips, M.F.; Tomasevic, G.; Johansson, B.B.; Wieloch, T.; McIntosh, T.K. The rotating pole test: Evaluation of its effectiveness in assessing functional motor deficits following experimental head injury in the rat. J. Neurosci. Methods 2000, 95, 75–82. [Google Scholar] [CrossRef]
- Beitchman, J.A.; Griffiths, D.R.; Hur, Y.; Ogle, S.B.; Hair, C.E.; Morrison, H.W.; Lifshitz, J.; Adelson, P.D.; Thomas, T.C. Experimental traumatic brain injury induces chronic glutamatergic dysfunction in amygdala circuitry known to regulate anxiety-like behavior. Front. Neurosci. 2019. [Google Scholar] [CrossRef]
- Herrera, J.J.; Bockhorst, K.; Kondraganti, S.; Stertz, L.; Quevedo, J.; Narayana, P.A. Acute white matter tract damage after frontal mild traumatic brain injury. J. Neurotrauma 2017, 34, 291–299. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar]
- Learoyd, A.E.; Lifshitz, J. Comparison of rat sensory behavioral tasks to detect somatosensory morbidity after diffuse brain-injury. Behav. Brain Res. 2012, 226, 197–204. [Google Scholar] [CrossRef]
- Thomas, T.C.; Hinzman, J.M.; Gerhardt, G.A.; Lifshitz, J. Hypersensitive glutamate signaling correlates with the development of late-onset behavioral morbidity in diffuse brain-injured circuitry. J. Neurotrauma 2012, 29, 187–200. [Google Scholar] [CrossRef]
- Krishna, G.; Ying, Z.; Gomez-Pinilla, F. Blueberry supplementation mitigates altered brain plasticity and behaviour after traumatic brain injury in rats. Mol. Nutr. Food Res. 2019, 63, 1801055. [Google Scholar] [CrossRef]
- Cramer, S.C.; Sur, M.; Dobkin, B.H.; O’brien, C.; Sanger, T.D.; Trojanowski, J.Q.; Rumsey, J.M.; Hicks, R.; Cameron, J.; Chen, D. Harnessing neuroplasticity for clinical applications. Brain 2011, 134, 1591–1609. [Google Scholar] [CrossRef]
- Wieloch, T.; Nikolich, K. Mechanisms of neural plasticity following brain injury. Curr. Opin. Neurobiol. 2006, 16, 258–264. [Google Scholar] [CrossRef]
- Povlishock, J.T.; Katz, D.I. Update of neuropathology and neurological recovery after traumatic brain injury. J. Head Trauma Rehabil. 2005, 20, 76–94. [Google Scholar] [CrossRef]
- Cheng, H.W.; Rafols, J.A.; Goshgarian, H.G.; Anavi, Y.; Tong, J.; McNeill, T.H. Differential spine loss and regrowth of striatal neurons following multiple forms of deafferentation: A Golgi study. Exp. Neurol. 1997, 147, 287–298. [Google Scholar] [CrossRef]
- Xing, C.; Hayakawa, K.; Lok, J.; Arai, K.; Lo, E.H. Injury and repair in the neurovascular unit. Neurol. Res. 2012, 34, 325–330. [Google Scholar] [CrossRef]
- Emery, D.L.; Royo, N.C.; Fischer, I.; Saatman, K.E.; McIntosh, T.K. Plasticity following injury to the adult central nervous system: Is recapitulation of a developmental state worth promoting? J. Neurotrauma 2003, 20, 1271–1292. [Google Scholar] [CrossRef]
- Giza, C.C.; Prins, M.L. Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev. Neurosci. 2006, 28, 364–379. [Google Scholar] [CrossRef]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Schmidt, R.H.; Grady, M.S. Regional patterns of blood–brain barrier breakdown following central and lateral fluid percussion injury in rodents. J. Neurotrauma 1993, 10, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.D.; Thomas, T.C.; Lass, E.; Ai, J.; Wan, H.; Lifshitz, J.; Baker, A.J.; Macdonald, R.L. Platelet-mediated changes to neuronal glutamate receptor expression at sites of microthrombosis following experimental subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Andrus, P.K.; Yonkers, P.A.; Smith, S.L.; Zhang, J.R.; Taylor, B.M.; Sun, F.F. Generation and detection of hydroxyl radical following experimental head injury. Ann. N. Y. Acad. Sci. 1994, 738, 15–24. [Google Scholar] [CrossRef]
- Hillered, L.; Persson, L.; Carlson, H.; Ungerstedt, U.; Ronne-Engström, E.; Nilsson, P. Studies on excitatory amino acid receptor-linked brain disorders in rat and man using in vivo microdialysis. Clin. Neuropharmacol. 1992, 15, 695A–696A. [Google Scholar] [CrossRef]
- Vespa, P.; Prins, M.; Ronne-Engstrom, E.; Caron, M.; Shalmon, E.; Hovda, D.A.; Martin, N.A.; Becker, D.P. Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: A microdialysis study. J. Neurosurg. 1998, 89, 971–982. [Google Scholar] [CrossRef]
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. 2010, 113, 564–570. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.; Algamal, M.; Leary, P.; Lynch, C.; Abdullah, L.; Evans, J.; Mullan, M.; Bachmeier, C.; Stewart, W. Chronic repetitive mild traumatic brain injury results in reduced cerebral blood flow, axonal injury, gliosis, and increased t-tau and tau oligomers. J. Neuropathol. Exp. Neurol. 2016, 75, 636–655. [Google Scholar] [CrossRef]
- Katayama, Y.; Becker, D.P.; Tamura, T.; Hovda, D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 1990, 73, 889–900. [Google Scholar] [CrossRef]
- Levenson, J.; Weeber, E.; Selcher, J.C.; Kategaya, L.S.; Sweatt, J.D.; Eskin, A. Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat. Neurosci. 2002, 5, 155. [Google Scholar] [CrossRef]
- Zipfel, G.J.; Babcock, D.J.; Lee, J.M.; Choi, D.W. Neuronal apoptosis after CNS injury: The roles of glutamate and calcium. J. Neurotrauma 2000, 17, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA A receptors. Nat. Rev. Neurosci. 2005, 6, 215. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra Rao, V.L.; Dhodda, V.K.; Song, G.; Bowen, K.K.; Dempsey, R.J. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J. Neurosci. Res. 2003, 71, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kang, I.; Jeong, H.; Park, S.; Hong, H.; Kim, J.; Kim, J.; Edden, R.; Lyoo, I.K.; Yoon, S. Low prefrontal GABA levels are associated with poor cognitive functions in professional boxers. Front. Hum. Neurosci. 2019, 13, 193. [Google Scholar] [CrossRef]
- Yasen, A.L.; Smith, J.; Christie, A.D. Glutamate and GABA concentrations following mild traumatic brain injury: A pilot study. J. Neurophysiol. 2018, 120, 1318–1322. [Google Scholar] [CrossRef]
- De Beaumont, L.; Tremblay, S.; Poirier, J.; Lassonde, M.; Théoret, H. Altered bidirectional plasticity and reduced implicit motor learning in concussed athletes. Cereb. Cortex 2011, 22, 112–121. [Google Scholar] [CrossRef]
- De Beaumont, L.; Lassonde, M.; Leclerc, S.; Théoret, H. Long-term and cumulative effects of sports concussion on motor cortex inhibition. Neurosurgery 2007, 61, 329–337. [Google Scholar] [CrossRef]
- Hossain, I.; Tan, C.; Dutta, G.; Siddiqui, S.; Iasemidis, L.D.; Arumugam, P.U. A novel electrochemical microbiosensor microarray for real-time continuous in vitro monitoring of gamma-aminobutyric acid. Front. Neurosci. 2018, 12, 500. [Google Scholar] [CrossRef]
- Burmeister, J.J.; Price, D.A.; Pomerleau, F.; Huettl, P.; Quintero, J.E.; Gerhardt, G.A. Challenges of simultaneous measurements of brain extracellular GABA and glutamate in vivo using enzyme-coated microelectrode arrays. J. Neurosci. Methods 2020, 329, 108435. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, K.H.; King, A.I. Comparison of brain responses between frontal and lateral impacts by finite element modeling. J. Neurotrauma 2001, 18, 21–30. [Google Scholar] [CrossRef]
- Horn, A.S. Dopamine uptake: A review of progress in the last decade. Prog. Neurobiol. 1990, 34, 387–400. [Google Scholar] [CrossRef]
- Kraus, M.F.; Smith, G.S.; Butters, M.; Donnell, A.J.; Dixon, E.; Yilong, C.; Marion, D. Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: A study using positron emission tomography (PET). Brain Inj. 2005, 19, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.K.; Scanion, J.M.; Becker, C.R.; Ritter, A.C.; Niyonkuru, C.; Dixon, C.E.; Conley, Y.P.; Price, J.C. The influence of genetic variants on striatal dopamine transporter and D2 receptor binding after TBI. J. Cereb. Blood Flow Metab. 2014, 34, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Donnemiller, E.; Brenneis, C.; Wissel, J.; Scherfler, C.; Poewe, W.; Riccabona, G.; Wenning, G.K. Impaired dopaminergic neurotransmission in patients with traumatic brain injury: A SPET study using 123 I-β-CIT and 123 I-IBZM. Eur. J. Nucl. Med. 2000, 27, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G. Enzymatic oxidation of dopamine: The role of prostaglandin H synthase. J. Neurochem. 1995, 64, 919–924. [Google Scholar] [CrossRef]
- Jenkins, P.O.; De Simoni, S.; Bourke, N.J.; Fleminger, J.; Scott, G.; Towey, D.J.; Svensson, W.; Khan, S.; Patel, M.; Greenwood, R. Dopaminergic abnormalities following traumatic brain injury. Brain 2018, 141, 797–810. [Google Scholar] [CrossRef]
- Van Bregt, D.R.; Thomas, T.C.; Hinzman, J.M.; Cao, T.; Liu, M.; Bing, G.; Gerhardt, G.A.; Pauly, J.R.; Lifshitz, J. Substantia nigra vulnerability after a single moderate diffuse brain injury in the rat. Exp. Neurol. 2012, 234, 8–19. [Google Scholar] [CrossRef]
- Reeves, T.M.; Phillips, L.L.; Povlishock, J.T. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp. Neurol. 2005, 196, 126–137. [Google Scholar] [CrossRef]
- Massucci, J.L.; Kline, A.E.; Ma, X.; Zafonte, R.D.; Dixon, C.E. Time dependent alterations in dopamine tissue levels and metabolism after experimental traumatic brain injury in rats. Neurosci. Lett. 2004, 372, 127–131. [Google Scholar] [CrossRef]
- Kobori, N.; Clifton, G.L.; Dash, P.K. Enhanced catecholamine synthesis in the prefrontal cortex after traumatic brain injury: Implications for prefrontal dysfunction. J. Neurotrauma 2006, 23, 1094–1102. [Google Scholar] [CrossRef]
- Wagner, A.K.; Sokoloski, J.E.; Ren, D.; Chen, X.; Khan, A.S.; Zafonte, R.D.; Michael, A.C.; Dixon, C.E. Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. J. Neurochem. 2005, 95, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.K.; Drewencki, L.L.; Chen, X.; Santos, F.R.; Khan, A.S.; Harun, R.; Torres, G.E.; Michael, A.C.; Dixon, C.E. Chronic methylphenidate treatment enhances striatal dopamine neurotransmission after experimental traumatic brain injury. J. Neurochem. 2009, 108, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bachstetter, A.D.; Cass, W.A.; Lifshitz, J.; Bing, G. Pioglitazone attenuates neuroinflammation and promotes dopaminergic neuronal survival in the nigrostriatal system of rats after diffuse brain injury. J. Neurotrauma 2017, 34, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.P.; Noda, Y.; Nabeshima, T. Involvement of activation of dopamineraic neuronal system in learning and memory deficits associated with experimental mild traumatic brain injury. Eur. J. Neurosci. 1997, 9, 1720–1727. [Google Scholar] [CrossRef]
- Arciniegas, D.B.; Held, K.; Wagner, P. Cognitive impairment following traumatic brain injury. Curr. Treat. Options Neurol. 2002, 4, 43–57. [Google Scholar] [CrossRef]
- Bales, J.W.; Wagner, A.K.; Kline, A.E.; Dixon, C.E. Persistent cognitive dysfunction after traumatic brain injury: A dopamine hypothesis. Neurosci. Biobehav. Rev. 2009, 33, 981–1003. [Google Scholar] [CrossRef]
- Cools, R.; D’Esposito, M. Inverted-U–shaped dopamine actions on human working memory and cognitive control. Biol. Psychiatry 2011, 69, e113–e125. [Google Scholar] [CrossRef]
- Boyeson, M.G.; Feeney, D.M. Intraventricular norepinephrine facilitates motor recovery following sensorimotor cortex injury. Pharmacol. Biochem. Behav. 1990, 35, 497–501. [Google Scholar] [CrossRef]
- Goldstein, L.B.; Coviello, A.; Miller, G.D.; Davis, J.N. Norepinephrine depletion impairs motor recovery following sensorimotor cortex injury in the rat. Restor. Neurol. Neurosci. 1991, 3, 41–47. [Google Scholar] [CrossRef]
- McIntosh, T.K.; Yu, T.; Gennarelli, T.A. Alterations in regional brain catecholamine concentrations after experimental brain injury in the rat. J. Neurochem. 1994, 63, 1426–1433. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.; Shijun, P.; Levin, B.E. Focal traumatic brain injury causes widespread reductions in rat brain norepinephrine turnover from 6 to 24 h. Brain Res. 1994, 660, 88–95. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Hassanain, M.; Levin, B.E. Norepinephrine and traumatic brain injury: A possible role in post-traumatic edema. Brain Res. 1998, 800, 245–252. [Google Scholar] [CrossRef]
- Levin, B.E.; Brown, K.L.; Pawar, G.; Dunn-Meynell, A. Widespread and lateralized effects of acute traumatic brain injury on norepinephrine turnover in the rat brain. Brain Res. 1995, 674, 307–313. [Google Scholar] [CrossRef]
- Fujinaka, T.; Kohmura, E.; Yuguchi, T.; Yoshimine, T. The morphological and neurochemical effects of diffuse brain injury on rat central noradrenergic system. Neurol. Res. 2003, 25, 35–41. [Google Scholar] [CrossRef]
- Ramos, B.P.; Arnsten, A.F. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef]
- Arnsten, A.F.; Li, B.M. Neurobiology of executive functions: Catecholamine influences on prefrontal cortical functions. Biol. Psychiatry 2005, 57, 1377–1384. [Google Scholar] [CrossRef]
- Bonnelle, V.; Leech, R.; Kinnunen, K.M.; Ham, T.E.; Beckmann, C.F.; De Boissezon, X.; Greenwood, R.J.; Sharp, D.J. Default mode network connectivity predicts sustained attention deficits after traumatic brain injury. J. Neurosci. 2011, 31, 13442–13451. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Arciniegas, D.; Olincy, A.; Topkoff, J.; McRae, K.; Cawthra, E.; Filley, C.M.; Reite, M.; Adler, L.E. Impaired auditory gating and P50 nonsuppression following traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 2000, 12, 77–85. [Google Scholar] [CrossRef]
- Tower, D.B.; McEachern, D. Acetylcholine and neuronal activity in craniocerebral trauma. J. Clin. Investig. 1948, 27, 558. [Google Scholar]
- Tower, D.B.; McEachern, D. Acetylcholine and neuronal activity: I. Cholinesterase patterns and acetylcholine in the cerebrospinal fluids of patients with craniocerebral trauma. Can. J. Res. 1949, 27, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, D.B. Cholinergic dysfunction and cognitive impairment after traumatic brain injury. Part 2: Evidence from basic and clinical investigations. J. Head Trauma Rehabil. 2011, 26, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Sachs, E. Acetylcholine and serotonin in the spinal fluid. J. Neurosurg. 1957, 14, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Lyeth, B.G.; Jiang, J.Y.; Robinson, S.E.; Guo, H.; Jenkins, L.W. Hypothermia blunts acetylcholine increase in CSF of traumatically brain injured rats. Mol. Chem. Neuropathol. 1993, 18, 247–256. [Google Scholar] [CrossRef]
- Dewar, D.; Graham, D.I. Depletion of choline acetyltransferase activity but preservation of Ml and M2 muscarinic receptor binding sites in temporal cortex following head injury: A preliminary human postmortem study. J. Neurotrauma 1996, 13, 181–187. [Google Scholar] [CrossRef]
- Salmond, C.H.; Chatfield, D.A.; Menon, D.K.; Pickard, J.D.; Sahakian, B.J. Cognitive sequelae of head injury: Involvement of basal forebrain and associated structures. Brain 2004, 128, 189–200. [Google Scholar] [CrossRef]
- Zhang, L.; Plotkin, R.C.; Wang, G.; Sandel, M.E.; Lee, S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch. Phys. Med. Rehabil. 2004, 85, 1050–1055. [Google Scholar] [CrossRef]
- Becker, R.E.; Kapogiannis, D.; Greig, N.H. Does traumatic brain injury hold the key to the Alzheimer’s disease puzzle? Alzheimers Dement. 2018, 14, 431–443. [Google Scholar] [CrossRef]
- H. Ferreira-Vieira, T.; M. Guimaraes, I.; R. Silva, F.; M. Ribeiro, F. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Gorman, L.K.; Fu, K.; Hovda, D.A.; Becker, D.P.; Katayama, Y. Analysis of acetylcholine release following concussive brain injury in the rat. J. Neurotrauma 1989, 6, 1–2. [Google Scholar]
- Saija, A.; Hayes, R.L.; Lyeth, B.G.; Dixon, C.E.; Yamamoto, T.; Robinson, S.E. The effect of concussive head injury on central cholinergic neurons. Brain Res. 1988, 452, 303–311. [Google Scholar] [CrossRef]
- Arciniegas, D.B. The cholinergic hypothesis of cognitive impairment caused by traumatic brain injury. Curr. Psychiatry Rep. 2003, 5, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Everitt, B.J.; Robbins, T.W. Central cholinergic systems and cognition. Annu. Rev. Psychol. 1997, 48, 649–684. [Google Scholar] [CrossRef] [PubMed]
- Gorman, L.K.; Fu, K.; Hovda, D.A.; Murray, M.; Traystman, R.J. Effects of traumatic brain injury on the cholinergic system in the rat. J. Neurotrauma 1996, 13, 457–463. [Google Scholar] [CrossRef]
- Dixon, C.E.; Bao, J.; Bergmann, J.S.; Johnson, K.M. Traumatic brain injury reduces hippocampal high-affinity [3H] choline uptake but not extracellular choline levels in rats. Neurosci. Lett. 1994, 180, 127–130. [Google Scholar] [CrossRef]
- Dixon, C.E.; Bao, J.; Long, D.A.; Hayes, R.L. Reduced evoked release of acetylcholine in the rodent hippocampus following traumatic brain injury. Pharmacol. Biochem. Behav. 1996, 53, 679–686. [Google Scholar] [CrossRef]
- Dixon, C.E.; Ma, X.; Marion, D.W. Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury. Brain Res. 1997, 749, 127–130. [Google Scholar] [CrossRef]
- Dixon, C.E.; Bao, J.; Johnson, K.M.; Yang, K.; Whitson, J.; Clifton, G.L.; Hayes, R.L. Basal and scopolamine-evoked release of hippocampal acetylcholine following traumatic brain injury in rats. Neurosci. Lett. 1995, 198, 111–114. [Google Scholar] [CrossRef]
- Leonard, J.R.; Maris, D.O.; Grady, M.S. Fluid percussion injury causes loss of forebrain choline acetyltransferase and nerve growth factor receptor immunoreactive cells in the rat. J. Neurotrauma 1994, 11, 379–392. [Google Scholar] [CrossRef]
- Ciallella, J.R.; Yan, H.Q.; Ma, X.; Wolfson, B.M.; Marion, D.W.; DeKosky, S.T.; Dixon, C.E. Chronic effects of traumatic brain injury on hippocampal vesicular acetylcholine transporter and M2 muscarinic receptor protein in rats. Exp. Neurol. 1998, 152, 11–19. [Google Scholar] [CrossRef]
- Dixon, C.E.; Kochanek, P.M.; Yan, H.Q.; Schiding, J.K.; Griffith, R.G.; Baum, E.; Marion, D.W.; DeKosky, S.T. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J. Neurotrauma 1999, 16, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Ruhé, H.G.; Mason, N.S.; Schene, A.H. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: A meta-analysis of monoamine depletion studies. Mol. Psychiatry 2007, 12, 331. [Google Scholar] [CrossRef] [PubMed]
- Busto, R.; Dietrich, W.D.; Globus, M.Y.T.; Alonso, O.; Ginsberg, M.D. Extracellular release of serotonin following fluid-percussion brain injury in rats. J. Neurotrauma 1997, 14, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Shimada, R.; Okada, Y.; Kibayashi, K. Traumatic brain injury decreases serotonin transporter expression in the rat cerebrum. Neurol. Res. 2016, 38, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rasmussen, L.; Saraswati, M.; Koehler, R.C.; Robertson, C.; Kannan, S. Traumatic injury leads to inflammation and altered tryptophan metabolism in the juvenile rabbit brain. J. Neurotrauma 2018, 36, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Kreutzer, J.S.; Seel, R.T.; Gourley, E. The prevalence and symptom rates of depression after traumatic brain injury: A comprehensive examination. Brain Inj. 2001, 15, 563–576. [Google Scholar] [CrossRef]
- Yue, J.; Burke, J.; Upadhyayula, P.; Winkler, E.; Deng, H.; Robinson, C.; Pirracchio, R.; Suen, C.; Sharma, S.; Ferguson, A. Selective serotonin reuptake inhibitors for treating neurocognitive and neuropsychiatric disorders following traumatic brain injury: An evaluation of current evidence. Brain Sci. 2017, 7, 93. [Google Scholar] [CrossRef]
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Schroeder, L.F.; Mank, M.; Muller, A.; Taylor, P.; Griesbeck, O.; Kleinfeld, D. An in vivo biosensor for neurotransmitter release and in situ receptor activity. Nat. Neurosci. 2010, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Venton, B.J.; Kennedy, R.T. In vivo measurements of neurotransmitters by microdialysis sampling. Anal. Chem. 2006, 78, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Heien, M.L.; Johnson, M.A.; Wightman, R.M. Resolving neurotransmitters detected by fast-scan cyclic voltammetry. Anal. Chem. 2004, 76, 5697–5704. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Michael, A.C. Invasive consequences of using micro-electrodes and microdialysis probes in the brain. TrAC Trends Anal. Chem. 2003, 22, 503–508. [Google Scholar] [CrossRef]
- Hascup, K.N.; Hascup, E.R.; Littrell, O.M.; Hinzman, J.M.; Werner, C.E.; Davis, V.A.; Burmeister, J.J.; Pomerleau, F.; Quintero, J.E.; Huettl, P. Microelectrode array fabrication and optimization for selective neurochemical detection. In Microelectrode Biosensors; Springer: Berlin, Germany, 2013; pp. 27–54. [Google Scholar]
- Wall, M.J.; Usowicz, M.M. Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur. J. Neurosci. 1997, 9, 533–548. [Google Scholar] [CrossRef]
- Kullmann, D.M.; Asztely, F. Extrasynaptic glutamate spillover in the hippocampus: Evidence and implications. Trends Neurosci. 1998, 21, 8–14. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Diao, L. Regulation of extracellular adenosine in rat hippocampal slices is temperature dependent: Role of adenosine transporters. Neuroscience 1999, 95, 81–88. [Google Scholar] [CrossRef]
- Diamond, M.E.; Arabzadeh, E. Whisker sensory system–from receptor to decision. Prog. Neurobiol. 2013, 103, 28–40. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-term consequences of traumatic brain injury: Current status of potential mechanisms of injury and neurological outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Simons, D.J.; Carvell, G.E. Thalamocortical response transformation in the rat vibrissa/barrel system. J. Neurophysiol. 1989, 61, 311–330. [Google Scholar] [CrossRef]
- Urbain, N.; Deschênes, M. Motor cortex gates vibrissal responses in a thalamocortical projection pathway. Neuron 2007, 56, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.D.; Lindholm, E.P. Organization of motor and somatosensory neocortex in the albino rat. Brain Res. 1974, 66, 23–38. [Google Scholar] [CrossRef]
- Welker, C. Receptive fields of barrels in the somatosensory neocortex of the rat. J. Comp. Neurol. 1976, 166, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alamancos, M.A.; Connors, B.W. Thalamocortical synapses. Prog. Neurobiol. 1997, 51, 581–606. [Google Scholar] [CrossRef]
- Lo, F.S.; Erzurumlu, R.S. Neonatal sensory nerve injury-induced synaptic plasticity in the trigeminal principal sensory nucleus. Exp. Neurol. 2016, 275, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Martini, F.J.; Moreno-Juan, V.; Filipchuk, A.; Valdeolmillos, M.; Lopez-Bendito, G. Impact of thalamocortical input on barrel cortex development. Neuroscience 2018, 368, 246–255. [Google Scholar] [CrossRef]
- Sofroniew, N.J.; Vlasov, Y.A.; Hires, S.A.; Freeman, J.; Svoboda, K. Neural coding in barrel cortex during whisker-guided locomotion. eLife 2015, 4, e12559. [Google Scholar] [CrossRef]
- McNamara, K.C.; Lisembee, A.M.; Lifshitz, J. The whisker nuisance task identifies a late-onset, persistent sensory sensitivity in diffuse brain-injured rats. J. Neurotrauma 2010, 27, 695–706. [Google Scholar] [CrossRef]
- Lifshitz, J.; Lisembee, A.M. Neurodegeneration in the somatosensory cortex after experimental diffuse brain injury. Brain Struct. Funct. 2012, 217, 49–61. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; Krahe, T.E.; Povlishock, J.T. Moderately elevated intracranial pressure after diffuse traumatic brain injury is associated with exacerbated neuronal pathology and behavioral morbidity in the rat. J. Cereb. Blood Flow Metab. 2014, 34, 1628–1636. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; McGinn, M.J.; Povlishock, J.T. Increased intracranial pressure after diffuse traumatic brain injury exacerbates neuronal somatic membrane poration but not axonal injury: Evidence for primary intracranial pressure-induced neuronal perturbation. J. Cereb. Blood Flow Metab. 2012, 32, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Alwis, D.S.; Yan, E.B.; Morganti-Kossmann, M.C.; Rajan, R. Sensory cortex underpinnings of traumatic brain injury deficits. PLoS ONE 2012, 7, e52169. [Google Scholar] [CrossRef] [PubMed]
- Bhalala, O.G.; Rahman, M.M.; Sun, L. Whisker Nuisance Test: A Valuable Tool to Assess Tactile Hypersensitivity in Mice. Available online: https://bio-protocol.org/e3331 (accessed on 13 January 2020).
- Bohnen, N.; Twijnstra, A.; Wijnen, G.; Jolles, J. Tolerance for light and sound of patients with persistent post-concussional symptoms 6 months after mild head injury. J. Neurol. 1991, 238, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Stern, C.D. Photophobia, light, and color in acquired brain injury. Multidiscip. Care Patient Brain Inj. 2016, 283. [Google Scholar]
- Hinzman, J.M.; Thomas, T.C.; Burmeister, J.J.; Quintero, J.E.; Huettl, P.; Pomerleau, F.; Gerhardt, G.A.; Lifshitz, J. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: An enzyme-based microelectrode array study. J. Neurotrauma 2010, 27, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Hinzman, J.M.; Thomas, T.C.; Quintero, J.E.; Gerhardt, G.A.; Lifshitz, J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. J. Neurotrauma 2012, 29, 1197–1208. [Google Scholar] [CrossRef]
- Rutherford, E.C.; Pomerleau, F.; Huettl, P.; Strömberg, I.; Gerhardt, G.A. Chronic second-by-second measures of l-glutamate in the central nervous system of freely moving rats. J. Neurochem. 2007, 102, 712–722. [Google Scholar] [CrossRef]
- Hogan, B.L.; Lunte, S.M.; Stobaugh, J.F.; Lunte, C.E. Online coupling of in vivo microdialysis sampling with capillary electrophoresis. Anal. Chem. 1994, 66, 596–602. [Google Scholar] [CrossRef]
- Kennedy, R.T. Emerging trends in in vivo neurochemical monitoring by microdialysis. Curr. Opin. Chem. Biol. 2013, 17, 860–867. [Google Scholar] [CrossRef]
- Hascup, E.R.; Hascup, K.N.; Stephens, M.; Pomerleau, F.; Huettl, P.; Gratton, A.; Gerhardt, G.A. Rapid microelectrode measurements and the origin and regulation of extracellular glutamate in rat prefrontal cortex. J. Neurochem. 2010, 115, 1608–1620. [Google Scholar] [CrossRef]
- Chefer, V.I.; Thompson, A.C.; Zapata, A.; Shippenberg, T.S. Overview of brain microdialysis. Curr. Protoc. Neurosci. 2009, 47, 7.1.1–7.1.28. [Google Scholar] [CrossRef]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.-W.; Bargmann, C.I. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; LeDue, J.; Ko, R.W.Y.; Mahler, J.; Murphy, T.H.; MacVicar, B.A. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat. Commun. 2016, 7, 13441. [Google Scholar] [CrossRef] [PubMed]
- Bosche, B.; Dohmen, C.; Graf, R.; Neveling, M.; Staub, F.; Kracht, L.; Sobesky, J.; Lehnhardt, F.G.; Heiss, W.D. Extracellular concentrations of non–transmitter amino acids in peri-infarct tissue of patients predict malignant middle cerebral artery infarction. Stroke 2003, 34, 2908–2913. [Google Scholar] [CrossRef]
- Burmeister, J.J.; Moxon, K.; Gerhardt, G.A. Ceramic-based multisite microelectrodes for electrochemical recordings. Anal. Chem. 2000, 72, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Peters, J.L.; Michael, A.C. Coupled effects of mass transfer and uptake kinetics on in vivo microdialysis of dopamine. J. Neurochem. 1998, 71, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Weltin, A.; Kieninger, J.; Urban, G.A. Microfabricated, amperometric, enzyme-based biosensors for in vivo applications. Anal. Bioanal. Chem. 2016, 408, 4503–4521. [Google Scholar] [CrossRef]
- Burmeister, J.J.; Pomerleau, F.; Palmer, M.; Day, B.K.; Huettl, P.; Gerhardt, G.A. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J. Neurosci. Methods 2002, 119, 163–171. [Google Scholar] [CrossRef]
- Yu, P.; He, X.; Zhang, L.; Mao, L. Dual recognition unit strategy improves the specificity of the adenosine triphosphate (ATP) aptamer biosensor for cerebral ATP assay. Anal. Chem. 2014, 87, 1373–1380. [Google Scholar] [CrossRef]
- Ledo, A.; Lourenço, C.F.; Laranjinha, J.; Brett, C.M.; Gerhardt, G.A.; Barbosa, R.M. Ceramic-based multisite platinum microelectrode arrays: Morphological characteristics and electrochemical performance for extracellular oxygen measurements in brain tissue. Anal. Chem. 2017, 89, 1674–1683. [Google Scholar] [CrossRef]
- Hascup, E.R.; af Bjerkén, S.; Hascup, K.N.; Pomerleau, F.; Huettl, P.; Strömberg, I.; Gerhardt, G.A. Histological studies of the effects of chronic implantation of ceramic-based microelectrode arrays and microdialysis probes in rat prefrontal cortex. Brain Res. 2009, 1291, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Hascup, K.N.; Hascup, E.R. Electrochemical techniques for subsecond neurotransmitter detection in live rodents. Comp. Med. 2014, 64, 249–255. [Google Scholar] [PubMed]
- Hinzman, J.M.; Gibson, J.L.; Tackla, R.D.; Costello, M.S.; Burmeister, J.J.; Quintero, J.E.; Gerhardt, G.A.; Hartings, J.A. Real-time monitoring of extracellular adenosine using enzyme-linked microelectrode arrays. Biosens. Bioelectron. 2015, 74, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, J.J.; Pomerleau, F.; Huettl, P.; Gash, C.R.; Werner, C.E.; Bruno, J.P.; Gerhardt, G.A. Ceramic-based multisite microelectrode arrays for simultaneous measures of choline and acetylcholine in CNS. Biosens. Bioelectron. 2008, 23, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, J.J.; Palmer, M.; Gerhardt, G.A. L-lactate measures in brain tissue with ceramic-based multisite microelectrodes. Biosens. Bioelectron. 2005, 20, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, C.F.; Ledo, A.; Gerhardt, G.A.; Laranjinha, J.; Barbosa, R.M. Neurometabolic and electrophysiological changes during cortical spreading depolarization: Multimodal approach based on a lactate-glucose dual microbiosensor arrays. Sci. Rep. 2017, 7, 6764. [Google Scholar] [CrossRef]
- Hall, S.B.; Khudaish, E.A.; Hart, A.L. Electrochemical oxidation of hydrogen peroxide at platinum electrodes. Part 1. An adsorption-controlled mechanism. Electrochim. Acta 1998, 43, 579–588. [Google Scholar] [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [CrossRef]
- Hunsberger, H.C.; Setti, S.E.; Heslin, R.T.; Quintero, J.E.; Gerhardt, G.A.; Reed, M.N. Using enzyme-based biosensors to measure tonic and phasic glutamate in Alzheimer’s mouse models. J. Vis. Exp. 2017, 123, e55418. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Davis, V.A.; Whiteheart, S.W.; Vanaman, T.C.; Gerhardt, G.A.; Slevin, J.T. Kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes correlates with enhanced glutamate release. Epilepsia 2012, 53, 157–167. [Google Scholar] [CrossRef][Green Version]
- Hascup, E.R.; Hascup, K.N.; Pomerleau, F.; Huettl, P.; Hajos-Korcsok, E.; Kehr, J.; Gerhardt, G.A. An allosteric modulator of metabotropic glutamate receptors (mGluR2),(+)-TFMPIP, inhibits restraint stress-induced phasic glutamate release in rat prefrontal cortex. J. Neurochem. 2012, 122, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.L.; Quintero, J.E.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A. Age-related changes in glutamate release in the CA3 and dentate gyrus of the rat hippocampus. Neurobiol. Aging 2011, 32, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.C.; Beitchman, J.A.; Pomerleau, F.; Noel, T.; Jungsuwadee, P.; Butterfield, D.A.; Clair, D.K.S.; Vore, M.; Gerhardt, G.A. Acute treatment with doxorubicin affects glutamate neurotransmission in the mouse frontal cortex and hippocampus. Brain Res. 2017, 1672, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hascup, K.N.; Hascup, E.R.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A. Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. J. Pharmacol. Exp. Ther. 2008, 324, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, J.; Gerhardt, G.A.; Zahniser, N.R. Acute cocaine differentially alters accumbens and striatal dopamine clearance in low and high cocaine locomotor responders: Behavioral and electrochemical recordings in freely moving rats. J. Pharmacol. Exp. Ther. 2002, 302, 1201–1211. [Google Scholar] [CrossRef]
- Wassum, K.M.; Tolosa, V.M.; Tseng, T.C.; Balleine, B.W.; Monbouquette, H.G.; Maidment, N.T. Transient extracellular glutamate events in the basolateral amygdala track reward-seeking actions. J. Neurosci. 2012, 32, 2734–2746. [Google Scholar] [CrossRef]
- Dash, M.B.; Douglas, C.L.; Vyazovskiy, V.V.; Cirelli, C.; Tononi, G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J. Neurosci. 2009, 29, 620–629. [Google Scholar] [CrossRef]
- Miller, E.M.; Quintero, J.E.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A.; Glaser, P.E. Chronic methylphenidate alters tonic and phasic glutamate signaling in the frontal cortex of a freely-moving rat model of ADHD. Neurochem. Res. 2019, 44, 89–101. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, S.C.; Nicolelis, M.A. Acquiring local field potential information from amperometric neurochemical recordings. J. Neurosci. Methods 2009, 179, 191–200. [Google Scholar] [CrossRef]
- Burmeister, J.J.; Pomerleau, F.; Quintero, J.E.; Huettl, P.; Ai, Y.; Jakobsson, J.; Lundblad, M.; Heuer, A.; Slevin, J.T.; Gerhardt, G.A. In vivo electrochemical studies of optogenetic control of glutamate signaling measured using enzyme-based ceramic microelectrode arrays. In Biochemical Approaches for Glutamatergic Neurotransmission; Springer: Berlin, Germany, 2018; pp. 327–351. [Google Scholar]
- Disney, A.A.; McKinney, C.; Grissom, L.; Lu, X.; Reynolds, J.H. A multi-site array for combined local electrochemistry and electrophysiology in the non-human primate brain. J. Neurosci. Methods 2015, 255, 29–37. [Google Scholar] [CrossRef][Green Version]
- Ledo, A.; Lourenco, C.F.; Laranjinha, J.; Gerhardt, G.A.; Barbosa, R.M. Concurrent measurements of neurochemical and electrophysiological activity with microelectrode arrays: New perspectives for constant potential amperometry. Curr. Opin. Electrochem. 2018, 12, 129–140. [Google Scholar] [CrossRef]
- Aldrin-Kirk, P.; Heuer, A.; Wang, G.; Mattsson, B.; Lundblad, M.; Parmar, M.; Björklund, T. DREADD modulation of transplanted DA neurons reveals a novel parkinsonian dyskinesia mechanism mediated by the serotonin 5-HT6 receptor. Neuron 2016, 90, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.R.; Boychuk, J.A.; Pomerleau, F.; Alcala, R.; Huettl, P.; Ai, Y.; Jakobsson, J.; Whiteheart, S.W.; Gerhardt, G.A.; Smith, B.N. Modulation of epileptogenesis: A paradigm for the integration of enzyme-based microelectrode arrays and optogenetics. Epilepsy Res. 2019, 159, 106244. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Manley, G.T. Brain tissue oxygen monitoring: Physiologic principles and clinical application. Oper. Tech. Neurosurg. 2004, 7, 2–9. [Google Scholar] [CrossRef]
- De Georgia, M.A. Brain tissue oxygen monitoring in neurocritical care. J. Intensive Care Med. 2015, 30, 473–483. [Google Scholar] [CrossRef]
- Cannestra, A.F.; Pouratian, N.; Bookheimer, S.Y.; Martin, N.A.; Becker, D.P.; Toga, A.W. Temporal spatial differences observed by functional MRI and human intraoperative optical imaging. Cereb. Cortex 2001, 11, 773–782. [Google Scholar] [CrossRef]
- Connell, E.; Krishna, G.; Hair, C.; Adelson, P.D.; Thomas, T.C. A novel approach to measuring real-time circuit dysregulation after diffuse brain injury. J. Neurotrauma. 2019, 36, A26. [Google Scholar]
- Deadwyler, S.A.; Hampson, R.E.; Song, D.; Opris, I.; Gerhardt, G.A.; Marmarelis, V.Z.; Berger, T.W. A cognitive prosthesis for memory facilitation by closed-loop functional ensemble stimulation of hippocampal neurons in primate brain. Exp. Neurol. 2017, 287, 452–460. [Google Scholar] [CrossRef]
- Stephens, M.L.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A.; Zhang, Z. Real-time glutamate measurements in the putamen of awake rhesus monkeys using an enzyme-based human microelectrode array prototype. J. Neurosci. Methods 2010, 185, 264–272. [Google Scholar] [CrossRef]
- Geyer, E.D.; Shetty, P.A.; Suozzi, C.J.; Allen, D.Z.; Benavidez, P.P.; Liu, J.; Hollis, C.N.; Gerhardt, G.A.; Quintero, J.E.; Burmeister, J.J. Adaptation of microelectrode array technology for the study of anesthesia-induced neurotoxicity in the intact piglet brain. J. Vis. Exp. 2018, 135, e57391. [Google Scholar] [CrossRef]
- Kasasbeh, A.; Lee, K.; Bieber, A.; Bennet, K.; Chang, S.Y. Wireless neurochemical monitoring in humans. Stereotact. Funct. Neurosurg. 2013, 91, 141–147. [Google Scholar] [CrossRef]
- Ferguson, M.; Sharma, D.; Ross, D.; Zhao, F. A Critical review of microelectrode arrays and strategies for improving neural interfaces. Adv. Healthc. Mater. 2019, 8, 1900558. [Google Scholar] [CrossRef]
- Klose, M.; Feldt-Rasmussen, U. Chronic endocrine consequences of traumatic brain injury—What is the evidence? Nat. Rev. Endocrinol. 2018, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Moreau, O.K.; Yollin, E.; Merlen, E.; Daveluy, W.; Rousseaux, M. Lasting pituitary hormone deficiency after traumatic brain injury. J. Neurotrauma 2012, 29, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Kreitschmann-Andermahr, I.; Ghigo, E.; Stalla, G.K.; Agha, A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. JAMA 2007, 298, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, G.S.; Hovda, D.A.; Tio, D.L.; Taylor, A.N. Heightening of the stress response during the first weeks after a mild traumatic brain injury. Neuroscience 2011, 178, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.K.; Rumney, B.M.; May, H.G.; Permana, P.; Adelson, P.D.; Harman, S.M.; Lifshitz, J.; Thomas, T.C. Diffuse traumatic brain injury affects chronic corticosterone function in the rat. Endocr. Connect. 2016, 5, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.L.; Richardson, M.R.; Bauman, B.M.; Hernandez, I.M.; Saperstein, S.; Handa, R.J.; Wu, T.J. Differential responses of the HPA axis to mild blast traumatic brain injury in male and female mice. Endocrinology 2018, 159, 2363–2375. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Rowe, R.K.; Muccigrosso, M.M.; Reddaway, J.T.; Adelson, P.D.; Godbout, J.P.; Lifshitz, J. Aging with a traumatic brain injury: Could behavioral morbidities and endocrine symptoms be influenced by microglial priming? Brain. Behav. Immun. 2017, 59, 1–7. [Google Scholar] [CrossRef]
- Frugier, T.; Morganti-Kossmann, M.C.; O’Reilly, D.; McLean, C.A. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma 2010, 27, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, K.P.; Mazure, C.M.; Staley, J.K. Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol. Psychiatry 2007, 62, 847–855. [Google Scholar] [CrossRef] [PubMed]
- De Vries, G.J. Sex differences in neurotransmitter systems. J. Neuroendocrinol. 1990, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.D.; Lieberman, A.R. Sexual dimorphism in the mammalian limbic system. Prog. Neurobiol. 1995, 45, 275–333. [Google Scholar] [CrossRef]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]
- Wunderle, M.K.; Hoeger, K.M.; Wasserman, M.E.; Bazarian, J.J. Menstrual phase as predictor of outcome after mild traumatic brain injury in women. J. Head Trauma Rehabil. 2014, 29, E1. [Google Scholar] [CrossRef]
- Yokomaku, D.; Numakawa, T.; Numakawa, Y.; Suzuki, S.; Matsumoto, T.; Adachi, N.; Nishio, C.; Taguchi, T.; Hatanaka, H. Estrogen enhances depolarization-induced glutamate release through activation of phosphatidylinositol 3-kinase and mitogen-activated protein kinase in cultured hippocampal neurons. Mol. Endocrinol. 2003, 17, 831–844. [Google Scholar] [CrossRef]
- Yankova, M.; Hart, S.A.; Woolley, C.S. Estrogen increases synaptic connectivity between single presynaptic inputs and multiple postsynaptic CA1 pyramidal cells: A serial electron-microscopic study. Proc. Natl. Acad. Sci. 2001, 98, 3525–3530. [Google Scholar] [CrossRef]
- Kao, C.H.; ChangLai, S.P.; Chieng, P.U.; Yen, T.C. Gastric emptying in head-injured patients. Am. J. Gastroenterol. 1998, 93, 1108. [Google Scholar] [CrossRef]
- Ott, L.; Young, B.; Phillips, R.; McClain, C.; Adams, L.; Dempsey, R.; Tibbs, P.; Ryo, U.Y. Altered gastric emptying in the head-injured patient: Relationship to feeding intolerance. J. Neurosurg. 1991, 74, 738–742. [Google Scholar] [CrossRef]
- Olsen, A.B.; Hetz, R.A.; Xue, H.; Aroom, K.R.; Bhattarai, D.; Johnson, E.; Bedi, S.; Cox, C.S., Jr.; Uray, K. Effects of traumatic brain injury on intestinal contractility. Neurogastroenterol. Motil. 2013, 25, 593-e463. [Google Scholar] [CrossRef] [PubMed]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus nerve as modulator of the brain–gut axis in psychiatric and inflammatory disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Neren, D.; Johnson, M.D.; Legon, W.; Bachour, S.P.; Ling, G.; Divani, A.A. Vagus nerve stimulation and other neuromodulation methods for treatment of traumatic brain injury. Neurocrit. Care 2016, 24, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Mahony, S.M. The microbiome-gut-brain axis: From bowel to behavior. Neurogastroenterol. Motil. 2011, 23, 187–192. [Google Scholar] [CrossRef]
- Treangen, T.J.; Wagner, J.; Burns, M.P.; Villapol, S. Traumatic brain injury in mice induces acute bacterial dysbiosis within the fecal microbiome. Front. Immunol. 2018, 9, 2757. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.E.; Watts, L.T.; Burmeister, D.M.; Merrill, D.; Scroggins, S.; Zou, Y.; Lai, Z.; Grandhi, R.; Lewis, A.M.; Newton, L.M. Moderate traumatic brain injury alters the gastrointestinal microbiome in a time-dependent manner. Shock 2018, 52, 240–248. [Google Scholar] [CrossRef]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255-e119. [Google Scholar] [CrossRef]
- Lyte, M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: Microbial endocrinology in the design and use of probiotics. Bioessays 2011, 33, 574–581. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701. [Google Scholar] [CrossRef]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Ozogul, F.; Kuley, E.; Ozogul, Y.; Ozogul, I. The function of lactic acid bacteria on biogenic amines production by food-borne pathogens in arginine decarboxylase broth. Food Sci. Technol. Res. 2012, 18, 795–804. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Barrett, E.; Ross, R.P.; O’toole, P.W.; Fitzgerald, G.F.; Stanton, C. γ-Aminobutyric acid production by culturable bacteria from the human intestine. J. Appl. Microbiol. 2012, 113, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Mohapatra, S.; Mohapatra, S.S. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J. Neuroinflammation 2012, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Utagawa, A.; Truettner, J.S.; Dietrich, W.D.; Bramlett, H.M. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp. Neurol. 2008, 211, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Kryndushkin, D.; Balarezo, M.G.; Campbell, A.M.; Saavedra, J.M.; Shewmaker, F.P.; Symes, A.J. Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am. J. Pathol. 2015, 185, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, Z.; Fang, Z.; Lin, W.; Wu, S.; Yang, F.; Li, Y.; Fu, H.; Gao, H.; Li, S. Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J. Neuroinflammation 2018, 15, 310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yan, T.; Li, L.; Chopp, M.; Venkat, P.; Qian, Y.; Li, R.; Wu, R.; Li, W.; Lu, M. Immune response mediates cardiac dysfunction after traumatic brain injury. J. Neurotrauma 2019, 36, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Felten, S.Y.; Olschowka, J. Noradrenergic sympathetic innervation of the spleen: II. Tyrosine hydroxylase (TH)-positive nerve terminals form synaptic like contacts on lymphocytes in the splenic white pulp. J. Neurosci. Res. 1987, 18, 37–48. [Google Scholar] [CrossRef]
- Das, M.; Leonardo, C.C.; Rangooni, S.; Pennypacker, K.R.; Mohapatra, S.; Mohapatra, S.S. Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J. Neuroinflammation 2011, 8, 148. [Google Scholar] [CrossRef]
- Straub, R.H. Complexity of the bi-directional neuroimmune junction in the spleen. Trends Pharmacol. Sci. 2004, 25, 640–646. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishna, G.; Beitchman, J.A.; Bromberg, C.E.; Currier Thomas, T. Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research. Int. J. Mol. Sci. 2020, 21, 588. https://doi.org/10.3390/ijms21020588
Krishna G, Beitchman JA, Bromberg CE, Currier Thomas T. Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research. International Journal of Molecular Sciences. 2020; 21(2):588. https://doi.org/10.3390/ijms21020588
Chicago/Turabian StyleKrishna, Gokul, Joshua A. Beitchman, Caitlin E. Bromberg, and Theresa Currier Thomas. 2020. "Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research" International Journal of Molecular Sciences 21, no. 2: 588. https://doi.org/10.3390/ijms21020588
APA StyleKrishna, G., Beitchman, J. A., Bromberg, C. E., & Currier Thomas, T. (2020). Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research. International Journal of Molecular Sciences, 21(2), 588. https://doi.org/10.3390/ijms21020588