Role of Sulfonylurea Receptor 1 and Glibenclamide in Traumatic Brain Injury: A Review of the Evidence

 , ,
, ,
Abstract
1. Introduction
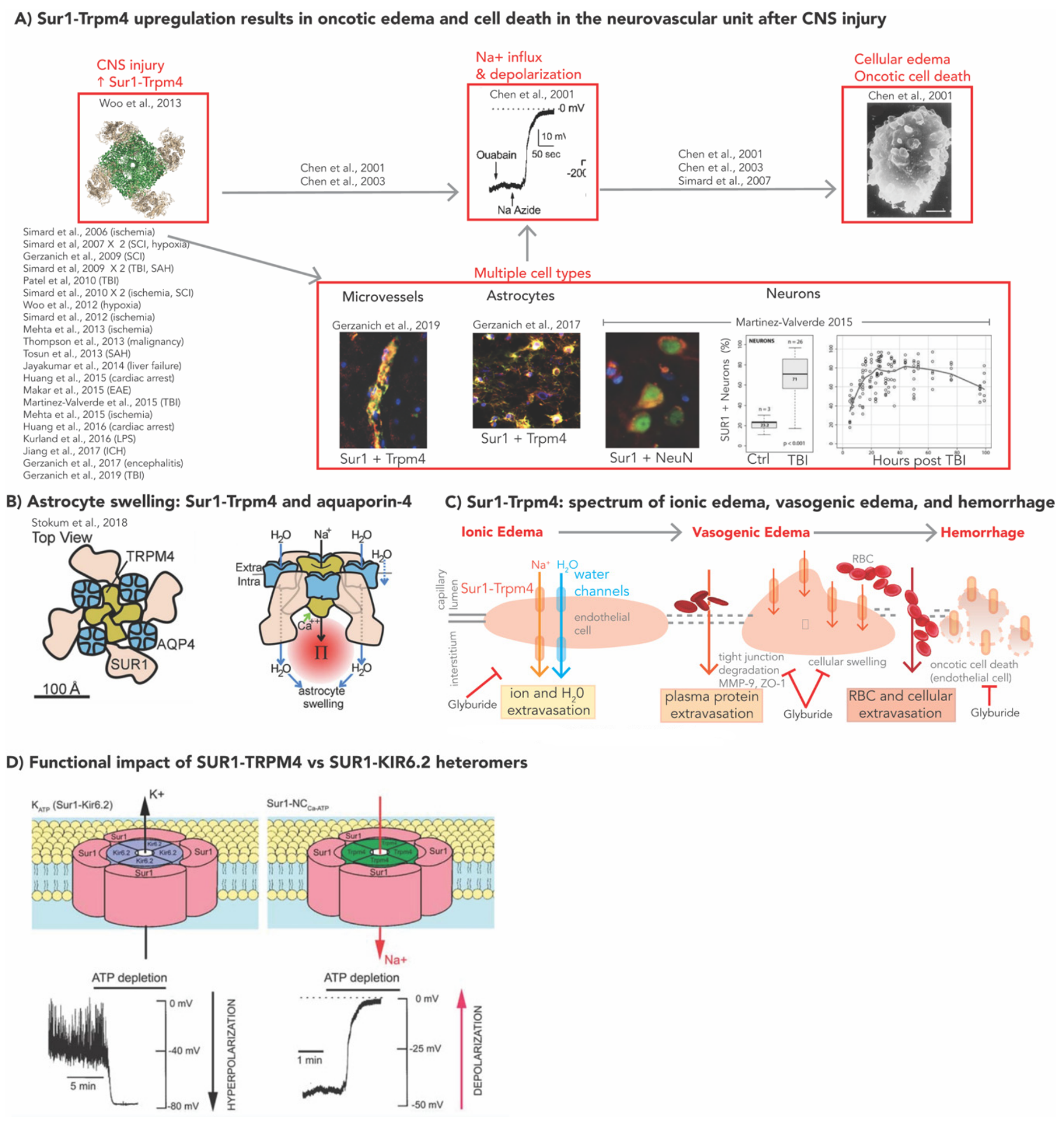
2. Molecular Mechanisms of SUR1-TRPM4 in Models of CNS Injury
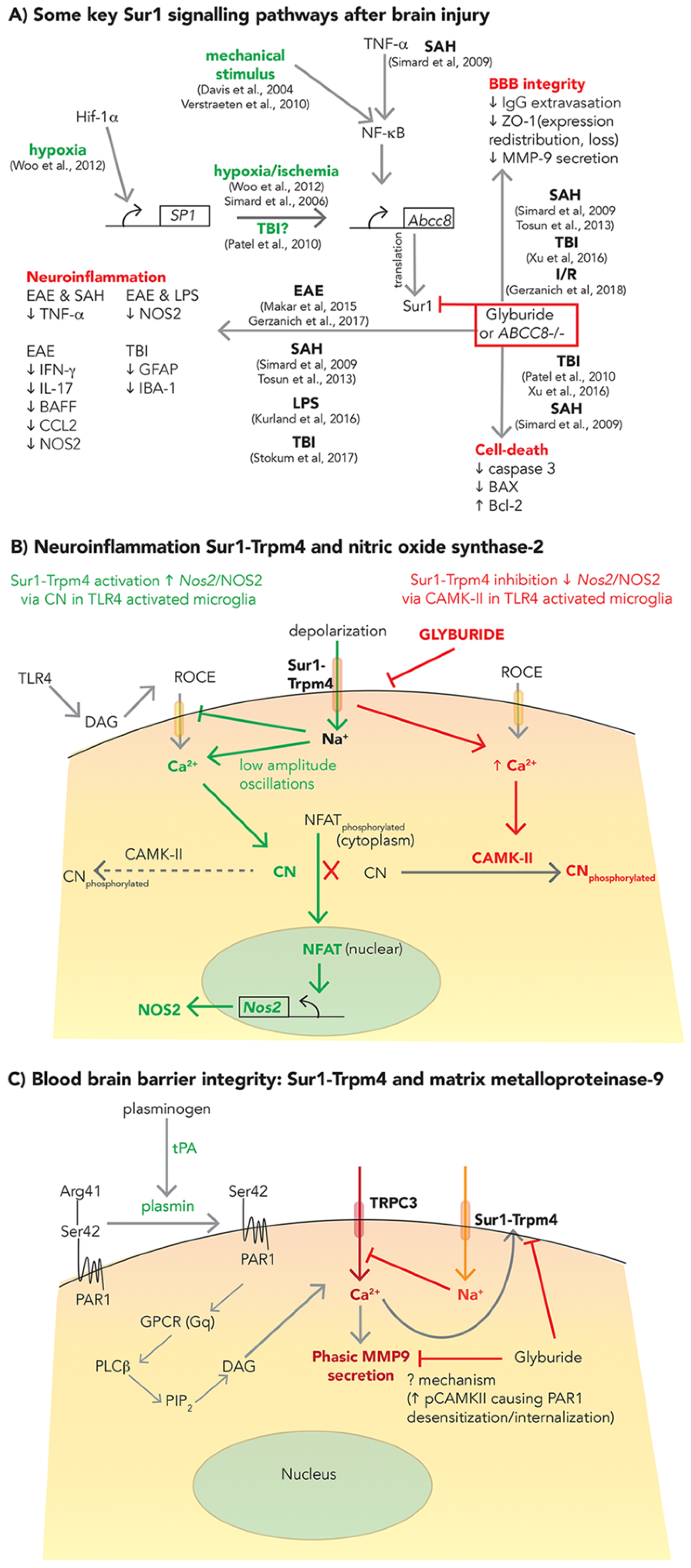
2.1. SUR1-TRPM4, GLI, and Cerebral Edema
2.2. SUR1-TRPM4: Neuroinflammation and BBB Integrity
2.2.1. Neuroinflammation
2.2.2. BBB Integrity
3. SUR1-TRPM4 Expression in TBI
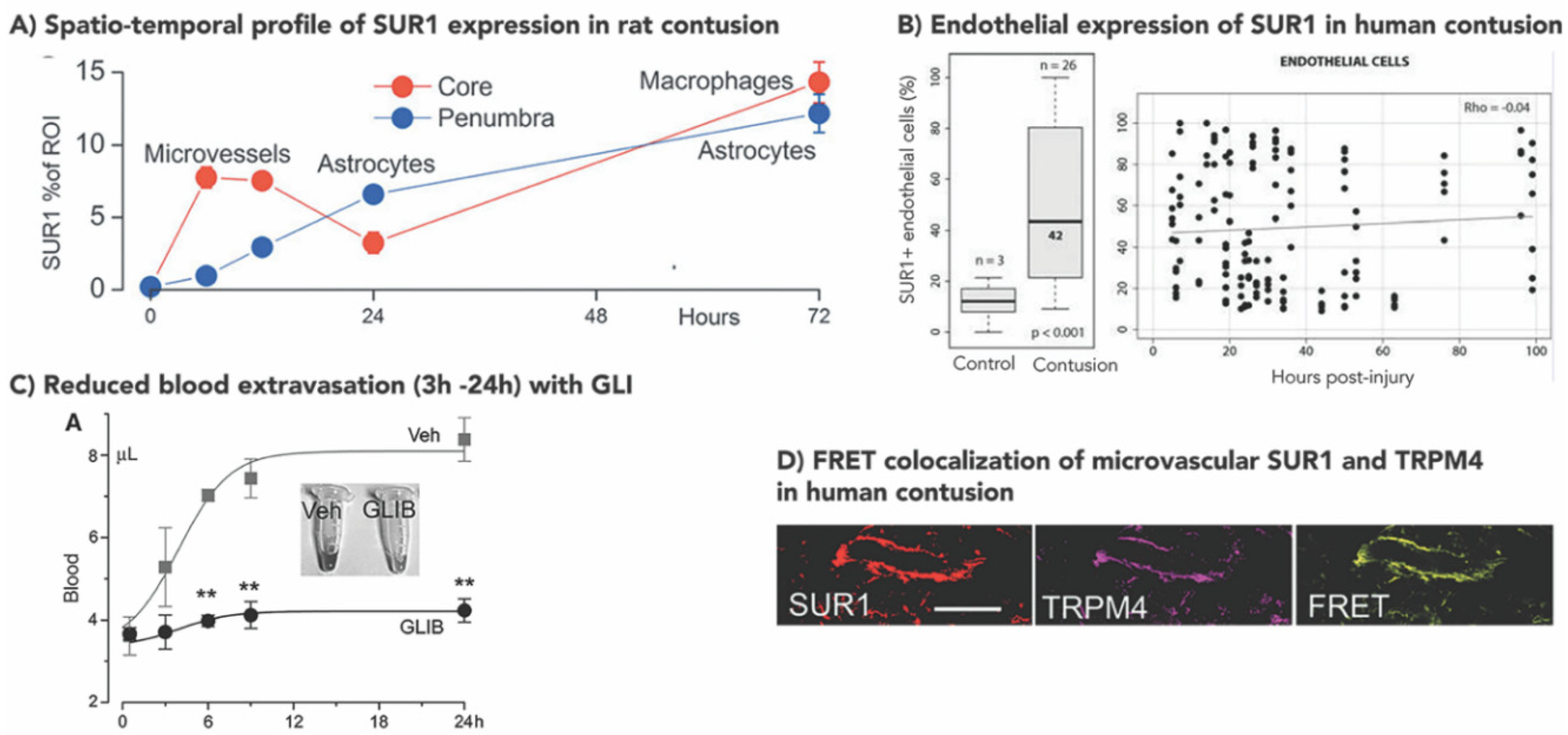
3.1. Increased SUR1±TRPM4 Expression in Rodent Models of TBI
3.2. Increased Human SUR1±TRPM4 Expression in TBI
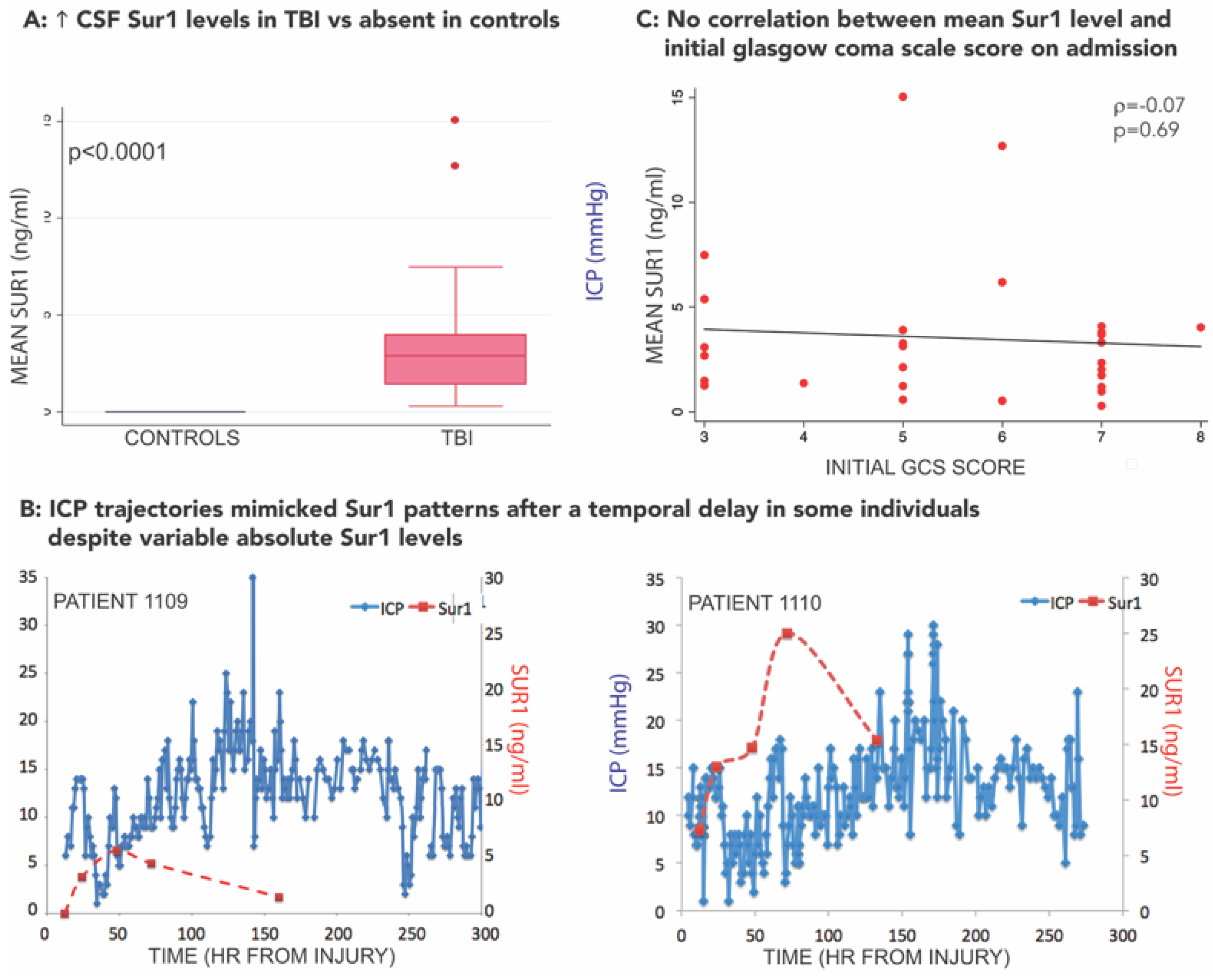
3.3. SUR1-TRPM4 Theranostic Biomarker Potential after TBI
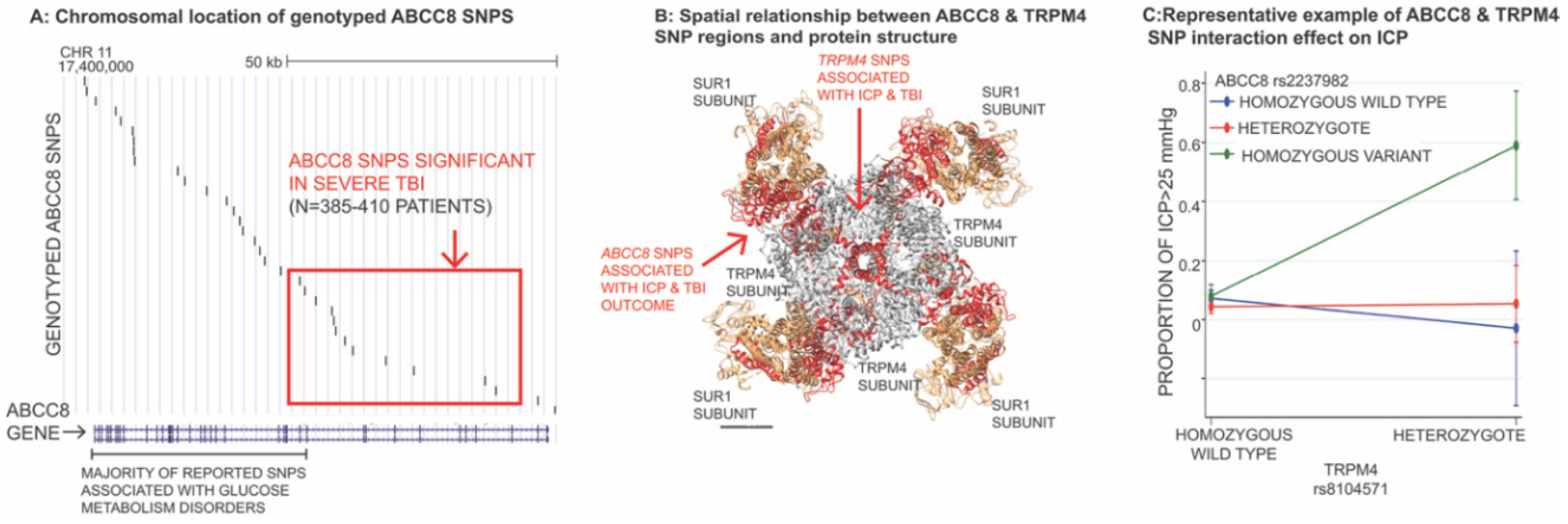
3.4. Impact of Genetic Variation in ABCC8 (Encoding SUR1) and TRPM4 in TBI
3.5. Preclinical Studies of SUR1 Inhibition in TBI
3.6. Clinical Studies of GLI in TBI
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AQP4 | aquaporin 4 |
| ATP | adenosine triphosphate |
| BAX | Bcl-associated X protein |
| BBB | blood brain barrier |
| CAMKII | calmodulin dependent protein kinase-II |
| CCI | controlled cortical impact |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| CT | computed tomography |
| DAI | diffuse axonal injury |
| FPI | fluid percussion injury |
| FRET | Förster resonance energy transfer |
| GCS | Glasgow Coma Scale |
| GFAP | glial fibrillary acidic protein |
| GLI | glibenclamide |
| GOS | Glasgow Outcome Scale |
| IMPACT | International Mission for Prognosis and Clinical Trial design in TBI |
| LHI | large hemispheric infarction |
| MMP-9 | matrix metalloproteinase-9 |
| MRI | magnetic resonance imaging |
| NFAT | nuclear factor of activated T-cells |
| NO | nitric oxide |
| NOS2 | nitric oxide synthase-2 |
| OBTT | operation brain trauma therapy |
| PAR1 | protease activated receptor 1 |
| SAH | subarachnoid hemorrhage |
| SCI | spinal cord injury |
| Sp1 | specificity protein 1 |
| SUR1 | Sulfonylurea receptor 1 |
| TBI | traumatic brain injury |
| TLR4 | toll like receptor 4 |
| tPA | tissue plasminogen activator |
| TRPM4 | transient receptor potential melastatin 4 |
| SUR1±TRPM4 | SUR1 with or without TRPM4 |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| TXA | tranexamic acid |
| UCHL1 | ubiquitin C-terminal hydrolase-L1 |
| ZO-1 | zona occludens 1 |
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. InTBIR participants and investigators traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Lingsma, H.F.; Roozenbeek, B.; Steyerberg, E.W.; Murray, G.D.; Maas, A.I.R. Early prognosis in traumatic brain injury: From prophecies to predictions. Lancet Neurol. 2010, 9, 543–554. [Google Scholar] [CrossRef]
- Murray, G.D.; Butcher, I.; McHugh, G.S.; Lu, J.; Mushkudiani, N.A.; Maas, A.I.R.; Marmarou, A.; Steyerberg, E.W. Multivariable prognostic analysis in traumatic brain injury: Results from the IMPACT study. J. Neurotrauma 2007, 24, 329–337. [Google Scholar] [CrossRef]
- Narayan, R.K.; Maas, A.I.R.; Servadei, F.; Skolnick, B.E.; Tillinger, M.N.; Marshall, L.F.; Traumatic Intracerebral Hemorrhage Study Group. Progression of traumatic intracerebral hemorrhage: A prospective observational study. J. Neurotrauma 2008, 25, 629–639. [Google Scholar] [CrossRef]
- O’Phelan, K.H.; Park, D.; Efird, J.T.; Johnson, K.; Albano, M.; Beniga, J.; Green, D.M.; Chang, C.W.J. Patterns of increased intracranial pressure after severe traumatic brain injury. Neurocrit. Care 2009, 10, 280–286. [Google Scholar] [CrossRef]
- Kurland, D.; Hong, C.; Aarabi, B.; Gerzanich, V.; Simard, J.M. Hemorrhagic progression of a contusion after traumatic brain injury: A review. J. Neurotrauma 2012, 29, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Oertel, M.; Kelly, D.F.; McArthur, D.; Boscardin, W.J.; Glenn, T.C.; Lee, J.H.; Gravori, T.; Obukhov, D.; McBride, D.Q.; Martin, N.A. Progressive hemorrhage after head trauma: Predictors and consequences of the evolving injury. J. Neurosurg. 2002, 96, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.F.; Meeker, M.; Holland, M.C. Acute traumatic intraparenchymal hemorrhage: Risk factors for progression in the early post-injury period. Neurosurgery 2006, 58, 647–656. [Google Scholar] [CrossRef]
- Tong, W.-S.; Zheng, P.; Xu, J.-F.; Guo, Y.-J.; Zeng, J.-S.; Yang, W.-J.; Li, G.-Y.; He, B.; Yu, H. Early CT signs of progressive hemorrhagic injury following acute traumatic brain injury. Neuroradiology 2011, 53, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Juratli, T.A.; Zang, B.; Litz, R.J.; Sitoci, K.-H.; Aschenbrenner, U.; Gottschlich, B.; Daubner, D.; Schackert, G.; Sobottka, S.B. Early hemorrhagic progression of traumatic brain contusions: Frequency, correlation with coagulation disorders, and patient outcome: A prospective study. J. Neurotrauma 2014, 31, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, J.A.; Segar, D.J.; Powers, A.Y.; Shah, M.; Doberstein, C.; Drapcho, B.; Morrison, J.F.; Williams, J.R.; Collins, S.; Monteiro, K.; et al. Blossoming contusions: Identifying factors contributing to the expansion of traumatic intracerebral hemorrhage. J. Neurosurg. 2018, 129, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Ding, J.; Chen, H.; Guo, Y.; Wang, G.; Gao, W.-W.; Chen, S.-W.; Tian, H.-L. Predicting progressive hemorrhagic injury after traumatic brain injury: Derivation and validation of a risk score based on admission characteristics. J. Neurotrauma 2012, 29, 2137–2142. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, H.M.; Gary, H.E.; Aldrich, E.F.; Saydjari, C.; Turner, B.; Foulkes, M.A.; Jane, J.A.; Marmarou, A.; Marshall, L.F.; Young, H.F. Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. J. Neurosurg. 1990, 73, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M. A precision medicine approach to cerebral edema and intracranial hypertension after severe traumatic brain injury: Quo vadis? Curr. Neurol. Neurosci. Rep. 2018, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.J.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 1993, 34, 216–222. [Google Scholar] [CrossRef]
- Feickert, H.J.; Drommer, S.; Heyer, R. Severe head injury in children: Impact of risk factors on outcome. J. Trauma 1999, 47, 33–38. [Google Scholar] [CrossRef]
- Feldmann, H.; Klages, G.; Gärtner, F.; Scharfenberg, J. The prognostic value of intracranial pressure monitoring after severe head injuries. In Proceedings of the 6th European Congress of Neurosurgery; Acta Neurochirurgica Book Series Volume 28; Brihaye, J., Clarke, P.R.R., Loew, F., Overgaard, J., Pásztor, E., Pertuiset, B., Schürmann, K., Symon, L., Eds.; Springer: Vienna, Austria, 1979; pp. 74–77. [Google Scholar]
- Hudak, A.M.; Peng, L.; Marquez de la Plata, C.; Thottakara, J.; Moore, C.; Harper, C.; McColl, R.; Babcock, E.; Diaz-Arrastia, R. Cytotoxic and vasogenic cerebral oedema in traumatic brain injury: Assessment with FLAIR and DWI imaging. Brain Inj. 2014, 28, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, C.; Schiavi, P.; Picetti, E.; Goldoni, M.; Cerasti, D.; Caspani, M.; Servadei, F. Patients with brain contusions: Predictors of outcome and relationship between radiological and clinical evolution. J. Neurosurg. 2014, 120, 908–918. [Google Scholar] [CrossRef]
- Marmarou, A.; Anderson, R.L.; Ward, J.D.; Choi, S.C.; Young, H.F.; Eisenberg, H.M.; Foulkes, M.A.; Marshall, L.F.; Jane, J.A. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J. Neurosurg. 1991, 75, S59–S66. [Google Scholar] [CrossRef]
- Marshall, L.F.; Smith, R.W.; Shapiro, H.M. The outcome with aggressive treatment in severe head injuries. Part I: The significance of intracranial pressure monitoring. J. Neurosurg. 1979, 50, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Becker, D.P.; Ward, J.D.; Sullivan, H.G.; Adams, W.E.; Rosner, M.J. Significance of intracranial hypertension in severe head injury. J. Neurosurg. 1977, 47, 503–516. [Google Scholar] [CrossRef]
- Saul, T.G.; Ducker, T.B. Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. J. Neurosurg. 1982, 56, 498–503. [Google Scholar] [CrossRef]
- Stocchetti, N.; Zanaboni, C.; Colombo, A.; Citerio, G.; Beretta, L.; Ghisoni, L.; Zanier, E.R.; Canavesi, K. Refractory intracranial hypertension and “second-tier” therapies in traumatic brain injury. Intensive Care Med. 2008, 34, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Aston, J.; Dines, M.; Caraman, E.; Yacyshyn, M.; McCarthy, M.; Olson, J.E. Early brain edema is a predictor of in-hospital mortality in traumatic brain injury. J. Emerg. Med. 2017, 53, 18–29. [Google Scholar] [CrossRef]
- Jha, R.M.; Elmer, J.; Zusman, B.E.; Desai, S.; Puccio, A.M.; Okonkwo, D.O.; Park, S.Y.; Shutter, L.A.; Wallisch, J.S.; Conley, Y.P.; et al. Intracranial pressure trajectories: A novel approach to informing severe traumatic brain injury phenotypes. Crit. Care Med. 2018, 46, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Griffin, A.D.; Turtzo, L.C.; Parikh, G.Y.; Tolpygo, A.; Lodato, Z.; Moses, A.D.; Nair, G.; Perl, D.P.; Edwards, N.A.; Dardzinski, B.J.; et al. Traumatic microbleeds suggest vascular injury and predict disability in traumatic brain injury. Brain 2019, 142, 3550–3564. [Google Scholar] [CrossRef]
- Izzy, S.; Mazwi, N.L.; Martinez, S.; Spencer, C.A.; Klein, J.P.; Parikh, G.; Glenn, M.B.; Greenberg, S.M.; Greer, D.M.; Wu, O.; et al. Revisiting grade 3 diffuse axonal injury: Not all brainstem microbleeds are prognostically equal. Neurocrit. Care 2017, 27, 199–207. [Google Scholar] [CrossRef]
- Yuh, E.L.; Mukherjee, P.; Lingsma, H.F.; Yue, J.K.; Ferguson, A.R.; Gordon, W.A.; Valadka, A.B.; Schnyer, D.M.; Okonkwo, D.O.; Maas, A.I.R.; et al. TRACK-TBI investigators magnetic resonance imaging improves 3-month outcome prediction in mild traumatic brain injury. Ann. Neurol. 2013, 73, 224–235. [Google Scholar] [CrossRef]
- Beauchamp, M.H.; Beare, R.; Ditchfield, M.; Coleman, L.; Babl, F.E.; Kean, M.; Crossley, L.; Catroppa, C.; Yeates, K.O.; Anderson, V. Susceptibility weighted imaging and its relationship to outcome after pediatric traumatic brain injury. Cortex 2013, 49, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.K.; Kishore, P.R.; Becker, D.P.; Ward, J.D.; Enas, G.G.; Greenberg, R.P.; Domingues Da Silva, A.; Lipper, M.H.; Choi, S.C.; Mayhall, C.G.; et al. Intracranial pressure: To monitor or not to monitor? A review of our experience with severe head injury. J. Neurosurg. 1982, 56, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.W.; Nyström, H.; MacCallum, R.M.; Thornquist, B.; Lilja, A.; Bellander, B.-M.; Rudehill, A.; Wanecek, M.; Weitzberg, E. Extended analysis of early computed tomography scans of traumatic brain injured patients and relations to outcome. J. Neurotrauma 2010, 27, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Steyerberg, E.W.; Butcher, I.; Dammers, R.; Lu, J.; Marmarou, A.; Mushkudiani, N.A.; McHugh, G.S.; Murray, G.D. Prognostic value of computerized tomography scan characteristics in traumatic brain injury: Results from the IMPACT study. J. Neurotrauma 2007, 24, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Pergakis, M.; Badjatia, N.; Chaturvedi, S.; Cronin, C.A.; Kimberly, W.T.; Sheth, K.N.; Simard, J.M. BIIB093 (IV glibenclamide): An investigational compound for the prevention and treatment of severe cerebral edema. Expert Opin. Investig. Drugs 2019, 28, 1–10. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Central nervous system trauma: Pharmacological and therapeutic approaches. In Encyclopedia of Molecular Cell Biology—Molecular Pharmacology; in press.
- Donkin, J.J.; Vink, R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr. Opin. Neurol. 2010, 23, 293–299. [Google Scholar] [CrossRef]
- Hawryluk, G.W.J.; Aguilera, S.; Buki, A.; Bulger, E.; Citerio, G.; Cooper, D.J.; Arrastia, R.D.; Diringer, M.; Figaji, A.; Gao, G.; et al. A management algorithm for patients with intracranial pressure monitoring: The Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Intensive Care Med. 2019. [Google Scholar] [CrossRef]
- Hutchinson, P.J.; Kolias, A.G.; Timofeev, I.S.; Corteen, E.A.; Czosnyka, M.; Timothy, J.; Anderson, I.; Bulters, D.O.; Belli, A.; Eynon, C.A.; et al. RESCUEicp trial collaborators trial of decompressive craniectomy for traumatic intracranial hypertension. N. Engl. J. Med. 2016, 375, 1119–1130. [Google Scholar] [CrossRef]
- Shutter, L.A.; Timmons, S.D. Intracranial pressure rescued by decompressive surgery after traumatic brain injury. N. Engl. J. Med. 2016, 375, 1183–1184. [Google Scholar] [CrossRef]
- Cooper, D.J.; Rosenfeld, J.V.; Murray, L.; Arabi, Y.M.; Davies, A.R.; D’Urso, P.; Kossmann, T.; Ponsford, J.; Seppelt, I.; Reilly, P.; et al. DECRA trial investigators; Australian and New Zealand intensive care society clinical trials group decompressive craniectomy in diffuse traumatic brain injury. N. Engl. J. Med. 2011, 364, 1493–1502. [Google Scholar] [CrossRef]
- Andrews, P.J.D.; Sinclair, H.L.; Rodriguez, A.; Harris, B.A.; Battison, C.G.; Rhodes, J.K.J.; Murray, G.D.; Eurotherm3235 Trial Collaborators. Hypothermia for intracranial hypertension after traumatic brain injury. N. Engl. J. Med. 2015, 373, 2403–2412. [Google Scholar] [CrossRef]
- CRASH-3 trial collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): A randomised, placebo-controlled trial. Lancet 2019, 394, 1713–1723. [Google Scholar] [CrossRef]
- Weng, S.; Wang, W.; Wei, Q.; Lan, H.; Su, J.; Xu, Y. Effect of tranexamic acid in patients with traumatic brain injury: A systematic review and meta-analysis. World Neurosurg. 2019, 123, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Schwartzbauer, G.T.; Gerzanich, V. Sulfonylurea receptor 1 in central nervous system injury: A focused review. J. Cereb. Blood Flow Metab. 2012, 32, 1699–1717. [Google Scholar] [CrossRef] [PubMed]
- Aittoniemi, J.; Fotinou, C.; Craig, T.J.; de Wet, H.; Proks, P.; Ashcroft, F.M. Review. SUR1: A unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Proks, P.; Reimann, F.; Green, N.; Gribble, F.; Ashcroft, F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002, 51, S368–S376. [Google Scholar] [CrossRef]
- Haider, S.; Antcliff, J.F.; Proks, P.; Sansom, M.S.P.; Ashcroft, F.M. Focus on Kir6.2: A key component of the ATP-sensitive potassium channel. J. Mol. Cell Cardiol. 2005, 38, 927–936. [Google Scholar] [CrossRef]
- Gerzanich, V.; Stokum, J.A.; Ivanova, S.; Woo, S.K.; Tsymbalyuk, O.; Sharma, A.; Akkentli, F.; Imran, Z.; Aarabi, B.; Sahuquillo, J.; et al. Sulfonylurea receptor 1, transient receptor potential cation channel subfamily M member 4, and Kir6.2: Role in hemorrhagic progression of contusion. J. Neurotrauma 2019, 36, 1060–1079. [Google Scholar] [CrossRef]
- Castro, L.; Noelia, M.; Vidal-Jorge, M.; Sánchez-Ortiz, D.; Gándara, D.; Martínez-Saez, E.; Cicuéndez, M.; Poca, M.-A.; Simard, J.M.; Sahuquillo, J. Kir6.2, the pore-forming subunit of ATP-sensitive K+ channels, is overexpressed in human posttraumatic brain contusions. J. Neurotrauma 2018, 36. [Google Scholar] [CrossRef]
- Chen, M.; Simard, J.M. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J. Neurosci. 2001, 21, 6512–6521. [Google Scholar] [CrossRef]
- Chen, M.; Dong, Y.; Simard, J.M. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J. Neurosci. 2003, 23, 8568–8577. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Ivanov, A.; Gerzanich, V.; Simard, J.M. The sulfonylurea receptor 1 (SUR1)-transient receptor potential melastatin 4 (TRPM4) channel. J. Biol. Chem. 2013, 288, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Kwon, M.S.; Woo, S.K.; Tsymbalyuk, O.; Vennekens, R.; Gerzanich, V.; Simard, J.M. SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia 2018, 66, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Chen, M.; Tarasov, K.V.; Bhatta, S.; Ivanova, S.; Melnitchenko, L.; Tsymbalyuk, N.; West, G.A.; Gerzanich, V. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat. Med. 2006, 12, 433–440. [Google Scholar] [CrossRef]
- Simard, J.M.; Woo, S.K.; Tsymbalyuk, N.; Voloshyn, O.; Yurovsky, V.; Ivanova, S.; Lee, R.; Gerzanich, V. Glibenclamide-10-h treatment window in a clinically relevant model of stroke. Transl. Stroke Res. 2012, 3, 286–295. [Google Scholar] [CrossRef]
- Mehta, R.I.; Tosun, C.; Ivanova, S.; Tsymbalyuk, N.; Famakin, B.M.; Kwon, M.S.; Castellani, R.J.; Gerzanich, V.; Simard, J.M. SUR1-TRPM4 cation channel expression in human cerebral infarcts. J. Neuropathol. Exp. Neurol. 2015, 74, 835–849. [Google Scholar] [CrossRef]
- Mehta, R.I.; Ivanova, S.; Tosun, C.; Castellani, R.J.; Gerzanich, V.; Simard, J.M. Sulfonylurea receptor 1 expression in human cerebral infarcts. J. Neuropathol. Exp. Neurol. 2013, 72, 871–883. [Google Scholar] [CrossRef]
- Patel, A.D.; Gerzanich, V.; Geng, Z.; Simard, J.M. Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J. Neuropathol. Exp. Neurol. 2010, 69, 1177–1190. [Google Scholar] [CrossRef]
- Simard, J.M.; Kilbourne, M.; Tsymbalyuk, O.; Tosun, C.; Caridi, J.; Ivanova, S.; Keledjian, K.; Bochicchio, G.; Gerzanich, V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J. Neurotrauma 2009, 26, 2257–2267. [Google Scholar] [CrossRef]
- Martínez-Valverde, T.; Vidal-Jorge, M.; Martínez-Saez, E.; Castro, L.; Arikan, F.; Cordero, E.; Rădoi, A.; Poca, M.-A.; Simard, J.M.; Sahuquillo, J. Sulfonylurea receptor 1 in humans with post-traumatic brain contusions. J. Neurotrauma 2015, 32, 1478–1487. [Google Scholar] [CrossRef]
- Simard, J.M.; Tsymbalyuk, O.; Ivanov, A.; Ivanova, S.; Bhatta, S.; Geng, Z.; Woo, S.K.; Gerzanich, V. Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J. Clin. Investig. 2007, 117, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Norenberg, M.D.; Tosun, C.; Chen, Z.; Ivanova, S.; Tsymbalyuk, O.; Bryan, J.; Landsman, D.; Gerzanich, V. Brief suppression of Abcc8 prevents autodestruction of spinal cord after trauma. Sci. Transl. Med. 2010, 2, 28ra29. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Li, L.; Chen, Q.; Tao, Y.; Yang, L.; Zhang, B.; Zhang, J.H.; Feng, H.; Chen, Z.; Tang, J.; et al. Role of glibenclamide in brain injury after intracerebral hemorrhage. Transl. Stroke Res. 2017, 8, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Tosun, C.; Kurland, D.B.; Mehta, R.; Castellani, R.J.; deJong, J.L.; Kwon, M.S.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Inhibition of the SUR1-TRPM4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke 2013, 44, 3522–3528. [Google Scholar] [CrossRef]
- Simard, J.M.; Geng, Z.; Woo, S.K.; Ivanova, S.; Tosun, C.; Melnichenko, L.; Gerzanich, V. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2009, 29, 317–330. [Google Scholar] [CrossRef]
- Thompson, E.M.; Pishko, G.L.; Muldoon, L.L.; Neuwelt, E.A. Inhibition of SUR1 decreases the vascular permeability of cerebral metastases. Neoplasia 2013, 15, 535–543. [Google Scholar] [CrossRef]
- Huang, K.; Gu, Y.; Hu, Y.; Ji, Z.; Wang, S.; Lin, Z.; Li, X.; Xie, Z.; Pan, S. Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats. Crit. Care Med. 2015, 43, e341–e349. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Z.; Gu, Y.; Hu, Y.; Ji, Z.; Wang, S.; Lin, Z.; Li, X.; Xie, Z.; Pan, S. Glibenclamide is comparable to target temperature management in improving survival and neurological outcome after asphyxial cardiac arrest in rats. J. Am. Heart Assoc. 2016, 5, e003465. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Valdes, V.; Tong, X.Y.; Shamaladevi, N.; Gonzalez, W.; Norenberg, M.D. Sulfonylurea receptor 1 contributes to the astrocyte swelling and brain edema in acute liver failure. Transl. Stroke Res. 2014, 5, 28–37. [Google Scholar] [CrossRef]
- Makar, T.K.; Gerzanich, V.; Nimmagadda, V.K.C.; Jain, R.; Lam, K.; Mubariz, F.; Trisler, D.; Ivanova, S.; Woo, S.K.; Kwon, M.S.; et al. Silencing of Abcc8 or inhibition of newly upregulated SUR1-TRPM4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis. J. Neuroinflammation 2015, 12, 210. [Google Scholar] [CrossRef]
- Gerzanich, V.; Makar, T.K.; Guda, P.R.; Kwon, M.S.; Stokum, J.A.; Woo, S.K.; Ivanova, S.; Ivanov, A.; Mehta, R.I.; Morris, A.B.; et al. Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis. J. Neuroinflammation 2017, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Schattling, B.; Steinbach, K.; Thies, E.; Kruse, M.; Menigoz, A.; Ufer, F.; Flockerzi, V.; Brück, W.; Pongs, O.; Vennekens, R.; et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2012, 18, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef]
- Hosier, H.; Peterson, D.; Tsymbalyuk, O.; Keledjian, K.; Smith, B.R.; Ivanova, S.; Gerzanich, V.; Popovich, P.G.; Simard, J.M. A direct comparison of three clinically relevant treatments in a rat model of cervical spinal cord injury. J. Neurotrauma 2015, 32, 1633–1644. [Google Scholar] [CrossRef]
- Simard, J.M.; Tsymbalyuk, N.; Tsymbalyuk, O.; Ivanova, S.; Yurovsky, V.; Gerzanich, V. Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke. Stroke 2010, 41, 531–537. [Google Scholar] [CrossRef]
- Wali, B.; Ishrat, T.; Atif, F.; Hua, F.; Stein, D.G.; Sayeed, I. Glibenclamide administration attenuates infarct volume, hemispheric swelling, and functional impairments following permanent focal cerebral ischemia in rats. Stroke Res. Treat. 2012, 2012, 460909. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Z.; Gu, Y.; Ji, Z.; Lin, Z.; Wang, S.; Pan, S.; Wu, Y. Glibenclamide prevents water diffusion abnormality in the brain after cardiac arrest in rats. Neurocrit. Care 2018, 29, 128–135. [Google Scholar] [CrossRef]
- Nakayama, S.; Taguchi, N.; Isaka, Y.; Nakamura, T.; Tanaka, M. Glibenclamide and therapeutic hypothermia have comparable effect on attenuating global cerebral edema following experimental cardiac arrest. Neurocrit. Care 2018, 29, 119–127. [Google Scholar] [CrossRef]
- Zweckberger, K.; Hackenberg, K.; Jung, C.S.; Hertle, D.N.; Kiening, K.L.; Unterberg, A.W.; Sakowitz, O.W. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience 2014, 272, 199–206. [Google Scholar] [CrossRef]
- Xu, Z.-M.; Yuan, F.; Liu, Y.-L.; Ding, J.; Tian, H.-L. Glibenclamide attenuates blood-brain barrier disruption in adult mice after traumatic brain injury. J. Neurotrauma 2017, 34, 925–933. [Google Scholar] [CrossRef]
- Jha, R.M.; Molyneaux, B.J.; Jackson, T.C.; Wallisch, J.S.; Park, S.-Y.; Poloyac, S.; Vagni, V.A.; Janesko-Feldman, K.L.; Hoshitsuki, K.; Minnigh, M.B.; et al. Glibenclamide produces region-dependent effects on cerebral edema in a combined injury model of traumatic brain injury and hemorrhagic shock in mice. J. Neurotrauma 2018, 35, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.; Yan, H.; Dixon, C.E.; Poloyac, S.; Jackson, T.; Hoshitsuki, K.; Ma, X.; Henchir, J.; Janesko-Feldman, K.; Kochanek, P. Evaluation of glibenclamide in the Pittsburgh controlled cortical impact model of traumatic brain injury: An OBTT consortium study. J. Neurotrauma 2015, 32, 119. [Google Scholar]
- Simard, J.M.; Tsymbalyuk, O.; Keledjian, K.; Ivanov, A.; Ivanova, S.; Gerzanich, V. Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury. Exp. Neurol. 2012, 233, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Popovich, P.G.; Tsymbalyuk, O.; Gerzanich, V. Spinal cord injury with unilateral versus bilateral primary hemorrhage—Effects of glibenclamide. Exp. Neurol. 2012, 233, 829–835. [Google Scholar] [CrossRef]
- Popovich, P.G.; Lemeshow, S.; Gensel, J.C.; Tovar, C.A. Independent evaluation of the effects of glibenclamide on reducing progressive hemorrhagic necrosis after cervical spinal cord injury. Exp. Neurol. 2012, 233, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; del Zoppo, G.J.; Jacobson, S.; Gerzanich, V. Glibenclamide in cerebral ischemia and stroke. Neurocrit. Care 2014, 20, 319–333. [Google Scholar] [CrossRef]
- Nedergaard, M.; Kraig, R.P.; Tanabe, J.; Pulsinelli, W.A. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 1991, 260, R581–R588. [Google Scholar] [CrossRef]
- Sheth, K.N.; Elm, J.J.; Molyneaux, B.J.; Hinson, H.; Beslow, L.A.; Sze, G.K.; Ostwaldt, A.-C.; Del Zoppo, G.J.; Simard, J.M.; Jacobson, S.; et al. Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016, 15, 1160–1169. [Google Scholar] [CrossRef]
- Sheth, K.N.; Kimberly, W.T.; Elm, J.J.; Kent, T.A.; Yoo, A.J.; Thomalla, G.; Campbell, B.; Donnan, G.A.; Davis, S.M.; Albers, G.W.; et al. Exploratory analysis of glyburide as a novel therapy for preventing brain swelling. Neurocrit. Care 2014, 21, 43–51. [Google Scholar] [CrossRef]
- Kimberly, W.T.; Battey, T.W.K.; Pham, L.; Wu, O.; Yoo, A.J.; Furie, K.L.; Singhal, A.B.; Elm, J.J.; Stern, B.J.; Sheth, K.N. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit. Care 2014, 20, 193–201. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Sheth, K.N.; Kimberly, W.T.; Simard, J.M. Profile of intravenous glyburide for the prevention of cerebral edema following large hemispheric infarction: Evidence to date. Drug Des. Devel. Ther. 2018, 12, 2539–2552. [Google Scholar] [CrossRef]
- Simard, J.M.; Geng, Z.; Silver, F.L.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; Colucci, M.; Gerzanich, V. Does inhibiting SUR1 complement rt-PA in cerebral ischemia? Ann. N. Y. Acad. Sci. 2012, 1268, 95–107. [Google Scholar] [CrossRef]
- Simard, J.M.; Yurovsky, V.; Tsymbalyuk, N.; Melnichenko, L.; Ivanova, S.; Gerzanich, V. Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke J. Cereb. Circ. 2009, 40, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Geng, Z.; Chen, Z.; Ivanov, A.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J. Cereb. Blood Flow Metab. 2012, 32, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, S.V.; Mackenzie, G.G.; Oteiza, P.I. The plasma membrane plays a central role in cells response to mechanical stress. Biochim. Biophys. Acta 2010, 1798, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Grumbach, I.M.; Fukai, T.; Cutchins, A.; Harrison, D.G. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor κB binding. J. Biol. Chem. 2004, 279, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Kurland, D.B.; Gerzanich, V.; Karimy, J.K.; Woo, S.K.; Vennekens, R.; Freichel, M.; Nilius, B.; Bryan, J.; Simard, J.M. The SUR1-TRPM4 channel regulates NOS2 transcription in TLR4-activated microglia. J. Neuroinflam. 2016, 13, 130. [Google Scholar] [CrossRef]
- Stokum, J.A.; Keledjian, K.; Hayman, E.; Karimy, J.K.; Pampori, A.; Imran, Z.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Glibenclamide pretreatment protects against chronic memory dysfunction and glial activation in rat cranial blast traumatic brain injury. Behav. Brain Res. 2017, 333, 43–53. [Google Scholar] [CrossRef]
- Gerzanich, V.; Kwon, M.S.; Woo, S.K.; Ivanov, A.; Simard, J.M. SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells. PLoS ONE 2018, 13, e0195526. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Kumar, A.; Chen, S.-H.; Kadiiska, M.B.; Hong, J.-S.; Zielonka, J.; Kalyanaraman, B.; Mason, R.P. Inducible nitric oxide synthase is key to peroxynitrite-mediated, LPS-induced protein radical formation in murine microglial BV2 cells. Free Radic. Biol. Med. 2014, 73, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Béchade, C.; Colasse, S.; Diana, M.A.; Rouault, M.; Bessis, A. NOS2 expression is restricted to neurons in the healthy brain but is triggered in microglia upon inflammation. Glia 2014, 62, 956–963. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front. Cell Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 29, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Gorse, K.M.; Lantzy, M.K.; Lee, E.D.; Lafrenaye, A.D. Transient receptor potential melastatin 4 induces astrocyte swelling but not death after diffuse traumatic brain injury. J. Neurotrauma 2018, 35, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Puccio, A.M.; Chou, S.H.-Y.; Chang, C.-C.H.; Wallisch, J.S.; Molyneaux, B.J.; Zusman, B.E.; Shutter, L.A.; Poloyac, S.M.; Janesko-Feldman, K.L.; et al. Sulfonylurea receptor-1: A novel biomarker for cerebral edema in severe traumatic brain injury. Crit. Care Med. 2017, 45, e255–e264. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Yang, Z.; Zhu, T.; Shi, Y.; Rubenstein, R.; Tyndall, J.A.; Manley, G.T. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev. Mol. Diagn. 2018, 18, 165–180. [Google Scholar] [CrossRef]
- Brophy, G.M.; Mondello, S.; Papa, L.; Robicsek, S.A.; Gabrielli, A.; Tepas, J.; Buki, A.; Robertson, C.; Tortella, F.C.; Hayes, R.L.; et al. Biokinetic analysis of ubiquitin C-terminal hydrolase-L1 (UCH-L1) in severe traumatic brain injury patient biofluids. J. Neurotrauma 2011, 28, 861–870. [Google Scholar] [CrossRef]
- Roberts, D.J.; Jenne, C.N.; Léger, C.; Kramer, A.H.; Gallagher, C.N.; Todd, S.; Parney, I.F.; Doig, C.J.; Yong, V.W.; Kubes, P.; et al. A prospective evaluation of the temporal matrix metalloproteinase response after severe traumatic brain injury in humans. J. Neurotrauma 2013, 30, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Guilfoyle, M.R.; Carpenter, K.L.H.; Helmy, A.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J.A. Matrix metalloproteinase expression in contusional traumatic brain injury: A paired microdialysis study. J. Neurotrauma 2015, 32, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, F.A.; McFadyen, C.; Newcombe, V.F.J.; Synnot, A.; Donoghue, E.L.; Ripatti, S.; Steyerberg, E.W.; Gruen, R.L.; McAllister, T.W.; Rosand, J.; et al. Genetic influences on patient-oriented outcomes in traumatic brain injury: A living systematic review of non-apolipoprotein E single-nucleotide polymorphisms. J. Neurotrauma 2019. [CrossRef] [PubMed]
- Zeiler, F.A.; Thelin, E.P.; Donnelly, J.; Stevens, A.R.; Smielewski, P.; Czosnyka, M.; Hutchinson, P.J.; Menon, D.K. Genetic drivers of cerebral blood flow dysfunction in TBI: A speculative synthesis. Nat. Rev. Neurol. 2019, 15, 25–39. [Google Scholar] [CrossRef]
- McFadyen, C.A.; Zeiler, F.A.; Newcombe, V.; Synnot, A.; Steyerberg, E.; Gruen, R.L.; Rosand, J.; Palotie, A.; Maas, A.I.R.; Menon, D.K. Apolipoprotein E4 polymorphism and outcomes from traumatic brain injury: A living systematic review and meta-analysis. J. Neurotrauma 2019. [Google Scholar] [CrossRef]
- Yue, J.K.; Pronger, A.M.; Ferguson, A.R.; Temkin, N.R.; Sharma, S.; Rosand, J.; Sorani, M.D.; McAllister, T.W.; Barber, J.; Winkler, E.A.; et al. COBRIT investigators; TRACK-TBI investigators association of a common genetic variant within ANKK1 with six-month cognitive performance after traumatic brain injury. Neurogenetics 2015, 16, 169–180. [Google Scholar] [CrossRef]
- Winkler, E.A.; Yue, J.K.; Ferguson, A.R.; Temkin, N.R.; Stein, M.B.; Barber, J.; Yuh, E.L.; Sharma, S.; Satris, G.G.; McAllister, T.W.; et al. TRACK-TBI investigators COMT Val158Met polymorphism is associated with post-traumatic stress disorder and functional outcome following mild traumatic brain injury. J. Clin. Neurosci. 2017, 35, 109–116. [Google Scholar] [CrossRef]
- Winkler, E.A.; Yue, J.K.; McAllister, T.W.; Temkin, N.R.; Oh, S.S.; Burchard, E.G.; Hu, D.; Ferguson, A.R.; Lingsma, H.F.; Burke, J.F.; et al. TRACK-TBI investigators COMT Val158 Met polymorphism is associated with nonverbal cognition following mild traumatic brain injury. Neurogenetics 2016, 17, 31–41. [Google Scholar] [CrossRef]
- Yue, J.K.; Robinson, C.K.; Burke, J.F.; Winkler, E.A.; Deng, H.; Cnossen, M.C.; Lingsma, H.F.; Ferguson, A.R.; McAllister, T.W.; Rosand, J.; et al. TRACK-TBI investigators apolipoprotein E epsilon 4 (APOE-ε4) genotype is associated with decreased 6-month verbal memory performance after mild traumatic brain injury. Brain Behav. 2017, 7, e00791. [Google Scholar] [CrossRef]
- Jha, R.M.; Puccio, A.M.; Okonkwo, D.O.; Zusman, B.E.; Park, S.-Y.; Wallisch, J.; Empey, P.E.; Shutter, L.A.; Clark, R.S.B.; Kochanek, P.M.; et al. Abcc8 single nucleotide polymorphisms are associated with cerebral edema in severe TBI. Neurocrit. Care 2017, 26, 213–224. [Google Scholar] [CrossRef]
- Jha, R.M.; Koleck, T.A.; Puccio, A.M.; Okonkwo, D.O.; Park, S.-Y.; Zusman, B.E.; Clark, R.S.B.; Shutter, L.A.; Wallisch, J.S.; Empey, P.E.; et al. Regionally clustered Abcc8 polymorphisms in a prospective cohort predict cerebral oedema and outcome in severe traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Desai, S.M.; Zusman, B.E.; Koleck, T.A.; Puccio, A.M.; Okonkwo, D.O.; Park, S.-Y.; Shutter, L.A.; Kochanek, P.M.; Conley, Y.P. Downstream TRPM4 polymorphisms are associated with intracranial hypertension and statistically interact with Abcc8 polymorphisms in a prospective cohort of severe traumatic brain injury. J. Neurotrauma 2019, 36, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Paterakis, K.; Tsivgoulis, G.; Tsintou, M.; Hadjigeorgiou, G.F.; Dardioti, M.; Grigoriadis, S.; Simeonidou, C.; Komnos, A.; Kapsalaki, E.; et al. AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J. Neurotrauma 2014, 31, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Sorani, M.D.; Zador, Z.; Hurowitz, E.; Yan, D.; Giacomini, K.M.; Manley, G.T. Novel variants in human Aquaporin-4 reduce cellular water permeability. Hum. Mol. Genet. 2008, 17, 2379–2389. [Google Scholar] [CrossRef]
- Appelboom, G.; Bruce, S.; Duren, A.; Piazza, M.; Monahan, A.; Christophe, B.; Zoller, S.; LoPresti, M.; Connolly, E.S. Aquaporin-4 gene variant independently associated with oedema after intracerebral haemorrhage. Neurol. Res. 2015, 37, 657–661. [Google Scholar] [CrossRef]
- Bramlett, H.; Furones-Alonso, O.; Sanchez-Molano, J.; Sequiera, D.; Moreno, W.; Dietrich, W.D. Evaluation of Glibenclamide in the Miami Fluid Percussion Model of Traumatic Brain Injury: An OBTT Consortium Study. Abstracts from the 33rd Annual National Neurotrauma Symposium June 28–July 1, 2015 Santa Fe, New Mexico. J. Neurotrauma 2015, 32. [Google Scholar] [CrossRef]
- Jha, R.M.; Koleck, T.; Wu, Y.; Zusman, B.; Salamacha, N.; Wyman, S.; Janesko Feldman, K.; Kochanek, P. Evaluation of Glibenclamide in the Pittsburgh Controlled Cortical Impact Model of Traumatic Brain Injury: An OBTT Consortium Study. Abstracts from the 37th Annual National Neurotrauma Symposium June 29–July 3, 2019 Pittsburgh, Pennsylvania. J. Neurotrauma 2019, 36. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Bramlett, H.M.; Dixon, C.E.; Dietrich, W.D.; Mondello, S.; Wang, K.K.W.; Hayes, R.L.; Lafrenaye, A.; Povlishock, J.T.; Tortella, F.C.; et al. Operation brain trauma therapy: 2016 update. Mil. Med. 2018, 183, 303–312. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Bramlett, H.M.; Dixon, C.E.; Shear, D.A.; Dietrich, W.D.; Schmid, K.E.; Mondello, S.; Wang, K.K.W.; Hayes, R.L.; Povlishock, J.T.; et al. Approach to modeling, therapy evaluation, drug selection, and biomarker assessments for a multicenter pre-clinical drug screening consortium for acute therapies in severe traumatic brain injury: Operation brain trauma therapy. J. Neurotrauma 2016, 33, 513–522. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Bramlett, H.M.; Shear, D.A.; Dixon, C.E.; Mondello, S.; Dietrich, W.D.; Hayes, R.L.; Wang, K.K.W.; Poloyac, S.M.; Empey, P.E.; et al. Synthesis of findings, current investigations, and future directions: Operation brain trauma therapy. J. Neurotrauma 2016, 33, 606–614. [Google Scholar] [CrossRef]
- Zafardoost, P.; Ghasemi, A.A.; Salehpour, F.; Piroti, C.; Ziaeii, E. Evaluation of the effect of glibenclamide in patients with diffuse axonal injury due to moderate to severe head trauma. Trauma Mon 2016, 21, e25113. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Derakhshan, N.; Niakan, A.; Ghaffarpasand, F.; Salehi, M.; Eshraghian, H.; Shakibafard, A.; Zahabi, B. Effects of oral glibenclamide on brain contusion volume and functional outcome of patients with moderate and severe traumatic brain injuries: A randomized double-blind placebo-controlled clinical trial. World Neurosurg. 2017, 101, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, H.M.; Shenton, M.E.; Pasternak, O.; Simard, J.M.; Okonkwo, D.O.; Aldrich, C.; He, F.; Jain, S.; Hayman, E.G. Magnetic resonance imaging pilot study of intravenous glyburide in traumatic brain injury. J. Neurotrauma 2019. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Gerzanich, V.; Simard, J.M. Glibenclamide enhances erythrocyte phagocytosis and resolution of contusion-TBI. J. Neurotrauma. in preparation.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat Contusion | Human Contusion | |||||
|---|---|---|---|---|---|---|
| Increased (Y/N) | Peak (h Post Injury) | Ref | Increased (Y/N) | Peak (h Post Injury) | Ref | |
| SUR1 | ||||||
| Neurons | Y | 12–24 h | Simard et al., 2009 [61] Patel et al., 2010 [60] | Y | 24 h | Martinez-Valverde et al., 2015 [62] |
| GFAP+ Glia (Astrocytes) | Y | 72 h (penumbra) | Gerzanich et al., 2019 [50] | Y | ↑ with time (100 h) GFAP+ penumbra (prominent) | Martinez-Valverde et al., 2015 [62] Gerzanich et al., 2019 [50] |
| Microvessels | Y | 6–12 h (core) | Gerzanich et al., 2019 [50] | Y | stably ↑ GFAP- core | Martinez-Valverde et al., 2015 [62] Gerzanich et al., 2019 [50] |
| Microglia | Y | 72 h (core) | Gerzanich et al., 2019 [50] | Y | ↑ with time (100 h) GFAP- core | Martinez-Valverde et al., 2015 [62] Gerzanich et al., 2019 [50] |
| TRPM4 | ||||||
| Neurons | Unk | - | - | Unk | - | - |
| GFAP+ Glia (Astrocytes) | Y | 72 h (penumbra) | Gerzanich et al., 2019 [50] | Y | GFAP+ penumbra | Gerzanich et al., 2019 [50] |
| Microvessels | Y | 24 h | Gerzanich et al., 2019 [50] | Y | GFAP- core | Gerzanich et al., 2019 [50] |
| Microglia | Y | 72 h (core) | Gerzanich et al., 2019 [50] | Y | GFAP-core GFAP+ penumbra (some) | Gerzanich et al., 2019 [50] |
| KIR6.2 | ||||||
| Neurons | Unk | - | N | - | Castro et al., 2018 [51] Gerzanich et al., 2019 [50] | |
| GFAP+ Glia (Astrocytes) | Y | 72 h (penumbra) | Gerzanich et al., 2019 [50] | Y | Unk GFAP+ penumbra | Castro et al., 2018 [51] Gerzanich et al., 2019 [50] |
| Microvessels | N | - | Gerzanich et al., 2019 [50] | N | - | Castro et al., 2018 [51] Gerzanich et al., 2019 [50] |
| Microglia | Y | 72 h (core) | Gerzanich et al., 2019 [50] | Mixed Y | - (not different vs. controls) GFAP- core | Castro et al., 2018 [51] Gerzanich et al., 2019 [50] |
| Authors, Year | Study Title | TBI Model | GLI Dose | Results |
|---|---|---|---|---|
| Simard et al, 2009 [61] | Key Role of Sulfonylurea Receptor 1 in Progressive Secondary Hemorrhage after Brain Contusion | Rat Focal Cortical Contusion
|
|
|
| Patel et al., 2010 [60] | GLI Reduces Hippocampal Injury and Preserves Rapid Spatial Learning in a Model of TBI | Rat Cortical Impact Injury
|
|
|
| Zweckberger et al., 2014 [82] | GLI Reduces Secondary Brain Damage After Experimental TBI | Rat CCI
|
|
|
| Xu et al., 2016 [83] | GLI Attenuates Blood Brain Barrier Disruption in Adult Mice after TBI | Mouse CCI
|
|
|
| Stokum et al., 2017 [101] | GLI Pretreatment Protects Against Chronic Memory Dysfunction and Glial Activation in Rat Cranial Blast TBI | Rat Direct Cranial Blast TBI
|
|
|
| Jha et al., 2018 [84] | GLI Produces Region Dependent Effects on Cerebral Edema in a Combined Injury Model of TBI and Hemorrhagic Shock in Mice | Mouse CCI
|
|
|
| Gerzanich et al., 2019 [50] | SUR1, TRPM4 and KIR6.2 Role in Hemorrhagic Progression of Contusion | Rat CCI
|
|
|
| Bramlett et al [128] Deng-Bryant et al Jha et al., (abstracts 2015), [129] Jha et al (in preparation) | Operation Brain Trauma Therapy: GLI Treatment in TBI | Rat CCI Rat FPI I, 1.8–2.1 atm) Rat PBBI |
|
|
| Authors, Year | Study Title | Study Design | Sample Size | Outcome |
|---|---|---|---|---|
| Zafardoost et al., 2016 [133] | Evaluation of the Effect of Glibenclamide in Patients with DAI Due to Moderate to Severe Head Trauma | Randomized Controlled Trial
| N = 40 |
|
| Khalili et al., 2017 [134] | Effects of Oral Glibenclamide on Brain Contusion Volume and Functional Outcome of Patients with Moderate and Severe TBI: A Randomized Double Blind Placebo Controlled Clinical Trial | Double-Blind Randomized Controlled Trial
| N = 66 |
|
| Eisenberg et al., 2019 [135] | Magnetic Resonance Imaging Pilot Study of Intravenous Glyburide in TBI | Phase-2 Randomized Controlled Trial
| N = 28 (N = 14 contusion) |
|
| NCT03954041 | Antagonizing SUR1-TRPM4 To Reduce the progression of intracerebral hematoma And edema surrounding Lesions (ASTRAL) | Multicenter Double-Blind Multidose Placebo-Controlled Randomized Trial (Phase-2)
| Estimated N = 160 |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jha, R.M.; Bell, J.; Citerio, G.; Hemphill, J.C.; Kimberly, W.T.; Narayan, R.K.; Sahuquillo, J.; Sheth, K.N.; Simard, J.M. Role of Sulfonylurea Receptor 1 and Glibenclamide in Traumatic Brain Injury: A Review of the Evidence. Int. J. Mol. Sci. 2020, 21, 409. https://doi.org/10.3390/ijms21020409
Jha RM, Bell J, Citerio G, Hemphill JC, Kimberly WT, Narayan RK, Sahuquillo J, Sheth KN, Simard JM. Role of Sulfonylurea Receptor 1 and Glibenclamide in Traumatic Brain Injury: A Review of the Evidence. International Journal of Molecular Sciences. 2020; 21(2):409. https://doi.org/10.3390/ijms21020409
Chicago/Turabian StyleJha, Ruchira M., Josh Bell, Giuseppe Citerio, J. Claude Hemphill, W. Taylor Kimberly, Raj K. Narayan, Juan Sahuquillo, Kevin N. Sheth, and J. Marc Simard. 2020. "Role of Sulfonylurea Receptor 1 and Glibenclamide in Traumatic Brain Injury: A Review of the Evidence" International Journal of Molecular Sciences 21, no. 2: 409. https://doi.org/10.3390/ijms21020409
APA StyleJha, R. M., Bell, J., Citerio, G., Hemphill, J. C., Kimberly, W. T., Narayan, R. K., Sahuquillo, J., Sheth, K. N., & Simard, J. M. (2020). Role of Sulfonylurea Receptor 1 and Glibenclamide in Traumatic Brain Injury: A Review of the Evidence. International Journal of Molecular Sciences, 21(2), 409. https://doi.org/10.3390/ijms21020409