Abstract
The aim of the manuscript is to discuss the influence of plant polyphenols in overcoming multidrug resistance in four types of solid cancers (breast, colorectal, lung and prostate cancer). Effective treatment requires the use of multiple toxic chemotherapeutic drugs with different properties and targets. However, a major cause of cancer treatment failure and metastasis is the development of multidrug resistance. Potential mechanisms of multidrug resistance include increase of drug efflux, drug inactivation, detoxification mechanisms, modification of drug target, inhibition of cell death, involvement of cancer stem cells, dysregulation of miRNAs activity, epigenetic variations, imbalance of DNA damage/repair processes, tumor heterogeneity, tumor microenvironment, epithelial to mesenchymal transition and modulation of reactive oxygen species. Taking into consideration that synthetic multidrug resistance agents have failed to demonstrate significant survival benefits in patients with different types of cancer, recent research have focused on beneficial effects of natural compounds. Several phenolic compounds (flavones, phenolcarboxylic acids, ellagitannins, stilbens, lignans, curcumin, etc.) act as chemopreventive agents due to their antioxidant capacity, inhibition of proliferation, survival, angiogenesis, and metastasis, modulation of immune and inflammatory responses or inactivation of pro-carcinogens. Moreover, preclinical and clinical studies revealed that these compounds prevent multidrug resistance in cancer by modulating different pathways. Additional research is needed regarding the role of phenolic compounds in the prevention of multidrug resistance in different types of cancer.
1. Introduction
Cancer is one of the leading cause of death worldwide. It is usually caused by genome instability and mutations, which may be inherited, induced by environmental factors or represent a consequence of DNA replication errors [1]. The signature characteristics of cancer are represented by: a high rate cellular multiplication escaping growth inhibitors, cell migration inducing subsequent metastasis, stimulation of local new blood vessel formation (angiogenesis), the capacity to resist cell senescence and death signals leading to inflammation, and an almost unlimited self-replicating capacity [2].
The number of cancer cases is expected to increase rapidly as populations grow, age and adopt negative lifestyle behaviors (smoking, lack of physical activity, Western diet) that increase cancer risk [3,4]. Lung, breast, colorectal and prostate cancer are considered to be the most prevalent types of cancer among population [3].
For women, breast cancer is the most common diagnosed malignancy, followed by cervix or uterine cancer [3]. In Europe, it is estimated that breast cancer affects more than one in 10 women and accounts for more than 28% of female cancers [5]. Risk factors for breast cancer include unmodifiable factors and lifestyle factors. Among unmodifiable factors, age (above 40 years), family history of cancer in first-degree relatives, hormonal profile (late menopause, early menarche), dense breast tissue, race and genetics (mutation in breast cancer susceptibility genes—BCRA1 and BCRA2 genes, TP53, genetic polymorphisms in genes encoding enzymes involved in estrogen metabolism pathways COMT, CYP1A1, CYP1B1, estrogen receptors ERα/ERβ, CYP17A1 and CYP19A1) are of great importance. Lifestyle factors include nulliparity, use of birth control pills, induced abortion or obesity [6,7,8,9,10]. Although breast cancer usually appears in pre- and post-menopausal women, recently new cases have occurred even in young women, below 35 years. This represents a serious concern, due to higher incidence of advanced stages at diagnosis and poorer five-year survival rate [11] compared to older women. Breast cancer represents a heterogeneous disease and it is clinically divided into three basic subtypes: (I) based on the level of expression of estrogen and progesterone receptors, (II) based on the human epidermal growth factor 2 (HER2) and (III) a third subtype, when neither estrogen, progesterone or HER2 is expressed (triple negative breast cancer [12]. Breast tumors expressing hormone receptors (mainly estrogen) are classified as luminal breast type (luminal A and B). Luminal A subtype has a better prognosis compared to luminal-B type, which is more aggressive, has a higher recurrence and an increased expression of growth receptor signaling molecules, such as epidermal growth factor (EGF), fibroblast growth factor (FGF), nerve growth factor (NGF), hepatocyte growth factor receptor (HGFR/MET) and Wnt/β-catenin [13]. Increased growth receptor signaling genes is also observed for triple breast negative cancer [14]. Nowadays, mammography represents the golden standard for breast cancer screening [15].
Lung cancer is the most common cancer in men worldwide, and the fourth most frequent cancer in women [16]. Lung cancer is often divided into four major types due to distinct clinic-pathological features: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), which is further divided into squamous cell carcinoma (SCC), adenocarcinoma and large cell carcinoma [17]. Risk factors for lung cancer include smoking, environmental exposure to tobacco, radon, cooking oil vapors or hormonal factors (mainly in women). Moreover, genetic factors play a major role in lung cancer etiology [18,19,20].
Colorectal cancer is one of the most preventable and treatable cancers if detected early; however, it has a multifactorial etiology. The hallmark of colorectal cancer is the presence of serrated or adenomatous polyps (adenoma) that usually occur in proximal or distal colon [21]. Besides adenomas, patients with colorectal cancer have multiple aberrant crypt foci, which are microscopic mucosal abnormalities involved in early carcinogenesis [22]. Main risk factors include alterations of gut microbiota [23], Western diet [24], obesity, hormonal status or chronic inflammatory bowel diseases [25]. Genetic factors such as mutations in KRAS, BRAF, PI3K genes and polymorphisms in nucleic acid-binding protein 1, laminin γ 1, cyclin D2, T-box 3 are also involved in colorectal cancer etiology [26,27].
Prostate cancer is the second most prevalent type of cancer among men, besides lung cancer. The majority of prostate cancers originate from luminal cells and do not have a neuroendocrine origin [28]. Risk factors for prostate cancer include age, obesity, other diseases (diabetes), lifestyle behaviors (diet, lack of physical activity) and sexually transmitted diseases [29]. Main characteristics of prostate cancer include activation of androgen receptor signaling, elevated lymphocyte infiltration and activation of inflammatory pathways [30].
The above-mentioned cancer types have a common feature, which is represented by multidrug resistance (MDR) to chemotherapeutic treatments [13,28,31]. Due to toxicity and lack of specificity of synthetic MDR agents, recent researches have focused on beneficial effects of natural compounds in overcoming MDR in cancer. According to recent research, polyphenols might overcome MDR through various mechanisms, which will be further discussed in our work [32,33,34,35].
Polyphenols are considered as important dietary components with biological activity due to a wide range of health benefits: antioxidant, anti-inflammatory, anti-carcinogenic, immunomodulatory, etc. [36,37]. Epidemiological studies have shown that intake of food rich in phenolic compounds have chemopreventive effects for cardiovascular, neurodegenerative diseases, cancer, obesity or diabetes [38]. Cancer chemopreventive effects of polyphenols are the consequence of antioxidant capacity, inhibition of proliferation, survival, angiogenesis and metastasis, modulation of immune and inflammatory responses or inactivation of pro-carcinogens [39].
Polyphenols comprise a variety of compounds with a wide range of chemical structures, ranging from single molecules to high molecular weight polymers. Polyphenols have at least one aromatic ring and are classified as flavonoids and non-flavonoids in correlation with the number of aromatic ring [38,40]. Flavonoids share a C6-C3-C6 structural backbone and are further classified into flavones, flavonols, flavanones and flavan-3-ols [38]. Isoflavones, are also members of flavonoids family [38]. Non-flavonoid compounds include phenolcarboxylic acids (hydroxy-benzoic/hydroxy-cinnamic acids), ellagitannins, lignans, stilbenes and other phenolic compounds (curcumin, gingerol) [40]. A selective list of polyphenols, which are frequently studied for overcoming MDR in breast, lung, prostate and colorectal cancer, is presented in Table 1.

Table 1.
Main classes of phenolic compounds with representative members and sources, frequently investigated for overcoming MDR in cancer.
2. Mechanism of Multidrug Resistance in Cancer
Earlier papers reported only few mechanisms responsible for MDR in cancer (Figure 1), such as (i) increased drug efflux through membrane pumps, (ii) detoxification mechanisms based on glutathione transferases activity, (iii) DNA damage repair that initially may be considered as an ally and further can turn into a resistant tool, and (iv) drug inactivation [52]. However, recent papers described extended lists of mechanisms responsible for drug resistance in malignancy (Figure 1) such as modification of drug target, inhibition of cell death, involvement of cancer stem cells, tumor heterogeneity, tumor microenvironment, epithelial to mesenchymal transition, epigenetic variations, dysregulation of miRNAs and modulation of reactive oxygen species [53,54,55].
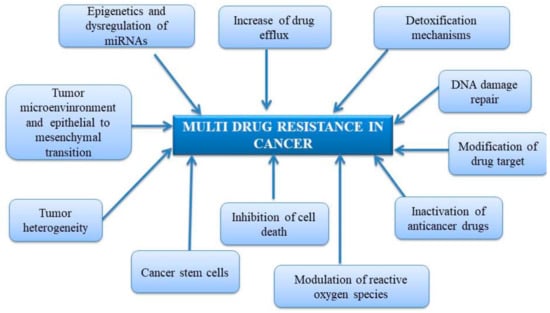
Figure 1.
Mechanisms of multidrug resistance in cancer.
2.1. Increase of Drug Efflux
At the plasma membrane level, the large family of ATP-binding cassette (ABC) transporter proteins is responsible mainly for the drug efflux [56]. ABC transporters consist of two transmembrane domains and two intracellular nucleotide-binding domains. It is the nucleotide binding domains that bind ATP and hydrolyze it to ADP providing the plasma membrane pump with energy required to export xenobiotic compounds [57]. There are 49 known ABC genes organized in subfamilies, from A to G, respectively 12 ABCA, 11 ABCB, 13 ABCC, 4 ABCD, 1 ABCE, 3 ABCF and 5 ABCG [58]. The most studied ABC transporters are multidrug-resistance protein 1 (MDR1)/permeability-glycoprotein (P-pg)/ABCB1, MDR-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP)/ABCG2 [56,59]. The majority of ABC transporters are localized in the liver, kidney, intestine, but they can have ubiquitous localization as well [56,59,60].
High levels of MDR1 are expressed in colorectal cancer [61], hepatocarcinoma [62], breast cancer [63], lung cancer [64] or prostate cancer [65]. Overexpression of ABC transporters in cancer is mediated by (i) increased activity of proteins involved in the MAPK (HRas, ERK1/2, JNK), PI3K/AKT, mTOR, JNK, PKC signaling pathways, (ii) activation of EGF/FGF growth factors [54,66,67,68], (iii) nuclear localization of Y-box binding protein 1 (YB-1) in solid tumors [69,70], (iv) increased COX-2 activity [71], (v) activation of VEGF2 (vascular endothelial growth factor receptor 2) by VEGF in tumor microenvironment [70], (vi) activation of nuclear receptors PXR and CAR [72,73,74] and (vii) hypoxia [75]. According to recent studies inhibition of ERK1/2, NF-κB pathways and increased sensitivity to all-trans retinoic acid (a ligand of retinoic acid receptors RARs) render cancer cells more sensitive to chemotherapeutic agents, due to reduced P-gp mediated efflux activity [54,76,77].
Moreover, extensive studies have shown a strong correlation between ABC transporters activity and TP53 tumor suppressor gene [78,79]. It is well known that TP53 mutations occur in almost 50% of cancers and are involved in inhibition of apoptosis [80]. According to Sullivan G. and his co-workers TP53 mutations become increasingly frequent as prostate cancer advances in stage and this is strongly correlated with increased MRP1 expression [79].
Several chemotherapeutic agents (doxorubicin, daunorubicin, vincristine, vinblastine, actinomycin D, paclitaxel, docetaxel, etoposide) and molecular targeted anticancer compounds (i.e., tyrosine kinase inhibitors, such as imatinib, erlotinib, sunitinib) are substrates for MDR1 [81,82,83,84] and this fact has negative impact on drug efflux in malignant cells. In this context, many attempts have been reported to overcome MDR.
Two main strategies have been employed to prevent drug resistance mediated by ABC protein transporters, namely (i) co-administration of MDR1 inhibitors with chemotherapeutical drugs with the aim to increase intracellular accumulation of drug and (ii) substrate competition by co-administration of MDR1 substrate together with the anticancer drug [85]. Some of the first modulators of MDR1 identified are calcium influx blockers (i.e., verapamil, nicardipine nifedipine), which increased the cytotoxicity of anticancer drugs in cancer cell lines [86,87,88]. Regrettably, the results from preclinical studies were difficult to apply in clinical trials for several reasons (i) necessity of higher concentrations, which in turn induced systemic toxicity, (ii) low selectivity and specificity due to the expression of the target in different tissues or (iii) low efficiency due to functional redundancy of ABC protein transporter family [85]. Recently, PPAR δ ligands (rosiglitazone and pioglitazone) were found to inhibit drug resistance in breast cancer cells by internalization of ABCG2 to cytoplasm [89]. Further research studies are needed to understand the molecular mechanism and to identify the optimal doses of MDR1 inhibitors for the development of new inhibitors of ABC protein transporters.
2.2. Detoxification Mechanisms and Inactivation of Anticancer Drugs
Downregulation or mutations in the proteins or enzymes involved in activation of chemotherapeutic agents can be responsible for drug resistance [90]. For example, in tumor cells resistant to capecitabine, the gene responsible for the synthesis of thymidine phosphorylase, an enzyme responsible for generation of the nucleotides, can be inactivated by hypermethylation [91]. Carbonyl reduction of doxorubicin induced by aldo-keto reductase is responsible for transformation of doxorubicin into doxorubicinol, which is an inactive form. Administration of both chemotherapeutic drugs and inhibitors of aldo-keto reductase is recommended to overcome inactivation of doxorubicin and to increase its therapeutic activity [92].
Other important pathways of drug inactivation involve the CYP450 system (mainly CYP2B6, CYP2C9, CYP2C19, CYP2D6), glutathione-S-transferase (GST) superfamily or uridine diphospho-glucuronosyltransferase (UGT) superfamily [54]. For example, CYP2D6 polymorphism is involved in tamoxifen variability among patients with breast cancer, since CYP2D6 is involved in tamoxifen metabolization to 4-hydroxytamoxifen and endoxifen, both of which display higher anti-estrogenic activity [93]. Some of the first reports, reconfirmed later on, indicated that resistance to platinum could occur through drug inactivation by thiol glutathione, which activates the detoxification system (GST) [94,95]. It was reported that resistance to other chemotherapeutic agents (doxorubicin, tamoxifen, epirubicin), commonly used to treat breast cancer, is mediated by the polymorphisms in UGT superfamily [96].
2.3. DNA Damage Repair
Several chemotherapeutic drugs interfere with DNA synthesis with the aim to induce senescence, apoptosis or cell cycle arrest in cancer cells [97]. DNA-damaging compounds with anticancer properties can act through different mechanisms such as inducing DNA crosslinking (i.e., cisplatin, carboplatin, oxaliplatin), preventing DNA synthesis (i.e., antimetabolites that inhibit the activity of dihydropholate reductase) or inhibiting topoisomerase activity (i.e., doxorubicin, daunorubicin) [98]. Nevertheless, these compounds do not have a specific tumor target and the selectivity of anticancer drugs is based on the rate of cell cycling. Tumor cells have a rapid cycling compared to normal cells and DNA damage response proteins (DDR) do not have enough time to repair DNA lesions [99]. The major mechanisms of DNA repair pathways in response to chemotherapy are elegantly and thoroughly explained elsewhere [99]. Briefly, these processes include (i) mismatch repair (MMR) mechanisms which remove mis-incorporated nucleotides during DNA replication [100]; (ii) nucleotide excision repair (NER) which removes bulky DNA lesions, such as DNA adducts [101]; (iii) base excision repair (BER) that corrects small base lesions which occur after DNA damage produced by oxidation, deamination or alkylation [102]; (iv) homologous recombination (HR) which repairs DNA double-stranded breaks and inter-strand crosslinks [103]; (v) non-homologous end-joining (NHEJ) with the aim to repair double-stranded breaks [104].
Recent reports demonstrate that MDR to platinum drugs in cancer cell lines, implicates multiple DDR pathways including HR, transcription-coupled NER and BER [105]. MutL homolog 1 (MLH1) and MutL homolog 2 (MLH2)—proteins belonging to MMR system—have been evaluated by immunohistochemistry from patients with colorectal cancer and 10% of these patients presented MMR deficiency. Administration of 5-fluorouracil induced the improvement of survival only in patients without MMR deficiency, demonstrating the association between dysregulation in MMR processes and multidrug resistance [106]. Due to constantly improving technology, the researchers might carry out genomic screening with the aim to identify potential DNA therapeutic targets responsible for MDR in malignancies.
2.4. Modification of Drug Target
A drug’s efficacy strongly depends on its molecular target. Alteration of these targets by means of different mechanisms (i.e., mutations) may lead to drug resistance [54]. One of the most studied mechanisms of drug resistance in respect with modification of the drug target is focused on epidermal growth factor receptor (EGFR) [107]. In non-small-cell lung cancer (NSCLC) activation mutations of EGFR in the tyrosine kinase domain had been identified. Small molecule inhibitors such as gefitinib and erlotinib are known to neutralize these modifications [107]. Nevertheless, after two years of gefitinib treatment the disease can relapse, due to occurrence of secondary mutation (T790M) in EGFR [108]. Second generation of EGFR tyrosine inhibitors (i.e., ponatinib) had been created to act against EGFR(T790M), but increased toxicity caused withdrawal of the drug from the market [109]. Due to ability of cancer cells to survive by occurrence of additional mutations, new generations of tyrosine kinase inhibitors (TKI) against EGFR or other molecular targets are needed to be developed to overcome MDR and side effects associated with anticancer therapy.
2.5. Inhibition of Cell Death
Cancer cells escape cell death using several mechanisms such as dysregulation of apoptosis, inhibition of other non-apoptotic processes (i.e., autophagy, etc.) or stimulation of alternative survival pathways [53]. The most studied mechanisms, which allow cancer cells to evade cell death and to acquire MDR, are the disturbance of apoptosis and inhibition of autophagy. The main proteins involved in apoptosis are the caspases, which can be activated by both intrinsic (in the mitochondria) and extrinsic (through tumor necrosis family factors that bind to cell death receptors) pathways [93,110,111,112].
Mechanisms of drug resistance due to apoptosis deregulation include: (i) imbalance of Bcl-2 family members (downregulation of pro-apoptotic proteins Bax and upregulation of anti-apoptotic proteins BCL-XL, BCL-2), (ii) altered apoptotic regulators (downregulation of caspase−3, −8, −9 and upregulation of inhibitors of apoptosis proteins such as XIAP, FLIP, survivin), (iii) upregulation of ubiquitin binding proteins (sharpin), which regulates Bcl-2 and survivin [113], (iv) decreased activity of p53 and PTEN [80,90,93], (v) decreased activity of cytochrome C and Smac/DIABLO (which are responsible for caspases activation) [114,115], (vi) deregulated activity of cyclin-dependent kinases (CDK), protein tyrosine kinases (Her2,/neu, Her3, Her4) [116] or different signaling pathways (GSK-3; STAT3, PI3K/AKT, mTOR) [115,117,118] or (vii) amplification of gene expression of CYCLINS (A1, D1) [119]. Checkpoint kinases (Chk1, Chk2), which are modulated by serine/threonine protein kinases (ATR), also play a major role in apoptosis since they promote activation of p21 and p53, which induce cell cycle arrest [120].
Autophagy is involved in MDR through increased activity of AMP-protein kinase (AMPK), beclin-1 and activation of autophagy lysosomes systems (ALP) [75,93]. ALP in most tumors may enhance the MDR phenotype through a protein clearance mechanism [75]. Elevated autophagy lysosomes systems are involved in EGFR inhibitors (gefitinib, erlotinib), mTOR inhibitors (temsirolimus) or targeted therapy (imatinib) chemoresistance [75].
It is reasonable to assume that genes, mRNA and proteins involved in disturbed apoptotic and autophagy processes are considered optimal targets to overcome multidrug resistance in malignant tumors. Against anti-apoptotic BCL-2 proteins both antisense oligonucleotides (i.e., oblimersen sodium) that target BCL-2 mRNA and small molecules which can interact with BH3 domains have been developed [121,122]. The last category might be divided in small molecules with BH3 mimetic activity (i.e., ABT-737, navitoclax/ABT-263/oral version of ABT-737) and small molecules with BH3 putative mimetic action (i.e., gossypol, obatoclax/a pan-BCL-2 inhibitor, etc.) [123].
Nevertheless, several mechanisms of drug resistance developed by cancer cells hindered the successful application of anti-apoptotic drugs in patients. For instance, clinical studies on combinatorial administration of several chemotherapeutics (i.e., dacarbazine, fludarabine, cyclophosphamide) and oblimersen did not bring favorable results in patients [122,124]. Polymorphism of BCL-2-like protein 11 (BIM) with different splicing variants resulted in lack of BH3 domain and resistance to targeted therapy in NSCLC positive for EGFR [125].
Stimulation of pro-apoptotic death receptors (i.e., DR4, DR5) localized in plasma membrane demonstrated in vitro and in vivo anti-proliferative activity, but clinical results have been unsatisfactory [126,127]. Nevertheless, preclinical experiments with the aim to test synergism of combinatorial administration of death receptors agonists and other anti-cancer drugs are under evaluation [128,129]. Recently, inhibitors of CDK (roscovitine, terameprocol, flavopiridol) are under investigation in different MDR cancers [116].
Moreover, it was shown that PPAR δ agonists (rosiglitazone) sensitizes colorectal cancer cells to 5-FU by downregulation of Bcl-2 proteins and upregulation of Bax [130]. Inhibition of ALP using chloroquine and hydroxychloroquine is also under investigation in both preclinical and clinical studies [75].
Further preclinical experiments and successful clinical trials are needed to better understand the molecular mechanisms of anti-apoptotic/autophagy processes and to circumvent the drug resistance in cancer cells.
2.6. Cancer Stem Cells
There is increasing evidence that cancer stem cells (CSCs), a subpopulation of cells within the heterogenous tumor niche, are responsible for initiation of some primary tumors as well as metastasis and MDR [90,93]. CSCs are resistant to chemotherapy and radiotherapy given to their particular characteristics such as increased DNA damage repair, resistance to cell death mechanisms, evasion from immune response, adaptation to hypoxia and overexpression of MDR efflux pumps [93,131]. Several lines of action have been developed to overcome drug resistance in cancer stem cells. These include (i) new inhibitors against ABC transporters, (ii) antibodies conjugated with toxins or radioisotopes against ABC transporters, (iii) inhibitors of signaling pathways identified in cancer stem cells (i.e., Hedgehog signaling pathway) or (iv) activation of immune system against cancer stem cells [131,132]. In spite of the extensive efforts to address drug resistance in cancer stem cells there are still open questions needing to be answered. For instance, how is it possible that ABC transporters or Hedgehog signaling pathways can be targeted only in cancer stem cells and not in normal stem cells? In addition, recent papers underline the contribution of cancer niche as a crucial factor in drug resistance of CSCs [133,134]. Cancer associated fibroblasts stimulated 5-fluorouracil resistance in colon CSC by activating Wnt signaling [135] or autocrine generation of inflammatory factors, such as interleukin-6 induced trastuzumab resistance in HER2 positive breast CSC [136]. Besides addressing ABC transporters as therapeutic targets, CSC niche could represent a potential objective in further anticancer approaches with the aim to overcome MDR.
2.7. Tumor Heterogeneity
Genetic instability allows survival of the best adaptable clonal populations of malignant cells, and this heterogeneity represents one of the reasons for the failure of anticancer therapy [137,138]. It is already recognized that tumor heterogeneity implies two distinct types of processes, (i) tumor inter-heterogeneity, with tumors affecting the same organ, but with different characteristics in each patient, and (ii) tumor intra-heterogeneity, with two branches, spatial and temporal heterogeneity [139]. Spatial heterogeneity is present in the same patient and it is characterized by different genotypes and phenotypes of the malignant clones in the primary and metastatic sites, while temporal heterogeneity expresses the changes which are taking place in the same tumor over the time [139]. In cancer cells overexpressing hepatocyte growth factor receptor (HGFR/MET), heterogeneity occurred as a molecular mechanism of drug resistance after chemotherapy [140]. Thus, after two years of targeted therapy against MET, two additional changes have been identified, KRAS mutation and co-amplification of HER2 and/or EGFR genes [140]. Chronical administration of the chemotherapeutic drugs demonstrated that in one or two years the diseases relapsed due to the ability of cancer cells to generate new clones and to find alternative pathways to survive and proliferate [108,141].
In vitro and in vivo experiments have been performed to identify the culprit molecules or alternative pathways that confer drug resistance [142,143]. Escape of human epidermal growth factor receptor type 2 (HER2) from the inhibition with tyrosine kinase inhibitor (TKI) through alternative HER3 activation has been demonstrated in mammary cancer cell lines [142]. Not only in case of chemotherapy, but also in case of hormone therapy the existence of adaptive mechanisms and acquired resistance has been reported [144,145]. Increased survival and reduction of prostate serum antigen (PSA) levels are described after androgen deprivation by enzalutamide in prostate cancers [146]. However, secondary mutations are identified in castration-resistant prostate cancers after administration of enzalutamide [147]. Similar to hormone therapy against prostate cancer, first results about administration of tamoxifen in estrogen receptor (ER) positive breast cancer patients have been promising and there are recommendations to increase the administration from five to 10 years [148]. Notably, chronical administration of hormone therapy can cause resistance and most frequently alternative signaling pathways activated in estrogen resistant breast cancer are plasma membrane tyrosine kinase receptors, such as EGFR, HER2, IGF-1R or downstream kinases, such as ERK1/2, PI3K/AKT [144,149].
Increased exposure of the malignant cells to different anticancer agents amplifies the heterogeneity of the tumor and several overcoming therapies against drug resistance are proposed [139]. These include (i) combination therapy against single target (i.e., TKI afatinib against EGFR and monoclonal antibody cetuximab against EGFR) [150] or against multiple targets (i.e., a third generation TKI of EGFR(T790M) and navitoclax an inhibitor of ABC transporters) [151]; (ii) sequential therapy to reduce the toxicity induced by combination of chemotherapeutic agents [152] or (iii) targeted therapy after identification of genetic markers (i.e., patients with EGFR(T790M) mutation which can benefit from osimertinib treatment compared to patients with activating mutations in EGFR who can benefit by gefinitib/erolotinib/afatinib administration) [153]. New experimental studies and different therapeutic approaches are required to find the optimal way to interfere with development of tumor malignancy.
2.8. Tumor Microenvironment (TME)
In spite of the fact that TME is formed from non-malignant structures (i.e., cancer associated fibroblast, immune cells, adipocytes, extracellular matrix molecules, blood and lymphatic vessels, and mesenchymal cells), in most cases they are considered as tumor-promoting factors [154]. Main mechanisms involved in TME role in MDR are (i) abnormal tumor vasculature (promotion of angiogenesis and overexpression of VEGF), (ii) hypoxia, (iii) decreased pH (due to glycolysis), (iv) alterations in the expression of tumor suppressors and oncogenes [155,156,157,158] and (v) modulation of different signaling pathways (mTOR, ERK1/2) and growth-factors (FGF) [159]. Among TME factors, hypoxia plays a major role in lung, colorectal, breast and prostate cancers MDR [155,160,161,162,163]. Hypoxia induces HIF-1 (hypoxia-inducible factor 1) in tumor cells, upregulates the release of pro-angiogenic factors, increases the expression of growth-factor receptors (CXCR4) and MDR proteins (P-gp) [164]. Moreover, the relatively low pH values—a direct consequence of hypoxia—are responsible for reduced cellular uptake of chemotherapeutic agents [165].
Other important factors of TME which promote MDR are the overexpression of fatty acid synthase (FASN) and fatty acid-binding proteins (FBAP4, FBAP5, FBAP9) in breast/prostate tumor cells [166,167]. FSAN is required for de novo synthesis of fatty acids and is correlated with poor prognosis of cancer [166]. Overexpression of FASN may induce drug resistance by (i) altering the membrane composition, thus decreasing the influx of chemotherapeutic agents; (ii) upregulation of HER2 or (iii) inhibition of apoptosis [168,169].
According to recent research, the cellular components of the tumor stroma (fibroblasts, infiltrated immune cells or mesenchymal stromal cells) induce MDR through increased expression of cytokines (IL-6, IL-8, IL-18, IL-17), overexpression of HER2 and loss of PTEN (tumor suppressor gene) activity [54,170,171,172]. To date several small molecule inhibitors and antibodies against tumor stroma are in clinical trials (prinomastat, saridegib, bevacizumab, etc.) [171].
2.9. Epithelial to Mesenchymal Transition (EMT)
Tumor microenvironment plays a major role in cancer cells ability to develop further features such as cell transition from epithelial to mesenchymal phenotype. This transformation gives them the advantage to migrate to secondary sites [173]. EMT is considered to be an important mechanism by which tumors become metastatic and multidrug-resistant [54,174]. Drug resistance developed after administration of EGFR-target therapy (i.e., erlotinib and cetuximab) has been reported to be connected with EMT features [175].
The PI3K/AKT is one of the most important signaling pathways that mediates the process of EMT through (i) direct activation of transcription factors (twist 1, 2) which increases the expression of mesenchymal markers (N-cadherin), decreases the expression of epithelial markers (E-cadherin, claudin, occluding) and upregulates AKT gene, which is involved in drug resistance in breast cancer, (ii) increased activity of integrin-linked kinase (which downregulates E-cadherin) and (iii) activation of matrix-degrading proteases (MMP2, MMP9) [55,174]. Moreover, other factors are also involved in EMT activation such as growth factors (FGF, EGF, TGF-β), adhesion molecules (ICAM-1), signaling pathways (NF-κB, Wnt/β-catenin, Notch), overexpression of EMT transcription factors (slug, snail) and members of heat-shock proteins family (such as glucose regulated protein 78 (GRP78)) [53,172,174,176]. Notably, due to the correlation between drug resistance and acquisition of EMT phenotype (i.e., EMT modified cells appear similar to CSC as a result of their high levels of ABC transporters), targeting EMT might represent a new toll to circumvent drug resistance in cancer [177].
2.10. Epigenetic Variations
The main types of epigenetic mechanisms involved in cancer drug resistance are DNA methylation and histone alterations [54]. Aberrant DNA methylation is associated with genes encoding for proteins involved in cell differentiation, proliferation, apoptosis (MAPK, VEGF, Wnt/β-catenin, p15, p16, p53, APAF-1) or genes encoding drug transporters (MDR1) [90,93]. Moreover, epigenetic mechanisms can also affect the DNA repair system, since hypermethylation of hMLH1 gene is responsible for colorectal cancer [90].
Recently several studies revealed the important role of epigenetic regulator, polycomb repressive complex 2 catalytic component enhancer of zeste homolog 2 (EZH2), in neoplastic development and drug resistance in many types of cancer (gastric, lung, hepatic) [178]. According to Chang and co-workers, overexpression of EZH2 upregulates EMT transition and decreases sensitivity to several chemotherapeutic agents (i.e., cisplatin) [179]. Since epigenetic alterations might represent a viable anticancer and anti-drug resistance target, a large series of DNA methylation or histone deacetylases inhibitors have been generated. These comprise nucleoside analogs (i.e., 5-Azacytidine, zebularine) or non-nucleoside analogs (i.e., hydralazine) against DNA methylation or short fatty acids, hydroxy-cinnamic acids, cyclic tetrapeptides and benzamide against histone deacethylases [180,181]. Notably, a disadvantage of the drugs that act against epigenetic modification consists in lack of specificity. However, their systemic administration can activate oncogenes, which are involved in promotion of malignancy [182]. Besides the epigenetic inhibitors used to overcome drug resistance, Baylin proposed a mechanism based on withdrawal of the chronical drug administration, which in turn will reduce the number of cancer cells with epigenetic modifications and will increase the heterogeneity of the tumor cells, making them sensitive to other anticancer therapies [183]. All these studies and challenges make epigenetic alterations attractive candidates for further therapeutic applications.
2.11. Dysregulation of microRNA (miRNAs)
miRNAs are a family of small single-stranded non-coding RNAs of 20–25 nucleotides. Usually, their main function is downregulation of gene expression at post-transcriptional level [184]. The dysregulation of miRNAs in cancer cells can lead to drug resistance by abnormal modulation of genes expression responsible for MDR, such as (i) ABC transporter genes, (ii) genes related to apoptosis and autophagy, (iii) drug metabolism genes, (iv) DNA repair or (iv) redox system relating genes [93,184].
Regarding miRNAs role in regulation of MDR transporters, it was shown that downregulation of miR-38 and miR-200c led to doxorubicin resistance in breast cancer cells, through upregulation of BCRP protein [93]. Downregulation of miR-7 led to drug resistance in lung cancer, through upregulation of MRP1 [93]. Upregulation of several miRNA (miR-16, miR-17) sensitize resistant lung cancer cells to paclitaxel treatment through inhibition of beclin 1 and Bcl-2, promoting apoptosis. Moreover, it was shown that downregulation of miR-17-5p sensitizes colorectal cancer cells to chemotherapeutic agents (5-FU), through increased activity of PTEN [93].
miRNAs are also involved in chemotherapeutic agents metabolism; for example miR-27b negatively regulates CYP1B1 expression, while miR-892a regulates CYP1A1 activity and sensitize cells to a wide spectrum of chemotherapeutic agents [184]. Moreover, it was shown that miR-27a contributes to cisplatin resistance by modulation of GSH biosynthesis [184]. Several miRNAs modulate chemosensitivity of cancer cells through interfering with DNA repair mechanisms. For example, over–expression of miR-21 downregulated the expression of mismatch repair (MMR) proteins, thus reducing the therapeutic effect of 5-FU in colorectal cancer cells [184]. In conclusion, miRNAs can serve as therapeutic agents for overcoming MDR [90].
2.12. Modulation of Reactive Oxygen Species (ROS)
Modulating reactive oxygen species (ROS) represent a challenging approach to reverse MDR in cancer cells. It is well known that ROS level and the activity of antioxidant enzymes (glutathione peroxidase—GPX, glutathione-S-transferase, catalase, superoxide-dismutase—SOD, hem-oxygenase 1, NAD(P)H quinone oxidoreductase 1, glutamate/cysteine antiporter solute carrier family 7 member 11—xCT, etc.) in MDR cancer cells are overexpressed compared to non-MDR cells [185,186]. Overexpression of ROS facilitate MDR, through upregulation of different pathways (i.e., MAPK, JNK, Nf-kB, PI3K/AKT, Keap1-Nrf2-ARE) [55,75,185]. According to recent research, cancer cells expressing Nrf2 are resistant to chemotherapeutic agents (doxorubicin, etoposide, cisplatin) by increasing GSH production and upregulation of MRP1 [75]. According to Zeng et al., the transcriptional factor src/STAT3 also promotes MDR in cancer cells by promoting antioxidant feedback, through increased expression of GPX and SOD2 activity [187].
Usually ROS are produced by the highly reactive mitochondrial electron transport chain of aerobic respiration, oxido-reductase enzymes (xanthinoxidase, cyclooxygenase, NADPH oxidases—NOXs, etc.) or metal catalyzed oxidation [185]. Recent research has shown that mitochondrial functions are altered in cancer cells, due to imbalance between fusion/fission dynamics and increased mitophagy, which grants a rapid clearance of chemotherapeutic agents, increases ABC transporters activity (by providing ATP) and modifies mitochondrial membrane potential [75].
Several agents (current in preclinical or clinical studies) are involved in modulation of ROS in MDR by (i) disrupting mitochondrial electron transport chain (elesclomol), (ii) inhibition of NOXs (ampelopsin), (iii) depletion of intracellular GSH (APR246), (iv) inhibition of xCT, required for GSH synthesis (erastin, vorinostat) or (v) inhibition of Nrf2 pathway (camptothecin) [185].
3. Role of Polyphenols in MDR
3.1. In Vitro Studies
3.1.1. Flavonoid Compounds
Flavones
Flavonoid compounds were intensively tested for their capacity to enhance the effect of anti-cancer drugs and to combat MDR in different types of cancers. An experiment conducted on CD44+ prostate cancer stem cells provided relevant information that apigenin co-administrated with cisplatin stimulated the therapeutic effects of cisplatin by inducing a series of modulatory effects on the expression of essential proteins and enzymes [188]. The mechanism of apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) was studied in conjunction with flavonoids as potentiating agents, due to the high occurrence of TRAIL resistance in various cancer types. In this regard, Yang et al. demonstrated that wagonin showed the capacity to enhance apoptosis mediated by TRAIL in vitro through downregulating the expression levels of anti-apoptotic proteins [41].
According to Rao et al., luteolin overcomes MDR in breast cancer mitoxantrone resistant cells through increased apoptosis, DNA damage, activation of ATR/Chk2/p53 signaling pathways, inhibition of NF-κB signaling pathway and depletion of anti-apoptotic proteins [189].
Nucleoid factor erythroid-2 related factor 2 (Nrf2) is a transcription factor that regulates genes responsible for the synthesis of endogenous antioxidants (hemeoxygenase-1—HO-1), transporters (MRP1, MRP2) and detoxifying enzymes (glutathione-S-transferase) [190]. Recent research have demonstrated that Nrf2 is overexpressed in MDR cancer [190]. According to recent data, co-treatment of breast and lung cancer cells with luteolin and chemotherapeutic agents (oxaliplatin, doxorubicin, bleomycin) resulted in a higher percentage of cells death. The suggested mechanisms involve downregulation of NRF2 gene expression (MDR and HO1) and increased sensitization of the cells to chemotherapeutic treatment [190,191].
Flavonols
Quercetin was found to suppress effects of P-gp in breast cancer cells and to increase the disappearance of breast cancer stem cells. In this case, doxorubicin-resistant MCF-7 cells were evaluated for how they respond to different drugs (doxorubicin, paclitaxel and vincristine) in conjunction with quercetin. It was found that the co-administration of these drugs with quercetin potentiated their chemotherapeutic effect [192]. The potential of quercetin to reverse the MDR process through the inactivation of P-gp was also revealed on vincristine resistant human colorectal adenocarcinoma Caco-2 cells [193]. Another study performed on Caco-2 cells showed that quercetin as well as naringenin and genistein manifested inhibitory effects on cell elimination of cimetidine through P-gp activity [194]. Downregulation of P-gp by quercetin and other flavonoids such as naringenin, biochanin A, silymarin, genistein was successfully demonstrated in daunomycin resistant MCF-7 breast cancer cell lines. It was shown that these compounds not only stimulated the accumulation of the drug, but also substantially reduced its efflux [195].
It was also observed that fisetin—another dietary flavonoid compound—changed the MDR course of action leading to the chemosensitizing effects on colorectal cancer cells resistant to common chemotherapeutic drugs. Co-administration of fisetin with irinotecan and oxaliplatin induced apoptosis in cultured cells by increasing the activity of caspase-8 and caspase-3. Furthermore, this combined treatment triggered the efflux of cytochrome C and considerably reduced the phosphorylation mechanisms of IGF1R and AKT [196].
Flavanones
Hesperidin (hesperitin rutinoside) was able to increase the sensitivity of breast resistant cancer cells to doxorubicin, through decreased expression of P-gp [197]. Moreover, El-Readi M.Z. and his co-workers have shown that hesperidin had a significantly higher inhibitory effect of P-gp than nobiletin and stigmasterol but lower effect than limonin in overcoming MDR in colorectal cancer cells [198].
Flavan-3-ols
Impeding DNA damage repair processes through dietary flavonoids was also shown to be a successful endeavor in combating chemoresistance in cancer. It was found that quercetin, catechin and fisetin intensified the sensitivity of breast cancer cells to cisplatin by inhibiting ATR-Chk1 pathway [199]. Green tea polyphenols have also shown inhibitory properties towards efflux pumps (P-gp) [200]. The inhibitory effect decreased as follows epigallocatechingallate > epigallocatechin > catechin > epicatechin [201]. EGCG induces the reversal of MDR by regulating detoxification mechanisms and downregulation of Nrf2 pathway in breast cancer cells resistant to tamoxifen [202]. Moreover, according to La X. and co-workers, EGCG enhances the sensitivity of colorectal cancer cells to 5-fluorouracil by inhibiting GRP78/NF-κB/miR-155-5p/MDR1 pathway [203]. Green tea polyphenols (EGCG) associated with quercetin enhanced the therapeutic effect of docetaxel in metastatic and castration-resistant prostate cancer through downregulation of MRP expression, decreased percentage of CD44+/CD24− stem-like cells and induced inhibition of PI3K/AKT/STAT3 signaling pathway [204]. Receptor tyrosine kinase signaling pathway has been reported to promote cell proliferation, inhibit apoptosis and to play a major role in MDR. EGCG was shown to reverse MDR in cisplatin resistant lung cancer through downregulation of several receptor tyrosine kinases [205].
Isoflavones
Genistein—an isoflavone found mainly in soybeans—overcomes chemoresistance to doxorubicin in MDR breast cancer cells through increased accumulation of the chemotherapeutic agent, promotion of apoptosis and suppression of HER2 mRNA expression. However, it had no effect on MDR-1 expression [206]. According to Li and co-workers (2005), genistein pre-treatment of prostate and lung cancer cells inhibits NF-κB activity and contributes to increased growth inhibition and apoptosis induced by cisplatin and docetaxel [207]. Another isoflavone, daidzein, found in soybeans, inhibited BCRP and MRP1/2 drug transporters, therefore sensitizing breast cancer cells to chemotherapeutic agents (mitoxantrone, doxorubicin) [208].
3.1.2. Non-Flavonoid Compounds
Stilbenes
Resveratrol is a polyphenol commonly found in red wine and grapes that possesses strong antioxidant and anti-aging properties [209]. According to several studies co-administration of resveratrol and other therapeutic agents (paclitaxel, docetaxel, doxorubicin, rapamycin, gefitinib) reversed MDR in breast, lung and colorectal cancer through enhancement of chemotherapeutic agents bioavailability, increase drug retention time, stimulation of pro-apoptosis mechanisms, cell cycle arrest or downregulation of ABC transporters [209,210,211,212,213,214].
Lignans
Co-encapsulation of honokiol (a lignan isolated from the bark, stem and leaves of Magnolia sp.) and paclitaxel in pH-sensitive polymeric micelles suppressed MDR in breast cancer through downregulation of P-gp expression and increase of plasma membrane fluidity [43]. Moreover, honokiol radiosensitizes colorectal cancer cells due to higher levels of apoptosis (caspase-3 activation, increased Bax/Bcl-2 ratio) and reduced expression of cyclin A1 and D1 [215].
Other lignans, such as schizandrin A, isolated from Schisandra chinensis fruits enhanced chemosensitivity of colorectal carcinoma cells to 5-FU through upregulation of miR-195. In addition, upregulation of miR-195 inactivated NF-κB and PI3K/AKT signaling pathways [48]. Silybin is the major active constituent of silymarin (a mixture of flavonolignans) from milk thistle fruits. According to Molavi et al., silybin treatment of breast cancer cells resistant to doxorubicin/paclitaxel, sensitized cells to chemotherapeutic agents by suppressing the key oncogenic pathways STAT3, AKT and ERK [46]. According to recent research, a combination of flaxseed lignan (secoisolariciresinol) and its metabolite (enterolactone) enhanced the cytotoxic effects of docetaxel, carboplatin and doxorubicin in metastatic breast cancer cell lines, likely by inhibition of fatty acid synthase [47].
Ellagitannins
Ellagitannins and their metabolite, ellagic acid, overcome MDR in cancer, by inhibition of P-gp, MRP and BCRP proteins [216]. Ellagic acid sensitizes human colorectal cancer cells to 5-FU treatment through increased Bax/Bcl-2 ratio, activation of caspase-3 and loss of mitochondrial potential [217]. Ellagitannins and their metabolites play a key role for overcoming MDR in breast resistant cancer cell line [218]. Berdowska et al. have studied the effect of several ellagitannins (agrimoniin, sanguiin-H6, tellimagrandin I, rugosins A, D and pedunculagin) on doxorubicin-resistant breast cancer cells. Among the tested compounds, only sanguiin-H6 showed cytotoxic effects towards resistant MCF-7 cancer cells, probably due to the release of sanguisorbic acid dilactone, which inhibited ABC transporters, thus diminishing the ability of cells to extrude other products of sanguiin-H6 hydrolysis (ellagic acid, depsides), with cytotoxic effects [218].
Hydroxy-Benzoic Acids
Among phenolcarboxylic acids, gallic acid induces apoptosis, enhances the anticancer effect of cisplatin in human lung cancer and reverse MDR [219]. Mechanisms responsible for above-mentioned effects include induction of apoptosis by ROS generation, disruption of mitochondrial membrane potential, increase in the expression of Bax, APAF1, DIABLO and p53 and decrease in the expression of inhibitor of apoptosis protein 3 [219]. In addition, association between gallic acid and ECGC attenuated MDR in doxorubicin-resistant breast cancer cells through a concentration-dependent inhibition of metalloproteinases (MMP-2 and MMP-9). It is well known that metalloproteinases are involved in the degradation of extracellular matrix by metastatic cancer cells [220]. Another mechanism involved in gallic acid overcoming MDR is the inhibition of Src/STAT3-mediated signaling and the decrease in the expression of STAT3-regulated tumor-promoting genes, therefore inducing apoptosis and cell cycle arrest. It is well known that activation of STAT3 signaling pathway is associated with resistance to tyrosine kinase inhibitors, which are frequently used in lung cancer treatment [221].
Hydroxy-Cinnamic Acids
Ferulic acid and caffeic acid isolated from foxtail millet (a Chinese cereal food) reverse MDR in human colorectal cancer cells through decreased expression of MRP1, P-gp and BRCP [222].
Caffeic acid phenetyl ester (CAPE) is a strong inhibitor of human breast cancer stem cells by inhibition of cells’ renewal, progenitor formation and decrease in CD44+ cells content. CD44+ cells are responsible for tumor formation from a very few cells and are resistant to chemotherapy [223]. According to Khoram et al., CAPE augments the radio sensibility of breast cancer cells [224]. Moreover, CAPE shows beneficial effect in overcoming MDR in lung and prostate cancer through depleting intracellular stores of GSH (reduced glutathione), blocking NF-κB pathway, downregulation of apoptosis inhibitors (cIAP1, cIAP-2 and XIAP) and claudin-2 expression [225,226]. According to recent research, treatment of lung adenocarcinoma derived stem-like cells with cinnamic acid diminishes their proliferation and facilitates their differentiation into CD133 (a marker used for isolation of cancer stem cell population mainly from carcinomas) negative cells [227].
Other Compounds
Curcumin is the major active substance of the culinary spice turmeric (Curcuma longa) and has strong antioxidant, anti-inflammatory and anti-cancer effects [34,50,228,229]. Curcumin has been reported to attenuate oxaliplatin and 5-fluorouracil (5-FU) acquired resistance in colorectal and breast cancer cells through inhibition of NF-κB signaling cascade [230,231]. Moreover, association between curcumin and oxaliplatin downregulated the expression of NF-κB regulated gene products involved in inflammation (CXC-chemokines, which are highly overexpressed due to acquired resistance) and decreased the levels of p65 [230]. Recent research has shown that a curcumin-derivative (difluorinated curcumin) inhibits 5-FU and oxaliplatin resistant colorectal cancer cells through downregulation of miR-21. miR-21 downregulates PTEN, a tumor suppressor gene. Decreased activity of PTEN is involved in resistance to conventional therapy and recurrence of cancer initial treatment [232]. Moreover, PTEN downregulates Nrf2 activity and autophagy, which have been reported to play a protective role in cisplatin induced apoptotic cell death [233]. According to Gu et al., nanomicelles loaded with doxorubicin and curcumin alleviate MDR in lung cancer, due to increased cellular uptake of chemotherapeutic agents [234]. According to recent studies, curcumin reverses cisplatin resistance and promotes human lung adenocarcinoma apoptosis through increased apoptosis and down-regulation of HIF-1α [235]. It has been shown that curcumin inhibits mammalian target of rapamycin (mTOR)—a serin/threonine kinase—and downregulates the key epigenetic regulator enhancer of zeste homolog 2 (EZH2) in tamoxifen resistant breast cancer cells [236]. According to Thulasiraman, curcumin also restores sensitivity to retinoic acid in triple negative breast cancer cells by suppressing the expression level of fatty acid-binding protein 5 (FBAP5) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ) [237]. The combination of curcumin with other phenolic compounds (such as EGCG) showed synergistic effects in overcoming doxorubicin-resistant tumor breast cells through caspase-dependent apoptotic signaling pathways, downregulation of anti-apoptotic Bcl-2 and survivin, and enhancement of cellular incorporation of curcumin [238].
Gingerol represents the main active substance from dry or fresh ginger roots, a popular spice widely used in many diseases (nausea, diarrhea and cancer) [51]. According to Liu Chin-Ming and co-workers, 6-gingerol and 10-gingerol inhibited the proliferation of docetaxel resistant human prostate cancer cells through downregulation of MRP1 and GST [51]. According to recent research, 6-gingerol shows high anticancer potency in cyclophosphamide, 5-FU and doxorubicin-resistant breast cancer MCF-7 cell line, due to its antioxidant activity and regulation of different cellular pathways (Wnt-β catenin or glycogen synthase kinase 3—GSK3) [239].
In conclusion, recent in vitro studies (Table 2) have shown that phenolic compounds overcome MDR in different types of cancer (breast, lung, prostate, colorectal) by inhibition of efflux pumps (P-gp, MRP1, BCRP), increased apoptosis and decreased proliferation of cancer stem cells, increased cellular uptake of chemotherapeutic agents, downregulation of miR-27a, miR-195, miR-21, inactivation of DNA damage repair, decreased expression of anti-apoptotic proteins and modulation of important signaling pathways involved in carcinogenesis (PI3/Akt, Wnt-β catenin, GSK-3, NF-κB, mTOR, Nrf2, ERK, JNK, etc.).
Table 2.
Summary of in vitro experiments.
Considering the evidence provided by in vitro studies, continuous pharmacological research (pre-clinical and clinical studies) is needed in order to verify the potential beneficial effects of polyphenols in vivo and to discover new mechanisms of action for overcoming MDR.
3.1.3. Synergic and Pleiotropic Activity of Polyphenols
Recent data support the hypothesis that combined drug therapy might be more efficient than monotherapy (“one drug-one target” therapy). The synergistic effects of combined administration of polyphenols appears mainly at a molecular level, since they influence different pathways involved in multidrug resistance. For example, association between curcumin and EGCG showed synergistic effect in overcoming doxorubicin resistance in tumor breast cancer cells [238]. The synergistic effect occurs due to inhibition of P-gp expression by EGCG, thus increasing the incorporation of curcumin in breast cancer cells, leading to enhancement of apoptosis and regulation of apoptosis proteins [238]. A similar effect was observed for the association between EGCG and gallic acid in multidrug resistant MCF7/DOX breast cancer cells [220]. The inhibitory effect of EGCG upon P-gp increases gallic acid concentration in cancer cells leading to inhibition of matrix metaloproteinases (MMP-2, MMP-9). Regarding the combination of EGCG and quercetin in docetaxel resistant prostate cancer cells [204], both compounds are strong inhibitors of P-gp [240]. Consequently, both compounds have increased concentrations in prostate cancer cells and act by inhibition of PI3K/AKT, STAT3 signaling pathways and decreased cancer stem cells activity [204]. Since the data regarding the interactions between polyphenols in MDR models are promising but limited, this might represent starting points for future studies.
The pleiotropic effect of the polyphenols has already been acknowledged in the scientific publications [241,242]. Based on the reported data, polyphenols overcome multidrug resistance by affecting different pathways in different types of cancer [243]. For example: (i) quercetin increases apoptosis, inhibits angiogenesis (in colorectal cancer cells) [244], inhibits P-gp activity (in breast cancer cells) [245]; (ii) curcumin down-regulates P-gp and Hsp27, induces autophagy, reduces the markers of cancer stem cells (colon cancer cells) [246,247,248], inhibits the activity of ABCB4 pump, inhibits epithelial-mesenchymal transition (breast cancer cells) [249,250], inhibits JNK pathway, suppresses invasion by inhibition of STAT3 activity (prostate cancer) [251,252] or induces apoptosis (lung cancer cells) [253]; (iii) resveratrol down-regulates the expression of survivin (in prostate cancer cells) [254] and inhibits MAPK kinase in prostate and lung cancer cells [255]; (iv) EGCG inhibits drug efflux (in prostate cancer cells), increases drug concentration in cancer cells by inhibition of enzymes involved in drug metabolism (in colorectal cancer cells), increased ROS production (in colorectal cancer cells)—thus it is responsible for AMPK activation—and induces epigenetic restoration of estrogen receptors through histone modifications (in breast cancer cells) [256]. Nevertheless, based on reported data, some polyphenols can target the same molecule in different cancer cell lines. For instance, resveratrol can downregulate P-gp in breast, lung and colorectal cancer cells [210,211,212]. Taken together these data suggest that polyphenols are able to modulate different signaling pathways being cell-line-specific and to target certain molecules independent of cell type (Table 2).
3.2. In Vivo and Clinical Studies
3.2.1. Flavonoid Compounds
Flavones and Flavonols
Shin et al. published a study centered on the co-administration of tamoxifen with quercetin in rats, showing great evidence of the inhibition of P-gp, MRP2 and BCPR, as well as relevant data, which support the antioxidant property of quercetin through the reduction of CYP3A4 activity [257]. Experiments on animal models confirm the suppressing function of quercetin on ABC proteins involved in MDR.
Co-encapsulation of quercetin and doxorubicin in biotin receptor-targeting nanoparticles was more effectively taken up with less efflux due to downregulation of P-gp expression in nude mice bearing MCF-7 breast cancer cells resistant to adriamycin (doxorubicin) [258]. According to et al., applying wogonin and TRAIL in a mouse model of lung cancer enhances TRAIL’s antitumor activity and overcomes MDR through augmentation of apoptosis and decreased the expression of anti-apoptotic proteins (survivin, XIAP, etc.) [41].
Fisetin showed promising effects in a mouse model of lung cancer and prevented MDR through increased apoptosis and downregulation of AKT and IGFR1 phosphorylation levels [196].
Luteolin, another flavonoid, was analyzed for its potential beneficial role in reversing MDR in cancer. For this purpose, a group of researchers took into consideration the analysis of xenograft tumors of lung cancer, which were treated with luteolin, erlotinib and cisplatin for 15 days. They concluded that the group of mice treated with luteolin and cisplatin showed the most relevant reduction in the tumor mass. Moreover, luteolin was shown to sensitize tumor cells to erlotinib through downregulation of EGFR/PI3K/AKT/mTOR signaling pathway and increased apoptosis [259].
Flavan-3-ols
Combining EGCG with paclitaxel induced significant cell apoptosis in a murine model of breast carcinoma. Moreover, EGCG overcame MDR to paclitaxel by inhibiting GRP78 expression and inhibition of JNK phosphorylation [260]. In a rat model of breast carcinogenesis application of EGCG overcame MDR to paclitaxel through increased apoptosis, decrease of cancer stem cells, decreased VEGF expression and MMP-2 activity [261].
Isoflavones
The potential of genistein to cause inhibition of MDR in lung cancer was intensively studied. One representative case is the assessment of the genistein-cisplatin treatment of non-small cell lung cancer (NSCLC) in xenografted mice models, in order to prove the sensitization of drug-resistant cancer cells via enhanced activity of caspase-3, 8, 10 and suppression of PI3K/AKT activity [262]. The property of genistein to sensitize NSCLC cells was demonstrated for another chemotherapeutic agent, gefitinib. In this respect, it was acknowledged that the combinatory treatment using genistein and gefitinib increased apoptosis and downregulated EGFR and mTOR signaling pathways [263].
3.2.2. Non-Flavonoid Compounds
Stilbenes
Co-encapsulation of resveratrol and paclitaxel in a PEGylated liposome showed effective inhibitory effects in drug-resistant breast tumors in mice through increased cellular uptake of paclitaxel and decreased activity of efflux pumps (MRP, P-gp) [264]. According to Yang et al., resveratrol sensitized colorectal cancer cells to oxaliplatin, mainly by upregulation of miR-34c in correlation with increased levels of p53 and reduction of tumor growth in xenograft experiments [265]. Resveratrol significantly inhibited MDR in nude mouse models inoculated with human non-small cell lung cancer cells by downregulation of survivin and activation of caspase-3 [266].
Hydroxy-Cinammic Acids
Caffeic acid phenethyl ester (CAPE) reverses MDR in breast cancer mouse models due to downregulation of anti-apoptotic and cell proliferation genes, as well as NF-κB transcription factors. Moreover, it decreased MDR1-gene expression, so it might be used as an adjuvant to chemotherapeutic agents (paclitaxel) treatment [267].
Lignans
Podophyllotoxin, a lignan, found in the roots of Podophyllum peltatum L. exhibited significant activity against P-gp mediated MDR tumor cell lines [44]. However, due to its poor solubility, it cannot be used systemically. Nanoparticles composed of poldophyllotoxin and polyethylene glycol with acetylated carboxymethyl cellulose showed beneficial effects in breast and prostate resistant tumor models in mice through enhanced sensitization of tumor cells to chemotherapeutic agents and increased tumor penetration [44]. Moreover, the delivery of nanoparticles was highly selective to the tumors with minimal uptake in other tissues [44]. Another lignan, deoxypodophyllotoxin from the roots of Anthriscus sylvestris exhibited better efficacy to MDR in mouse models for breast cancer than paclitaxel [45]. According to Lou S. and co-workers a multifunctional nanosystem composed of doxorubicin, paclitaxel and silybin controlled drug release, decreased P-gp activity and synergistically inhibited breast tumors growth [268].
Other Compounds
In vivo studies have shown that curcumin sensitizes human colorectal cancer to capecitabine in an orthotopic mouse model, through inhibition of NF-κB, decreased expression of genes enconding for proteins involved in proliferation (COX-2), invasion (MMP-2, ICAM-1), metastasis (CXCR4), angiogenesis (VEGF) and anti-apoptotic gene products (Bcl-2, IAP-1 and survivin) [269]. Other authors reported that curcumin regulates colorectal cancer by inhibiting P-gp in in situ cancerous colon perfusion in a rat model. Inhibition of P-gp enhanced the cytotoxic effects of irinotecan [270]. According to Howells L. and co-workers curcumin also ameliorates oxaliplatin-induced chemoresistance in HCT-116 xenograft tumors by preventing oxaliplatin-induced upregulation of ALDH1 and decreased activity of excision nucleases, by which DNA lesions are repaired [271]. Administration of nanoparticles with docetaxel/doxorubicin and curcumin to mice inoculated with prostate cancer cells, overcame MDR to chemotherapeutic agents through enhanced cellular uptake of chemotherapeutic agents and inhibition of MDR1 and MRP [272,273]. Moreover, it was shown that curcumin decreases doxorubicin cardiotoxicity [273]. Besides, curcumin chemosensitizes prostate cancer cells to gemcitabine by downregulation of MDM2 oncogene through PI3K/mTOR/ETS2 pathway [274]. Cheng et al. investigated the effect of co-administration of curcumin and phospho-sulindac in a mouse xenograft model of human lung cancer. The results were promising, with improved phospho-sulindac pharmacokinetics and higher levels of the chemotherapeutic agent and its metabolites in the xenografts. It was observed that curcumin enhances phospho-sulindac accumulation in cancer tissues through inhibition of P-gp and MRPs [275]. Cui et al. demonstrated that administration of nanoparticles containing a pH-sensitive pro-drug transferrin-poly(ethylene glycol)-curcumin and doxorubicin exhibited higher cytotoxicity and sensitivity in breast cancer xenograft mouse model compared to the chemotherapeutic agent alone [276].
Few studies have investigated the effect of phenolic compounds for overcoming MDR in humans. According to Mahammedi et al., the combination of curcumin with docetaxel and prednisone showed a high-response rate, good tolerability and acceptability by patients with castration-resistant prostate cancer. It was shown that curcumin reverses docetaxel induced NF-κB activation [277]. Association between curcumin and docetaxel showed beneficial effects in women with advanced and metastatic breast cancer. Curcumin/docetaxel combination demonstrated significant anti-tumor activity, decreased levels of VEGF and other angiogenic growth factors (TGF-α). Moreover, curcumin improved docetaxel bioavailability and reversed drug resistance through downregulation of P-gp expression [278].
Taken together, these results shown that phenolic compounds overcome MDR in different types of solid cancer (breast, lung, prostate, colorectal) both in vivo and in clinical studies (Table 3). However, the data regarding clinical studies with polyphenols and multidrug resistance are very scarce. The mechanisms are generally the same, as previously reported for in vitro studies.
Table 3.
Summary of in vivo and clinical experiments.
3.2.3. Bioavailability and Toxicity of the Polyphenols
Although several studies have shown the beneficial effects of some plant polyphenols in overcoming multi-drug resistance in breast, colorectal, lung, prostate, most of the research was performed using only in vitro (cell lines) and in vivo (animal) models. However, data regarding clinical studies with polyphenols for overcoming chemoresistance are scarce. The extrapolation of the results from pre-clinical studies to humans is difficult and risky, keeping in mind that polyphenols bioavailability is complex and influenced by several factors: (i) chemical structure, (ii) liberation from the food/medicinal plant matrix, (iii) gastro-intestinal absorption, (iv) metabolism by gut microbiota, liver, enterocytes, (v) plasma transport, plasma concentration, (vi) distribution and elimination [40,279,280,281]. Polyphenols bioavailability is relatively low, due to low absorption, extensive biotransformation and rapid clearance from the body [281]. Still, polyphenols metabolites (produced by gut microbiota or liver) reach higher plasma concentrations compared to their parent compounds are considered responsible for polyphenols therapeutic effects. Several polyphenols metabolites such as urolithins (ellagitannins metabolites), enterolactone and enterodiol (lignans metabolites), equol (isoflavones metabolite) have shown a chemopreventive role in breast, prostate or colorectal cancer [282,283]. Taken together, clinical studies are imperative in order to demonstrate the beneficial role of polyphenols in overcoming multidrug resistance in various types of cancer.
In spite of promising results from laboratory experiments, implementation of them into the clinical trials might represent a challenge due to higher concentrations used in those studies. Nevertheless, several clinical studies validated the efficiency of polyphenols against different types of solid tumors [284,285,286]. Administration of regular cytostatic drugs is correlated with severe side effects, such as bone marrow modifications (leucopenia, thrombocytopenia, anemia), nausea, vomiting, alopecia, drug extravasation, hepatotoxicity or heart toxicity [287,288]. Conversely, the polyphenols toxicity is greatly reduced and the side effects could be constipation/diarrhea, dry mouth or flatulence [289]. For example, association of curcumin (0.5, 1, 2 g) for seven days prior to FOLFOX (5-fluorouracil, oxaliplatin, folinic acid) chemotheraphy (two-weekly cycles to a maximum of 12 cycles) in patients with colorectal cancer and liver metastasis, led to several side effects. The most common side effects, which were related to curcumin use (not with FOLFOX) were constipation, dry mouth and flatulence. One patient reported severe diarrhea, attributed to curcumin. Diarrhea was treated when curcumin dosage was changed from 2 g to 1 g and the dosage change did not affect the anticancer effect of curcumin [289].
As general considerations, if any of the cytotoxic effects are visible it is recommended to stop the treatment before the irreversible toxic effects occur. In addition, for better toleration of the treatment it is recommended to start the administration when the patient is in good physical condition [290]. Several general recommendations might be taken in account to reduce toxicity of the polyphenols:
(i) Combinatorial treatment. Administration of more than one polyphenols or the use of polyphenols as adjuvants in chemotherapy might reduce the concentration of the polyphenols when administrated. For instance, in human colon cancer cells with P-gp overexpression the synergism between DOX and EGCG/curcumin was demonstrated. Thus, lower concentration of DOX and polyphenols are required when co-administrated compared to single drug administration [291]. Similar synergism was seen in human colorectal cells treated with platinum-based compounds, such as oxaliplatin, cisplatin and EGCG [292].
(ii) Replacement of the natural compound with another one. In a clinical study performed in 49 patients with solid tumors (non-small cell lung cancer, head and neck cancer) the administration of capsules containing a green tea extract (GTE) (standardized in 26.9% total catechins – EGCG – 13.2%; epicatechin 2.2%; epicatechin gallate 3.3%; epigallocatechin 8.3% and 7% caffeine), at increasing dosages up to 8–10 g GTE once daily or 10–13 g distributed over three daily dosages for minimum four weeks to six months, several side effects occurred: nausea, abdominal bloating, headache, insomnia, tremor and palpitations. It was concluded that caffeine was responsible for the above-mentioned side effects. A possible solution to remedy these adverse effects would be the use of Polyphenon E (which is a decaffeinated GTE standardized in 65% EGCG), which was considered safe, when it was given to chronic lymphocytic leukemia patients (400–2000 mg orally twice a day) for one month [293,294]. However, Polyphenon E should be administered only with food and not after an overnight fast, due to higher EGCG plasma Cmax (seven-fold higher compared to EGCG administration with food) and high risk of hepatotoxicity [295]. Another polyphenols, resveratrol has shown kidney toxicity in clinical trials. According to Popat and co-workers the administration of a SRT501, a micronized oral formulation with resveratrol (5 g/day for 20 days in a 21 days cycle, up to 12 cycles followed by bortezomib) in patients with relapsed or refractory multiple myeloma, led to severe side effects (renal failure, nausea, anemia etc.). Renal failure occurred within the first two cycles of SRT501 monotherapy. However, it seems that SRT501 induces kidney failure only in myeloma patients, since the same dose of SRT501 was safe in diabetic patients or stroke-like episodes syndrome [296]. A solution to remedy renal failure in myeloma patients is the administration of a grape seed extract (rich in resveratrol but also other phenolic compounds. i.e. quercetin, proanthocyanidins), that have strong antioxidant effects and are able to protect the kidneys [297].
(iii) Validation the purity of the natural compound. The administration of a green tea extract (rich in catechins, mainly epigalocatechin gallate 11.8–4509 mcg/g extract), in a dosage of 5.9 g over five days to 240 g over 120 days was responsible for hepatic toxicity, mainly acute hepatocellular injury. Still, patients fully recovered with drug cessation [298,299]. According to some authors the observed hepatic toxicity of green tea extracts might be the consequence of contamination with pesticides (endosulfan), which is extensively used in green tea plantations [300].
(iv) Modes and route of administration. To increase specificity of polyphenols, they can be administrated as nanoparticles which have been coated with antibodies directed against molecular markers from the surface of the tumors [301,302]. In addition, local administration of the compound might be used whenever possible [301].
4. Conclusions
MDR has become the most important obstacle to the success of cancer chemotherapies. It implies several mechanisms, such as increased activity of efflux pumps (MRP 1/2, P-gp, BCRP), inhibition of cell death, cancer stem cells, epigenetic mechanisms, increased DNA repair, modification of drug target, inactivation of anticancer drugs, tumor cell heterogeneity, tumor microenvironment and epithelial to mesenchymal transition.
The use of natural compounds could overcome MDR through various mechanisms. Several studies have been performed using flavonoid (apigenin, luteolin, quercetin, genistein, epigallocatechin gallate, etc.) and non-flavonoid compounds (lignans, gallic acid, resveratrol, curcumin, etc.). In vitro and in vivo studies have revealed that administration of polyphenols (both from dietary sources and medicinal plants) overcome MDR to chemotherapeutic agents (paclitaxel, 5-fluorouracil, docetaxel, doxorubicin, gefitinib, etc.) in different types of cancer (breast, lung, prostate and colorectal) by downregulation of efflux pumps and anti-apoptotic proteins (survivin, XIAP), downregulation of NF-κB signaling cascade, decreased stem cells progenitor formation, increased cellular uptake of chemotherapeutic agents, epigenetic mechanisms, upregulation of apoptotic factors (DIABLO, APAF1) or modulation of several signaling pathways (Sonic-Hedgehog, EZH2, HER2, ERK, JNK, PI3K/AKT, STAT3, Wnt/β-catenin, etc.) and enzymes (FAS, GSK3, MMP2/MMP9, GST, etc.). However, few clinical studies demonstrated these effects. Therefore, we hope that this review will lead to continuous research regarding the role of phenolic compounds in overcoming multidrug resistance in various types of cancer.
Author Contributions
T.C., M.-M.M., O.C.V., L.-C.M. writing the manuscript, preparing the figures and tables, critical revising of the manuscript; M.-M.M., T.C. conceiving the concept, drafting, editing and critical revising of the manuscript, supervising the manuscript preparation. J.S., C.G. drafting, editing and critical revising of the manuscript, supervising the manuscript preparation. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by research grants from the National Research, Development and Innovation Office, Hungary (GINOP-2.3.2-15-2016-00050 and GINOP-2.3.3-15-2016-0003).
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| ↑ | upregulation |
| ↓ | downregulation |
| 5-FU | 5-fluorouracil |
| ABC | ATP-binding cassette transporter proteins |
| ABCB1, ABCG1 | isoforms of ATP-binding cassette transporter proteins |
| ARE | antioxidant response element |
| ABT-263 | small molecule that inhibits Bcl-2; 4-(4-{[2-(4-Chlorophenyl)-5,5-dimethyl-1-cyclohexen-1-yl]methyl}c-1-piperazinyl)-N-[(4-{[(2R)-4-(4-morpholinyl)-1-(phenylsulfanyl)-2-butanyl]amino}-3-[(trifluoromethyl)sulfonyl]phenyl)sulfonyl]benzamide |
| ABT-737 | small molecule that inhibits Bcl-2; 4-{4-[(4′-Chloro-2-biphenylyl)methyl]-1-piperazinyl}-N-[(4-{[(2R)-4-(dimethylamino)-1-(phenylsulfanyl)-2-butanyl]amino}-3-nitrophenyl)sulfonyl]benzamide |
| AKT | protein kinase B |
| ALDH | aldehyde dehydrogenase |
| ALP | autophagy lysosomes systems |
| AMPK | AMP-activated protein kinase |
| APAF1 | apoptotic protease activating factor 1 |
| APR-246 | drug that binds to p53 (restoring p53 function) and depletes glutathione; PRIMA-1, 2-hydroxymethyl-2-methoxymethyl-aza-bicyclo[2.2.2]octan-3-one |
| AR | androgen receptor |
| ATR | serine/threonine protein kinase |
| Axl, Tyro3 | receptors for tyrosine kinase |
| BASE | base excision repair |
| Bax | Bcl-2-associated X protein/Bcl-2-like protein 4 |
| Bcl-2 | B cell lymphoma 2 protein |
| Bcl-XL | B cell lymphoma extra-large protein |
| BCRA1, 2 | breast cancer susceptible genes |
| BCRP | breast cancer resistant protein |
| BER | base excision repair |
| BH | Bcl-2 homology domain |
| BIM | Bcl-2 like protein 11 |
| BNDQ | quercetin and doxorubicin co-encapsulated biotin receptor-targeting nanoparticles |
| BPIS | bound polyphenols of inner shell from foxtail millet bran |
| BRAF | serine/threonine-protein kinase B-Raf |
| C | catechin |
| CAB | carboplatin |
| CAPE | caffeic acid phenethyl ester |
| CAR | constitutive androstane receptor |
| caspase-3, 8, 9 | cysteine aspartic proteases-3, 8, 9 |
| CBZ | cabazitaxel |
| CD44, 24, 133 | cluster of differentiation 44, 24, 133 |
| CDF | difluorinated curcumin |
| CDK 2,4,6 | cyclin-dependent kinases 2,4,6 |
| CDPP | cisplatin |
| CEA | carcioembryonic antigen |
| cFLIP | regulator of caspase-8 activation; cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein |
| CgA | chromogranin |
| Chk1/2 | Check point kinase 1/2 |
| cIAP-1,2 | cellular inhibitor of apoptosis protein 1,2 |
| COMT | catechol-O-methyl transferase |
| COX-2 | ciclo-oxygenase 2 |
| CPT11 | irinotecan |
| CREB-1 | element binding protein-1 |
| CRPC | castration-resistant prostate cancer |
| CSC | cancer stem cells |
| CXCR4 | CXC chemokine receptor type 4 |
| CYP1A1, CYP1B1, CYP19A1, CYP17A1 | isoforms of cytochrome 450 |
| CYP3A4 | cytochrome P450 3A4 |
| DDR | DNA damage response |
| DIABLO | direct IAP-binding protein with Low pI |
| DMBA | 7,12-dimethylbenz[a] anthracene |
| DNA | deoxyribonucleic acid |
| DOC | docetaxel |
| DOX | doxorubicin (adriamycin) |
| DPPT | deoxypodophyllotoxin |
| DR4/5 | pro-apoptotic death receptors |
| EC | epicatechin |
| EGC | epigallocatechin |
| EGCG | epigallocatechingallate |
| EGF | epidermal growth factor |
| EGFR | epithelial growth factor receptor |
| EGFR(T790M) | epithelial growth factor receptor with a mutation that replace threonine by methionine at position 790 |
| EGR-1 | early growth response protein 1 |
| EMT | epithelial-mesenchymal transition |
| ENL | enterolactone |
| ER | estrogen receptors |
| ERα/ERβ | estrogen receptor alpha/estrogen receptor beta |
| ERK 1,2 | extracellular-signal regulated kinase |
| ETS2 | proto-oncogene 2, transcription factor (v-ets, Avian Erythroblastosis Virus E26 Oncogene Homolog 2) |
| EZH2 | enhancer of zeste homolog 2 (histone methyltransferase) |
| FASN | fatty acid synthase |
| FBAP5 | fatty acid-binding protein 5 |
| FGF | fibroblast growth factor |
| FOLFOX | 5-fluorouracil, oxaliplatin, folinic acid |
| GF | gefitinib |
| GRP78 | glucose regulated protein |
| GSH | reduced glutathione |
| GSK3 | glycogen synthase kinase 3 |
| GST | glutathione-S transferase |
| GPX | glutathione peroxidase |
| GTE | green tea extract |
| HER-2, 3 | human epidermal growth factor 2, 3 |
| Her2/neu | receptor tyrosine-proteinkinase erbB-2 |
| HGFR/MET | hepatocyte growth factor receptor |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| hMLH1 | mismatch repair gene of human mutL homolog 1 |
| HNK | honokiol |
| HO-1 | hemeoxygenase 1 |
| HR | homologous recombination |
| HRas | transforming protein p21 |
| IAP | inhibitors of apoptosis proteins |
| ICAM-1 | intercellular adhesion molecule 1 |
| IGF-1R | insulin growth factor receptor |
| IL-6, 8, 17, 18 | interleukin-6, 8, 17, 18 |
| i.p. | intraperitoneal administration |
| i.v | intravenous administration |
| JNK | c-Jun N-terminal kinase |
| Keap 1 | kelch-like ECH-associated protein 1 |
| KRAS | gene identified in Kirsten rat sarcoma |
| MAPK | mitogen activated protein kinase |
| MDM2 | mouse double minute 2 homolog |
| MDR | multidrug resistance |
| Meriva | turmeric/phospholipid formulation |
| MET | tyrosine-proteinkinase |
| MLH1,2 | human mutL homolog 1,2 |
| MMP | mitochondrial membrane potential |
| MMP-2,9 | metalloproteinases 2,9 |
| MMR | mismatch repair |
| MRP1/2 | multidrug resistance associated protein 1/2 |
| mRNA | messenger RNA |
| miRNA | microRNA |
| miR-16, 17, 21, 200c, 17-5p, 892c | microRNA-16, 17, 21, 200c, 17-5p, 892c |
| MSH 1/2 | DNA mismatch repair protein 1/2 |
| mTOR | mammalian target of rapamycin |
| NAD(P)H | reduced nicotinamide adenine dinucleotide phosphate |
| NER | nucleotide excision repair |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | nerve growth factor |
| NHEJ | non-homologous end-joining |
| NOX | NADPH oxidases |
| NPs | nanoparticles |
| Nrf2 | erythroid 2-related factor 2 |
| NSCLC | non-small cell lung cancer |
| NSE | neurospecific enolase |
| OX | oxaliplatin |
| p53 | tumor suppressor protein |
| P-gp (MDR1) | P-glycoprotein (multidrug resistance protein 1) |
| PI3K/AKT | phosphoinositide 3-kinase/protein kinase B |
| PKC | proteinkinase C |
| p.o. | oral administration |
| PPARβ/δ | peroxisome proliferator-activated receptor β/δ |
| PPT | podophyllotoxin |
| PS | phospho-sulindac |
| PSA | prostate serum antigen |
| PTEN | phosphatase and tensin homolog |
| PTX | paclitaxel |
| PXR | pregnane X receptor |
| RARs | retinoic acid receptors |
| RES | resveratrol |
| ROS | reactive oxygen species |
| SCC | squamous cell carcinoma |
| SChA | schizandrin A |
| SCLC | small cell lung cancer |
| SECO | secoisolariciresinol |
| Smac | second mitochondria-derived activator of caspase |
| SOD | superoxide-dismutase |
| Src | proto-oncogene tyrosine-protein kinase |
| SRT501 | small molecule, a form of resveratrol designed to target sirtuin 1 protein |
| STAT3 | signal transducer and activator of transcription 3 |
| T-box 3 | T-box transcription factor 3 |
| Tf-PEG-CUR | transferrin-poly(ethylene glycol)-curcumin |
| TGF-β | transforming growth factor |
| TKI | tyrosine kinase inhibitors |
| TME | tumor microenvironment |
| TP53 | gene coding tumor suppressor protein p53 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| UGT | uridine diphospho-glucuronosyltransferase |
| VCR | vincristine |
| VEGF | vascular endothelial growth factor |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
| Wnt/β-catenin | wingless-type MMTV integration site family member (MMTV, mouse mammary tumor virus)/beta-catenin signaling pathway |
| xCT | glutamate cysteine antiporter |
| XIAP | X-linked inhibitor of apoptosis protein |
| YB-1 | Y-box binding protein-1 |
References
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Andersson, E.; Ahlberg, I.; Nilbert, M.; Gerdtham, U. Socioeconomic inequalities in breast cancer incidence and mortality in Europe—A systematic review and meta-analysis. Eur. J. Public Health 2016, 26, 804–813. [Google Scholar] [CrossRef]
- Anderson, K.N.; Schwab, R.B.; Martinez, M.E. Reproductive risk factors and breast cancer subtypes: A review of the literature. Breast Cancer Res. Treat 2014, 144, 1–10. [Google Scholar] [CrossRef]
- Banin Hirata, B.K.; Oda, J.M.M.; Losi Guembarovski, R.; Ariza, C.B.; de Oliveira, C.E.; Watanabe, M.A.E. Molecular markers for breast cancer: Prediction on tumor behavior. Dis. Markers 2014. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, M.; Ciszewski, T.; Łopacka-Szatan, K.; Miotła, P.; Starosławska, E. Breast cancer risk factors. Prz. Menopauzalny 2015, 14, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Samavat, H.; Kurzer, M.S. Estrogen metabolism and breast cancer. Cancer Lett. 2015, 356, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Surakasula, A.; Nagarjunapu, G.C.; Raghavaiah, K.V. A comparative study of pre- and post-menopausal breast cancer: Risk factors, presentation, characteristics and management. J. Res. Pharm. Pr. 2014, 3, 12–18. [Google Scholar] [CrossRef]
- Farouk, O.; Ebrahim, M.A.; Senbel, A.; Emarah, Z.; Abozeed, W.; Seisa, M.O.; Mackisack, S.; Jalil, S.A.; Abdelhady, S. Breast cancer characteristics in very young Egyptian women ≤ 35 years. Breast Cancer: Targets Ther. 2016, 8, 53–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, H.L.; Smith, L.; Tomlinson, D.C. Multidrug-resistant breast cancer: Current perspectives. Targets Ther. 2014, 6, 1–13. [Google Scholar]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.G.; Prorok, P.C.; O’Malley, A.J.; Kramer, B.S. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N. Engl. J. Med. 2016, 375, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Ridge, C.A.; McErlean, A.M.; Ginsberg, M.S. Seminars in Interventional Radiology in Epidemiology of Lung Cancer; Thieme Medical Publishers: New York, NY, USA, 2013; Volume 30, pp. 093–098. [Google Scholar]
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, L.-Q.; Huang, J.-F.; Liu, K.; Chuai, Z.-R.; Yang, Z.; Wang, Y.-X.; Shi, D.-C.; Liu, Q.; Huang, Q. BRAF mutations in patients with non-small cell lung cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e101354. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Zhou, C. Lung Cancer in Never-Smokers: A Different Disease in IASLC Thoracic Oncology; Pass, H.I., Ball, D., Scagliotti, G.V., Eds.; Elsevier: Cambridge, MA, USA, 2018; pp. 23–29. [Google Scholar]
- Midha, A.; Dearden, S.; McCormack, R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 2015, 5, 2892–2911. [Google Scholar]
- Øines, M.; Helsingen, L.M.; Bretthauer, M.; Emilsson, L. Epidemiology and risk factors of colorectal polyps. Best Pr. Res. Clin. Gastroenterol. 2017, 31, 419–424. [Google Scholar] [CrossRef]
- Sakai, E.; Nakajima, A.; Kaneda, A. Accumulation of aberrant DNA methylation during colorectal cancer development. World J. Gastroenterol. 2014, 20, 978–987. [Google Scholar] [CrossRef]
- Wong, S.H.; Kwong, T.N.Y.; Wu, C.-Y.; Yu, J. Clinical applications of gut microbiota in cancer biology. Semin. Cancer Biol. 2019, 55, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Schwedhelm, C.; Hoffmann, G.; Knüppel, S.; Laure Preterre, A.; Iqbal, K.; Bechthold, A.; De Henauw, S.; Michels, N.; Devleesschauwer, B. Food groups and risk of colorectal cancer. Int. J. Cancer 2018, 142, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.-H.; Chay, W.Y.; Tan, M.-H.; Chow, K.Y.; Lim, W.-Y. Reproductive factors, obesity and risk of colorectal cancer in a cohort of Asian women. Cancer Epidemiol. 2019, 58, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Ward, H.A.; Jenab, M.; Rothwell, J.A.; Boutron-Ruault, M.-C.; Carbonnel, F.; Kvaskoff, M.; Kaaks, R.; Kühn, T.; Boeing, H.; et al. Heterogeneity of Colorectal Cancer Risk Factors by Anatomical Subsite in 10 European Countries: A Multinational Cohort Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Witold, K.; Anna, K.; Maciej, T.; Jakub, J. Adenomas–Genetic factors in colorectal cancer prevention. Rep. Pr. Oncol. Radiother. 2018, 23, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Heidegger, I.; Kretschmer, A.; Borgmann, H.; Gandaglia, G.; Briganti, A.; de Visschere, P.; Mathieu, R.; Valerio, M.; van den Bergh, R.; et al. Aggressive variants of prostate cancer—Are we ready to apply specific treatment right now? Cancer Treat. Rev. 2019, 75, 20–26. [Google Scholar] [CrossRef]
- Leitzmann, M.F.; Rohrmann, S. Risk factors for the onset of prostatic cancer: Age, location, and behavioral correlates. Clin. Epidemiol. 2012, 4, 1–11. [Google Scholar] [CrossRef]
- McAllister, M.J.; Underwood, M.A.; Leung, H.Y.; Edwards, J. A review on the interactions between the tumor microenvironment and androgen receptor signaling in prostate cancer. Transl. Res. 2019, 206, 91–106. [Google Scholar] [CrossRef]
- Nguyen, K.-S.H.; Neal, J.W.; Wakelee, H. Review of the current targeted therapies for non-small-cell lung cancer. World J. Clin. Oncol. 2014, 5, 576–587. [Google Scholar] [CrossRef]
- Eid, S.Y.; El-Readi, M.Z.; Fatani, S.H.; Eldin, E.E.M.N.; Wink, M. Natural products modulate the multifactorial multidrug resistance of cancer. Pharm. 2015, 6, 146–176. [Google Scholar] [CrossRef]
- Wang, P.; Yang, H.L.; Yang, Y.J.; Wang, L.; Lee, S.C. Overcome cancer cell drug resistance using natural products. Evid. Based Complement. Altern. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nabekura, T. Overcoming multidrug resistance in human cancer cells by natural compounds. Toxins 2010, 2, 1207–1224. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.W. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front. Oncol. 2019, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef] [PubMed]
- Tangney, C.C.; Rasmussen, H.E. Polyphenols, inflammation, and cardiovascular disease. Curr. Atheroscler. Rep. 2013, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Mrduljaš, N.; Krešić, G.; Bilušić, T. Polyphenols: Food Sources and Health Benefits in Functional Food-Improve Health through Adequate Food; Hueda, M.C., Ed.; IntechOpen: London, UK, 2017; Available online: https://www.intechopen.com/ (accessed on 2 January 2020). [CrossRef]
- Estrela, J.M.; Mena, S.; Obrador, E.; Benlloch, M.; Castellano, G.; Salvador, R.; Dellinger, R.W. Polyphenolic Phytochemicals in Cancer Prevention and Therapy: Bioavailability versus Bioefficacy. J. Med. Chem. 2017, 60, 9413–9436. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Q.; Li, D.; Zhou, Y.; Zheng, X.; Sun, H.; Yan, J.; Zhang, L.; Lin, Y.; Wang, X. Wogonin enhances antitumor activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo through ROS-mediated downregulation of cFLIPL and IAP proteins. Apoptosis 2013, 18, 618–626. [Google Scholar] [CrossRef]
- Kim, K.; Vance, T.M.; Chun, O.K. Estimated intake and major food sources of flavonoids among US adults: Changes between 1999–2002 and 2007–2010 in NHANES. Eur. J. Nutr. 2016, 55, 833–843. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Wang, D.; Zou, Y.; Qu, X.; He, C.; Deng, Y.; Jin, Y.; Zhou, Y.; Zhou, Y. Concurrently suppressing multidrug resistance and metastasis of breast cancer by co-delivery of paclitaxel and honokiol with pH-sensitive polymeric micelles. Acta Biomater. 2017, 62, 144–156. [Google Scholar] [CrossRef]
- Roy, A.; Ernsting, M.J.; Undzys, E.; Li, S.D. A highly tumor-targeted nanoparticle of podophyllotoxin penetrated tumor core and regressed multidrug resistant tumors. Biomaterials 2015, 52, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Wang, G.; Cai, Q.; Zheng, X.; Zhang, J.; Chen, Q.; Wu, B.; Zhu, X.; Hao, H.; Zhou, F. A Promising Microtubule Inhibitor Deoxypodophyllotoxin Exhibits Better Efficacy to Multidrug-Resistant Breast Cancer than Paclitaxel via Avoiding Efflux Transport. Drug Metab. Dispos. 2018, 46, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Molavi, O.; Narimani, F.; Asiaee, F.; Sharifi, S.; Tarhriz, V.; Shayanfar, A.; Hejazi, M.; Lai, R. Silibinin sensitizes chemo-resistant breast cancer cells to chemotherapy. Pharm. Biol. 2017, 55, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; De Silva, F.; Krol, E.S.; Alcorn, J. Flaxseed lignans enhance the cytotoxicity of chemotherapeutic agents against breast cancer cell lines MDA-MB-231 and SKBR3. Nutr. Cancer 2018, 70, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zhang, D.; Chu, X.; Wang, J. Schizandrin A enhances chemosensitivity of colon carcinoma cells to 5-fluorouracil through up-regulation of miR-195. Biomed. Pharm. 2018, 99, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.M.; Munekata, P.E.; Putnik, P.; Kovačević, D.B.; Muchenje, V.; Barba, F.J. Sources, Chemistry, and Biological Potential of Ellagitannins and Ellagic Acid Derivatives. In Studies in Natural Products Chemistry; Ur-Rahman, A., Ed.; Elsevier: Cambridge, MA, USA, 2018; Volume 60, pp. 189–221. [Google Scholar]
- Wei, Y.; Pu, X.; Zhao, L. Preclinical studies for the combination of paclitaxel and curcumin in cancer therapy. Oncol. Rep. 2017, 37, 3159–3166. [Google Scholar] [CrossRef]
- Liu, C.-M.; Kao, C.-L.; Tseng, Y.-T.; Lo, Y.-C.; Chen, C.-Y. Ginger phytochemicals inhibit cell growth and modulate drug resistance factors in docetaxel resistant prostate cancer cell. Molcules 2017, 22, 1477. [Google Scholar] [CrossRef]
- Harris, A.L.; Hochhauser, D. Mechanisms of multidrug resistance in cancer treatment. Acta Oncol. 1992, 31, 205–213. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Linn, S.C.; Giaccone, G. MDR1/P-glycoprotein expression in colorectal cancer. Eur. J. Cancer 1995, 31, 1291–1294. [Google Scholar] [CrossRef]
- Kong, X.B.; Yang, Z.K.; Liang, L.J.; Huang, J.F.; Lin, H.L. Overexpression of P-glycoprotein in hepatocellular carcinoma and its clinical implication. World J. Gastroenterol. 2000, 6, 134–135. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Trock, B. Multidrug resistance/P-glycoprotein and breast cancer: Review and meta-analysis. Semin. Oncol. 2005, 32, S9–S15. [Google Scholar] [CrossRef]
- Triller, N.; Korosec, P.; Kern, I.; Kosnik, M.; Debeljak, A. Multidrug resistance in small cell lung cancer: Expression of P-glycoprotein, multidrug resistance protein 1 and lung resistance protein in chemo-naive patients and in relapsed disease. Lung Cancer 2006, 54, 235–240. [Google Scholar] [CrossRef]
- Sanchez, C.; Mendoza, P.; Contreras, H.R.; Vergara, J.; McCubrey, J.A.; Huidobro, C.; Castellon, E.A. Expression of multidrug resistance proteins in prostate cancer is related with cell sensitivity to chemotherapeutic drugs. Prostate 2009, 69, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Aldonza, M.B.D.; Hong, J.-Y.; Bae, S.Y.; Song, J.; Kim, W.K.; Oh, J.; Shin, Y.; Lee, S.H.; Lee, S.K. Suppression of MAPK signaling and reversal of mTOR-dependent MDR1-associated multidrug resistance by 21α-methylmelianodiol in lung cancer cells. PLoS ONE 2015, 10, e0127841. [Google Scholar] [CrossRef]
- Xu, J.W.; Li, Q.Q.; Tao, L.L.; Cheng, Y.Y.; Yu, J.; Chen, Q.; Liu, X.P.; Xu, Z.D. Involvement of EGFR in the promotion of malignant properties in multidrug resistant breast cancer cells. Int. J. Oncol. 2011, 39, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/Pgp-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435. [Google Scholar] [CrossRef]
- Zhao, B.X.; Sun, Y.B.; Wang, S.Q.; Duan, L.; Huo, Q.L.; Ren, F.; Li, G.F. Grape seed procyanidin reversal of p-glycoprotein associated multi-drug resistance via down-regulation of NF-kappaB and MAPK/ERK mediated YB-1 activity in A2780/T cells. PLoS ONE 2013, 8, e71071. [Google Scholar] [CrossRef]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor Endothelial Cells Acquire Drug Resistance by MDR1 Up-Regulation via VEGF Signaling in Tumor Microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef]
- Mirzaei, S.A.; Dinmohammadi, F.; Alizadeh, A.; Elahian, F. Inflammatory pathway interactions and cancer multidrug resistance regulation. Life Sci. 2019, 235, 116825. [Google Scholar] [CrossRef]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef]
- Wang, X.; Sykes, D.B.; Miller, D.S. Constitutive androstane receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol. Pharm. 2010, 78, 376–383. [Google Scholar] [CrossRef]
- Banerjee, M.; Robbins, D.; Chen, T. Targeting xenobiotic receptors PXR and CAR in human diseases. Drug Discov. Today 2015, 20, 618–628. [Google Scholar] [CrossRef]
- Alexa-Stratulat, T.; Pešić, M.; Gašparović, A.Č.; Trougakos, I.P.; Riganti, C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? Looking beyond the tip of the iceberg. Drug Resist. Updates 2019, 46, 100643. [Google Scholar] [CrossRef] [PubMed]
- Sulova, Z.; Macejova, D.; Seres, M.; Sedlak, J.; Brtko, J.; Breier, A. Combined treatment of P-gp-positive L1210/VCR cells by verapamil and all-trans retinoic acid induces down-regulation of P-glycoprotein expression and transport activity. Toxicol. Vitr. 2008, 22, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Abd Ellah, N.H.; Taylor, L.; Ayres, N.; Elmahdy, M.M.; Fetih, G.N.; Jones, H.N.; Ibrahim, E.A.; Pauletti, G.M. NF-kappaB decoy polyplexes decrease P-glycoprotein-mediated multidrug resistance in colorectal cancer cells. Cancer Gene 2016, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.A.; Li, G. Cancer chemoresistance: The relationship between p53 and multidrug transporters. Int. J. Cancer 2002, 98, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.F.; Yang, J.M.; Vassil, A.; Yang, J.; Bash-Babula, J.; Hait, W.N. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J. Clin. Investig. 2000, 105, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, A.A. Cellular mechanisms of multidrug resistance of tumor cells. Biochem. Mosc. 2000, 65, 95–106. [Google Scholar]
- Wang, X.K.; Fu, L.W. Interaction of tyrosine kinase inhibitors with the MDR- related ABC transporter proteins. Curr. Drug Metab. 2010, 11, 618–628. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharm. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Increased accumulation of vincristine and adriamycin in drug-resistant P388 tumor cells following incubation with calcium antagonists and calmodulin inhibitors. Cancer Res. 1982, 42, 4730–4733. [Google Scholar]
- Tsuruo, T.; Iida, H.; Nojiri, M.; Tsukagoshi, S.; Sakurai, Y. Circumvention of vincristine and Adriamycin resistance in vitro and in vivo by calcium influx blockers. Cancer Res. 1983, 43, 2905–2910. [Google Scholar] [PubMed]
- Dalton, W.S.; Grogan, T.M.; Meltzer, P.S.; Scheper, R.J.; Durie, B.G.; Taylor, C.W.; Miller, T.P.; Salmon, S.E. Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: Detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J. Clin. Oncol. 1989, 7, 415–424. [Google Scholar] [CrossRef]
- To, K.K.; Tomlinson, B. Targeting the ABCG2-overexpressing multidrug resistant (MDR) cancer cells by PPARgamma agonists. Br. J. Pharm. 2013, 170, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kosuri, K.V.; Wu, X.; Wang, L.; Villalona-Calero, M.A.; Otterson, G.A. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem. Biophys. Res. Commun. 2010, 391, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Novotna, R.; Wsol, V.; Xiong, G.; Maser, E. Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol. Lett. 2008, 181, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef]
- Tew, K.D. Glutathione-Associated enzymes in anticancer drug resistance. Cancer Res. 2016, 76, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Starlard-Davenport, A.; Lyn-Cook, B.; Beland, F.A.; Pogribny, I.P. The role of UDP-glucuronosyltransferases and drug transporters in breast cancer drug resistance. Exp. Oncol. 2010, 32, 172–180. [Google Scholar] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- O’Hare, T.; Eide, C.A.; Deininger, M.W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Tian, Z.; Tang, J.; Yang, Q.; Li, X.; Zhu, J.; Wu, G. Atypical ubiquitin-binding protein SHARPIN promotes breast cancer progression. Biomed. Pharm. 2019, 119, 109414. [Google Scholar] [CrossRef]
- Baguley, B.C. Multiple drug resistance mechanisms in cancer. Mol. Biotechnol. 2010, 46, 308–316. [Google Scholar] [CrossRef]
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Bruyere, C.; Meijer, L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr. Opin. Cell Biol. 2013, 25, 772–779. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid Med. Cell Longev. 2017. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Wu, C.; You, M.; Kuo, M.T.; Feun, L.; Lampidis, T.; Savaraj, N. Inhibition of mTOR restores cisplatin sensitivity through down-regulation of growth and anti-apoptotic proteins. Eur. J. Pharm. 2008, 591, 124–127. [Google Scholar] [CrossRef]
- Sewify, E.M.; Afifi, O.A.; Mosad, E.; Zaki, A.H.; El Gammal, S.A. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin. Lymphoma Myeloma Leuk. 2014, 14, 215–222. [Google Scholar] [CrossRef]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of Multidrug Resistance in Cancer. In Multi-Drug Resistance in Cancer (Methods in Molecular Biology); Zhou, J., Ed.; Springer: New York, NY, USA, 2010; Volume 596, pp. 47–76. [Google Scholar]
- O’Brien, S.M.; Cunningham, C.C.; Golenkov, A.K.; Turkina, A.G.; Novick, S.C.; Rai, K.R. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 7697–7702. [Google Scholar] [CrossRef]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Bociek, R.G.; Pavletic, S.; et al. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2007, 25, 1114–1120. [Google Scholar] [CrossRef]
- Soderquist, R.S.; Eastman, A. BCL2 Inhibitors as Anticancer Drugs: A Plethora of Misleading BH3 Mimetics. Mol. Cancer 2016, 15, 2011–2017. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; Millward, M.; Pehamberger, H.; Conry, R.; Gore, M.; Trefzer, U.; Pavlick, A.C.; DeConti, R.; Hersh, E.M.; Hersey, P.; et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study Group. J. Clin. Oncol. 2006, 24, 4738–4745. [Google Scholar] [CrossRef]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Solary, E.; Micheau, O. TRAIL in cancer therapy: Present and future challenges. Expert Opin. Ther. Targets 2007, 11, 1299–1314. [Google Scholar] [CrossRef]
- Wu, G.S. TRAIL as a target in anti-cancer therapy. Cancer Lett. 2009, 285, 1–5. [Google Scholar] [CrossRef]
- Hetschko, H.; Voss, V.; Seifert, V.; Prehn, J.H.; Kogel, D. Upregulation of DR5 by proteasome inhibitors potently sensitizes glioma cells to TRAIL-induced apoptosis. FEBS J. 2008, 275, 1925–1936. [Google Scholar] [CrossRef]
- Hunter, T.B.; Manimala, N.J.; Luddy, K.A.; Catlin, T.; Antonia, S.J. Paclitaxel and TRAIL synergize to kill paclitaxel-resistant small cell lung cancer cells through a caspase-independent mechanism mediated through AIF. Anticancer Res. 2011, 31, 3193–3204. [Google Scholar]
- Zhang, Y.Q.; Tang, X.Q.; Sun, L.; Dong, L.; Qin, Y.; Liu, H.Q.; Xia, H.; Cao, J.G. Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29 cells by activating peroxisome proliferator-activated receptor gamma. World J. Gastroenterol. 2007, 13, 1534–1540. [Google Scholar] [CrossRef][Green Version]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharm. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 5416923. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Schmidt, F.; Efferth, T. Tumor Heterogeneity, Single-Cell Sequencing, and Drug Resistance. Pharmaceuticals 2016, 9, 33. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Kwak, E.L.; Ahronian, L.G.; Siravegna, G.; Mussolin, B.; Borger, D.R.; Godfrey, J.T.; Jessop, N.A.; Clark, J.W.; Blaszkowsky, L.S.; Ryan, D.P.; et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discov. 2015, 5, 1271–1281. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet Lond. Engl. 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Van Veggel, B.; de Langen, A.J.; Hashemi, S.M.S.; Monkhorst, K.; Heideman, D.A.M.; Thunnissen, E.; Smit, E.F. Afatinib and Cetuximab in Four Patients with EGFR Exon 20 Insertion-Positive Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1222–1226. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Sledge, G.W.; Neuberg, D.; Bernardo, P.; Ingle, J.N.; Martino, S.; Rowinsky, E.K.; Wood, W.C. Phase III trial of doxorubicin, paclitaxel, and the combination of doxorubicin and paclitaxel as front-line chemotherapy for metastatic breast cancer: An intergroup trial (E1193). J. Clin. Oncol. 2003, 21, 588–592. [Google Scholar] [CrossRef]
- Del Re, M.; Bordi, P.; Rofi, E.; Restante, G.; Valleggi, S.; Minari, R.; Crucitta, S.; Arrigoni, E.; Chella, A.; Morganti, R.; et al. The amount of activating EGFR mutations in circulating cell-free DNA is a marker to monitor osimertinib response. Br. J. Cancer 2018, 119, 1252–1258. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Milane, L.; Ganesh, S.; Shah, S.; Duan, Z.F.; Amiji, M. Multi-modal strategies for overcoming tumor drug resistance: Hypoxia, the Warburg effect, stem cells, and multifunctional nanotechnology. J. Control Release 2011, 155, 237–247. [Google Scholar] [CrossRef]
- Videira, M.; Reis, R.L.; Brito, M.A. Deconstructing breast cancer cell biology and the mechanisms of multidrug resistance. Biochim. Biophys. Acta 2014, 1846, 312–325. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour-stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Shee, K.; Yang, W.; Hinds, J.W.; Hampsch, R.A.; Varn, F.S.; Traphagen, N.A.; Patel, K.; Cheng, C.; Jenkins, N.P.; Kettenbach, A.N.; et al. Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. J. Exp. Med. 2018, 215, 895–910. [Google Scholar] [CrossRef]
- Shen, X.; Zhi, Q.; Wang, Y.; Li, Z.; Zhou, J.; Huang, J. Hypoxia Induces Multidrug Resistance via Enhancement of Epidermal Growth Factor-Like Domain 7 Expression in Non-Small Lung Cancer Cells. Chemotherapy 2017, 62, 172–180. [Google Scholar] [CrossRef]
- Faria, M.; Shepherd, P.; Pan, Y.; Chatterjee, S.S.; Navone, N.; Gustafsson, J.A.; Strom, A. The estrogen receptor variants beta2 and beta5 induce stem cell characteristics and chemotherapy resistance in prostate cancer through activation of hypoxic signaling. Oncotarget 2018, 9, 36273–36288. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, R.; de la Guardia, M.; Ahmadi, D.; Yousefi, B. Modulating tumor hypoxia by nanomedicine for effective cancer therapy. J. Cell Physiol. 2018, 233, 2019–2031. [Google Scholar] [CrossRef]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1α inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar] [CrossRef]
- Alexander, S.; Friedl, P. Cancer invasion and resistance: Interconnected processes of disease progression and therapy failure. Trends Mol. Med. 2012, 18, 13–26. [Google Scholar] [CrossRef]
- Tezcan, O.; Ojha, T.; Storm, G.; Kiessling, F.; Lammers, T. Targeting cellular and microenvironmental multidrug resistance. Expert Opin. Drug Deliv. 2016, 13, 1199–1202. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Zhang, J.-T. A new mechanism of drug resistance in breast cancer cells: Fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer 2008, 7, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Al Fayi, M.S.; Gou, X.; Forootan, S.S.; Al-Jameel, W.; Bao, Z.; Rudland, P.R.; Cornford, P.A.; Hussain, S.A.; Ke, Y. The increased expression of fatty acid-binding protein 9 in prostate cancer and its prognostic significance. Oncotarget 2016, 7, 82783–82797. [Google Scholar] [PubMed]
- Wu, X.; Qin, L.; Fako, V.; Zhang, J.T. Molecular mechanisms of fatty acid synthase (FASN)-mediated resistance to anti-cancer treatments. Adv. Biol. Regul. 2014, 54, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol Cancer 2019, 18, 67. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Butera, G.; Pacchiana, R.; Donadelli, M. Autocrine mechanisms of cancer chemoresistance. Semin. Cell Dev. Biol. 2018, 78, 3–12. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Fujii, T.; Dorfman, J.D.; Goodwin, J.M.; Zhu, A.X.; Lanuti, M.; Tanabe, K.K. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008, 68, 2391–2399. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molcules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Karantanos, T.; Bardhan, K.; Boussiotis, V.A. Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget 2016, 7, 85624–85640. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Gwak, S.Y.; Shim, G.A.; Liu, L.; Lim, Y.C.; Kim, J.M.; Jung, M.G.; Koo, B.S. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral Oncol. 2016, 52, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726. [Google Scholar] [CrossRef]
- An, X.; Sarmiento, C.; Tan, T.; Zhu, H. Regulation of multidrug resistance by microRNAs in anti-cancer therapy. Acta Pharm. Sinb. 2017, 7, 38–51. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Cen, J.; Zhang, L.; Liu, F.; Zhang, F.; Ji, B.S. Long-Term Alteration of Reactive Oxygen Species Led to Multidrug Resistance in MCF-7 Cells. Oxid Med. Cell Longev. 2016, 2016, 7053451. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Tang, Y.; Zhou, H.; Liu, Y.; Huang, J.; Li, L.; Liu, W.; Feng, Y.; Zhou, Y.; Chen, T.; et al. STAT3 mediates multidrug resistance of Burkitt lymphoma cells by promoting antioxidant feedback. Biochem. Biophys. Res. Commun. 2017, 488, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Turkekul, K.; Serttas, R.; Erdogan, Z. The natural flavonoid apigenin sensitizes human CD44(+) prostate cancer stem cells to cisplatin therapy. Biomed. Pharm. 2017, 88, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.; Satelli, A.; Moridani, M.; Jenkins, M.; Rao, U.S. Luteolin induces apoptosis in multidrug resistant cancer cells without affecting the drug transporter function: Involvement of cell line-specific apoptotic mechanisms. Int. J. Cancer 2012, 130, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Sabzichi, M.; Hamishehkar, H.; Ramezani, F.; Sharifi, S.; Tabasinezhad, M.; Pirouzpanah, M.; Ghanbari, P.; Samadi, N. Luteolin-loaded phytosomes sensitize human breast carcinoma MDA-MB 231 cells to doxorubicin by suppressing Nrf2 mediated signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5311–5316. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, Q.; Wang, B.; Yuan, S.; Wang, X.; Li, K. Quercetin reversed MDR in breast cancer cells through down-regulating P-gp expression and eliminating cancer stem cells mediated by YB-1 nuclear translocation. Phytother Res. 2018, 32, 1530–1536. [Google Scholar] [CrossRef]
- Chieli, E.; Romiti, N.; Rodeiro, I.; Garrido, G. In vitro effects of Mangifera indica and polyphenols derived on ABCB1/P-glycoprotein activity. Food Chem. Toxicol. 2009, 47, 2703–2710. [Google Scholar] [CrossRef]
- Taur, J.S.; Rodriguez-Proteau, R. Effects of dietary flavonoids on the transport of cimetidine via P-glycoprotein and cationic transporters in Caco-2 and LLC-PK1 cell models. Xenobiotica 2008, 38, 1536–1550. [Google Scholar] [CrossRef]
- Chung, S.Y.; Sung, M.K.; Kim, N.H.; Jang, J.O.; Go, E.J.; Lee, H.J. Inhibition of P-glycoprotein by natural products in human breast cancer cells. Arch. Pharm. Res. 2005, 28, 823–828. [Google Scholar] [CrossRef]
- Jeng, L.B.; Kumar Velmurugan, B.; Chen, M.C.; Hsu, H.H.; Ho, T.J.; Day, C.H.; Lin, Y.M.; Padma, V.V.; Tu, C.C.; Huang, C.Y. Fisetin mediated apoptotic cell death in parental and Oxaliplatin/irinotecan resistant colorectal cancer cells in vitro and in vivo. J. Cell Physiol. 2018, 233, 7134–7142. [Google Scholar] [CrossRef] [PubMed]
- Febriansah, R.; Putri, D.D.; Sarmoko; Nurulita, N.A.; Meiyanto, E.; Nugroho, A.E. Hesperidin as a preventive resistance agent in MCF-7 breast cancer cells line resistance to doxorubicin. Asian Pac. J. Trop. Biomed. 2014, 4, 228–233. [Google Scholar] [CrossRef]
- El-Readi, M.Z.; Hamdan, D.; Farrag, N.; El-Shazly, A.; Wink, M. Inhibition of P-glycoprotein activity by limonin and other secondary metabolites from Citrus species in human colon and leukaemia cell lines. Eur. J. Pharm. 2010, 626, 139–145. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Zupko, I.; Chang, F.R.; Hunyadi, A.; Wu, C.C.; Weng, T.S.; Wang, H.C. Dietary flavonoid derivatives enhance chemotherapeutic effect by inhibiting the DNA damage response pathway. Toxicol. Appl. Pharm. 2016, 311, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Knop, J.; Misaka, S.; Singer, K.; Hoier, E.; Muller, F.; Glaeser, H.; Konig, J.; Fromm, M.F. Inhibitory Effects of Green Tea and (-)-Epigallocatechin Gallate on Transport by OATP1B1, OATP1B3, OCT1, OCT2, MATE1, MATE2-K and P-Glycoprotein. PLoS ONE 2015, 10, e0139370. [Google Scholar] [CrossRef] [PubMed]
- Jodoin, J.; Demeule, M.; Beliveau, R. Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim. Biophys. Acta 2002, 1542, 149–159. [Google Scholar] [CrossRef]
- Esmaeili, M.A. Combination of siRNA-directed gene silencing with epigallocatechin-3-gallate (EGCG) reverses drug resistance in human breast cancer cells. J. Chem. Biol. 2016, 9, 41–52. [Google Scholar] [CrossRef]
- La, X.; Zhang, L.; Li, Z.; Li, H.; Yang, Y. (-)-Epigallocatechin Gallate (EGCG) Enhances the Sensitivity of Colorectal Cancer Cells to 5-FU by Inhibiting GRP78/NF-κB/miR-155–5p/MDR1 Pathway. J. Agric. Food Chem. 2019, 67, 2510–2518. [Google Scholar] [CrossRef]
- Wang, P.; Henning, S.M.; Heber, D.; Vadgama, J.V. Sensitization to docetaxel in prostate cancer cells by green tea and quercetin. J. Nutr. Biochem. 2015, 26, 408–415. [Google Scholar] [CrossRef]
- Kim, K.C.; Lee, C. Reversal of Cisplatin resistance by epigallocatechin gallate is mediated by downregulation of axl and tyro 3 expression in human lung cancer cells. Korean J. Physiol. Pharm. 2014, 18, 61–66. [Google Scholar] [CrossRef]
- Xue, J.P.; Wang, G.; Zhao, Z.B.; Wang, Q.; Shi, Y. Synergistic cytotoxic effect of genistein and doxorubicin on drug-resistant human breast cancer MCF-7/Adr cells. Oncol. Rep. 2014, 32, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor κB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef] [PubMed]
- Rigalli, J.P.; Scholz, P.N.; Tocchetti, G.N.; Ruiz, M.L.; Weiss, J. The phytoestrogens daidzein and equol inhibit the drug transporter BCRP/ABCG2 in breast cancer cells: Potential chemosensitizing effect. Eur. J. Nutr. 2019, 58, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Berger, S.M.; Kramer, M.Y.; Schwartz, N.S.; Holz, M.K. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J. Cell Biochem. 2015, 116, 450–457. [Google Scholar] [CrossRef]
- Huang, F.; Wu, X.-N.; Chen, J.; Wang, W.-X.; Lu, Z. Resveratrol reverses multidrug resistance in human breast cancer doxorubicin-resistant cells. Expther. Med. 2014, 7, 1611–1616. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.; Prasad, N. Resveratrol modulates expression of ABC transporters in non-small lung cancer cells: Molecular docking and gene expression studies. J. Cancer Sci. 2014, 6, 497–504. [Google Scholar] [CrossRef]
- Khaleel, S.A.; Al-Abd, A.M.; Ali, A.A.; Abdel-Naim, A.B. Didox and resveratrol sensitize colorectal cancer cells to doxorubicin via activating apoptosis and ameliorating P-glycoprotein activity. Sci. Rep. 2016, 6, 36855. [Google Scholar] [CrossRef]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef]
- Vinod, B.S.; Nair, H.H.; Vijayakurup, V.; Shabna, A.; Shah, S.; Krishna, A.; Pillai, K.S.; Thankachan, S.; Anto, R.J. Resveratrol chemosensitizes HER-2-overexpressing breast cancer cells to docetaxel chemoresistance by inhibiting docetaxel-mediated activation of HER-2–Akt axis. Cell Death Discov. 2015, 1, 15061. [Google Scholar] [CrossRef]
- He, Z.; Subramaniam, D.; Ramalingam, S.; Dhar, A.; Postier, R.G.; Umar, S.; Zhang, Y.; Anant, S. Honokiol radiosensitizes colorectal cancer cells: Enhanced activity in cells with mismatch repair defects. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 301, G929–G937. [Google Scholar] [CrossRef]
- Li, Y.; Revalde, J.; Paxton, J.W. The effects of dietary and herbal phytochemicals on drug transporters. Adv. Drug Deliv. Rev. 2017, 116, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.-Y.; Chung, Y.-C.; Hou, Y.-C.; Tsai, Y.-W.; Chen, C.-H.; Chang, H.-P.; Chou, J.-L.; Hsu, C.-P. Effects of ellagic acid on chemosensitivity to 5-fluorouracil in colorectal carcinoma cells. Anticancer Res. 2012, 32, 4413–4418. [Google Scholar] [PubMed]
- Berdowska, I.; Zieliński, B.; Saczko, J.; Sopel, M.; Gamian, A.; Fecka, I. Modulatory impact of selected ellagitannins on the viability of human breast cancer cells. J. Funct. Foods 2018, 42, 122–128. [Google Scholar] [CrossRef]
- Wang, R.; Ma, L.; Weng, D.; Yao, J.; Liu, X.; Jin, F. Gallic acid induces apoptosis and enhances the anticancer effects of cisplatin in human small cell lung cancer H446 cell line via the ROS-dependent mitochondrial apoptotic pathway. Oncol. Rep. 2016, 35, 3075–3083. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, A.; Tarasiuk, J. Comparative effects of selected plant polyphenols, gallic acid and epigallocatechin gallate, on matrix metalloproteinases activity in multidrug resistant MCF7/DOX breast cancer cells. Acta Biochim. Pol. 2016, 63, 571–575. [Google Scholar] [CrossRef]
- Phan, A.N.H.; Hua, T.N.M.; Kim, M.-K.; Vo, V.T.A.; Choi, J.-W.; Kim, H.-W.; Rho, J.K.; Kim, K.W.; Jeong, Y. Gallic acid inhibition of Src-Stat3 signaling overcomes acquired resistance to EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016, 7, 54702–54713. [Google Scholar] [CrossRef]
- Lu, Y.; Shan, S.; Li, H.; Shi, J.; Zhang, X.; Li, Z. Reversal effects of bound polyphenol from foxtail millet bran on multidrug resistance in human HCT-8/Fu colorectal cancer cell. J. Agric. Food Chem. 2018, 66, 5190–5199. [Google Scholar] [CrossRef]
- Omene, C.O.; Wu, J.; Frenkel, K. Caffeic Acid Phenethyl Ester (CAPE) derived from propolis, a honeybee product, inhibits growth of breast cancer stem cells. Investig. New Drugs 2012, 30, 1279–1288. [Google Scholar] [CrossRef]
- Khoram, N.M.; Bigdeli, B.; Nikoofar, A.; Goliaei, B. Caffeic acid phenethyl ester increases radiosensitivity of estrogen receptor-positive and-negative breast cancer cells by prolonging radiation-induced DNA damage. J. Breast Cancer 2016, 19, 18–25. [Google Scholar] [CrossRef]
- Ozturk, G.; Ginis, Z.; Akyol, S.; Erden, G.; Gurel, A.; Akyol, O. The anticancer mechanism of caffeic acid phenethyl ester (CAPE): Review of melanomas, lung and prostate cancers. Eur. Rev. Med. Pharm. Sci. 2012, 16, 2064–2068. [Google Scholar]
- Sonoki, H.; Tanimae, A.; Furuta, T.; Endo, S.; Matsunaga, T.; Ichihara, K.; Ikari, A. Caffeic acid phenethyl ester down-regulates claudin-2 expression at the transcriptional and post-translational levels and enhances chemosensitivity to doxorubicin in lung adenocarcinoma A549 cells. J. Nutr. Biochem. 2018, 56, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zeng, F.; Xu, L.; Zhou, J.; Liu, X.; Le, H. Anticancer effects of cinnamic acid in lung adenocarcinoma cell line h1299-derived stem-like cells. Oncol. Res. 2012, 20, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-D.; Qin, Y.; Yang, C.; Li, L. Effect of curcumin on human colon cancer multidrug resistance in vitro and in vivo. Clinics 2013, 68, 694–701. [Google Scholar] [CrossRef]
- Lu, Y.; Wei, C.; Xi, Z. Curcumin suppresses proliferation and invasion in non-small cell lung cancer by modulation of MTA1-mediated Wnt/β-catenin pathway. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 840–850. [Google Scholar] [CrossRef] [PubMed]
- De Porras, V.R.; Bystrup, S.; Martínez-Cardús, A.; Pluvinet, R.; Sumoy, L.; Howells, L.; James, M.I.; Iwuji, C.; Manzano, J.L.; Layos, L. Curcumin mediates oxaliplatin-acquired resistance reversion in colorectal cancer cell lines through modulation of CXC-Chemokine/NF-κB signalling pathway. Sci. Rep. 2016, 6, 24675. [Google Scholar] [CrossRef]
- Vinod, B.S.; Antony, J.; Nair, H.H.; Puliyappadamba, V.T.; Saikia, M.; Shyam Narayanan, S.; Bevin, A.; John Anto, R. Mechanistic evaluation of the signaling events regulating curcumin-mediated chemosensitization of breast cancer cells to 5-fluorouracil. Cell Death Discov. 2013, 4, e505. [Google Scholar] [CrossRef]
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P. Difluorinated-curcumin (CDF) restores PTEN expression in colon cancer cells by down-regulating miR-21. PLoS ONE 2013, 8, e68543. [Google Scholar] [CrossRef]
- Shen, J.; Chen, Y.-J.; Jia, Y.-W.; Zhao, W.-Y.; Chen, G.-H.; Liu, D.-F.; Chen, Y.-Y.; Zhang, C.; Liu, X.P. Reverse effect of curcumin on CDDP-induced drug-resistance via Keap1/p62-Nrf2 signaling in A549/CDDP cell. Asian Pac. J. Trop. Med. 2017, 10, 1190–1196. [Google Scholar] [CrossRef]
- Gu, Y.; Li, J.; Li, Y.; Song, L.; Li, D.; Peng, L.; Wan, Y.; Hua, S. Nanomicelles loaded with doxorubicin and curcumin for alleviating multidrug resistance in lung cancer. Int. J. Nanomed. 2016, 11, 5757–5770. [Google Scholar] [CrossRef]
- Ye, M.X.; Zhao, Y.L.; Li, Y.; Miao, Q.; Li, Z.-K.; Ren, X.L.; Song, L.Q.; Yin, H.; Zhang, J. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine 2012, 19, 779–787. [Google Scholar] [CrossRef]
- Jiang, M.; Huang, O.; Zhang, X.; Xie, Z.; Shen, A.; Liu, H.; Geng, M.; Shen, K. Curcumin induces cell death and restores tamoxifen sensitivity in the antiestrogen-resistant breast cancer cell lines MCF-7/LCC2 and MCF-7/LCC9. Molcules 2013, 18, 701–720. [Google Scholar] [CrossRef]
- Thulasiraman, P.; McAndrews, D.J.; Mohiudddin, I.Q. Curcumin restores sensitivity to retinoic acid in triple negative breast cancer cells. BMC Cancer 2014, 14, 724. [Google Scholar] [CrossRef]
- Wang, S.; Chen, R.; Zhong, Z.; Shi, Z.; Chen, M.; Wang, Y. Epigallocatechin-3-gallate potentiates the effect of curcumin in inducing growth inhibition and apoptosis of resistant breast cancer cells. Am. J. Chin. Med. 2014, 42, 1279–1300. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Sobh, M.A.; Eid, H.M.; Salem, A.; Elbelasi, H.H.; El-Naggar, M.H.; AbdelBar, F.M.; Sheashaa, H.; Sobh, M.A.; Badria, F.A. Gingerol-derivatives: Emerging new therapy against human drug-resistant MCF-7. Tumor Biol. 2014, 35, 9941–9948. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; Di Pietro, A.; Dumontet, C.; Barron, D. Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med. Res. Rev. 2002, 22, 512–529. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.Y.; Li, H.B.; Sui, Z.Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1428–1447. [Google Scholar] [CrossRef]
- Hussain, S.A.; Sulaiman, A.A.; Balch, C.; Chauhan, H.; Alhadidi, Q.M.; Tiwari, A.K. Natural Polyphenols in Cancer Chemoresistance. Nutr. Cancer 2016, 68, 879–891. [Google Scholar] [CrossRef]
- Czerwonka, A.; Maciolek, U.; Kalafut, J.; Mendyk, E.; Kuzniar, A.; Rzeski, W. Anticancer effects of sodium and potassium quercetin-5′-sulfonates through inhibition of proliferation, induction of apoptosis, and cell cycle arrest in the HT-29 human adenocarcinoma cell line. Bioorg. Chem. 2019, 94, 103426. [Google Scholar] [CrossRef]
- Scambia, G.; Ranelletti, F.O.; Panici, P.B.; De Vincenzo, R.; Bonanno, G.; Ferrandina, G.; Piantelli, M.; Bussa, S.; Rumi, C.; Cianfriglia, M.; et al. Quercetin potentiates the effect of adriamycin in a multidrug-resistant MCF-7 human breast-cancer cell line: P-glycoprotein as a possible target. Cancer Chemother. Pharm. 1994, 34, 459–464. [Google Scholar] [CrossRef]
- He, W.T.; Zhu, Y.H.; Zhang, T.; Abulimiti, P.; Zeng, F.Y.; Zhang, L.P.; Luo, L.J.; Xie, X.M.; Zhang, H.L. Curcumin Reverses 5-Fluorouracil Resistance by Promoting Human Colon Cancer HCT-8/5-FU Cell Apoptosis and Down-regulating Heat Shock Protein 27 and P-Glycoprotein. Chin. J. Integr. Med. 2019, 25, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhao, B.; Xiong, P.; Wang, C.; Zhang, J.; Tian, X.; Huang, Y. Curcumin Induces Autophagy via Inhibition of Yes-Associated Protein (YAP) in Human Colon Cancer Cells. Med. Sci. Monit. 2018, 24, 7035–7042. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Yang, Y.; Wang, G.; Chen, X.; Ju, Y. Curcumin attenuates resistance to irinotecan via induction of apoptosis of cancer stem cells in chemoresistant colon cancer cells. Int. J. Oncol. 2018, 53, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Fu, L.; Huang, J.; Dai, Y.; Wang, B.; Xu, G.; Wu, L.; Zhou, H. Curcumin reverses doxorubicin resistance via inhibition the efflux function of ABCB4 in doxorubicinresistant breast cancer cells. Mol. Med. Rep. 2019, 19, 5162–5168. [Google Scholar]
- Hu, C.; Li, M.; Guo, T.; Wang, S.; Huang, W.; Yang, K.; Liao, Z.; Wang, J.; Zhang, F.; Wang, H. Anti-metastasis activity of curcumin against breast cancer via the inhibition of stem cell-like properties and EMT. Phytomedicine 2019, 58, 152740. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, X.; Qi, G.; Guo, Y. Curcumin suppressed the prostate cancer by inhibiting JNK pathways via epigenetic regulation. J. Biochem. Mol. Toxicol. 2018, 32, e22049. [Google Scholar] [CrossRef]
- Lin, W.; Luo, J.; Sun, Y.; Lin, C.; Li, G.; Niu, Y.; Chang, C. ASC-J9((R)) suppresses prostate cancer cell invasion via altering the sumoylation-phosphorylation of STAT3. Cancer Lett. 2018, 425, 21–30. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Lu, G.H.; Wu, Y.Q.; Zheng, Y.; Xu, K.; Wu, L.J.; Jiang, Z.Y.; Feng, R.; Zhou, J.Y. Curcumin induces mitochondria pathway mediated cell apoptosis in A549 lung adenocarcinoma cells. Oncol. Rep. 2010, 23, 1285–1292. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004, 64, 337–346. [Google Scholar] [CrossRef]
- Lin, J.N.; Lin, V.C.; Rau, K.M.; Shieh, P.C.; Kuo, D.H.; Shieh, J.C.; Chen, W.J.; Tsai, S.C.; Way, T.D. Resveratrol modulates tumor cell proliferation and protein translation via SIRT1-dependent AMPK activation. J. Agric. Food Chem. 2010, 58, 1584–1592. [Google Scholar] [CrossRef]
- Lecumberri, E.; Dupertuis, Y.M.; Miralbell, R.; Pichard, C. Green tea polyphenol epigallocatechin-3-gallate (EGCG) as adjuvant in cancer therapy. Clin. Nutr. 2013, 32, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Choi, J.S.; Li, X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int. J. Pharm. 2006, 313, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Liu, C.; Chen, C.; Yu, X.; Chen, G.; Shi, Y.; Qin, F.; Ou, J.; Qiu, K.; Li, G. Quercetin and doxorubicin co-encapsulated biotin receptor-targeting nanoparticles for minimizing drug resistance in breast cancer. Oncotarget 2016, 7, 32184–32199. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Cao, X.; Li, N.; Zhang, Y.; Lan, L.; Zhou, Y.; Pan, X.; Shen, L.; Yin, Z.; Luo, L. Luteolin is effective in the non-small cell lung cancer model with L 858 R/T 790 M EGF receptor mutation and erlotinib resistance. Br. J. Pharm. 2014, 171, 2842–2853. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Wang, J.; Yin, Y.; Hua, H.; Jing, J.; Sun, X.; Li, M.; Zhang, Y.; Jiang, Y. (-)-Epigallocatechin gallate sensitizes breast cancer cells to paclitaxel in a murine model of breast carcinoma. Breast Cancer Res. 2010, 12, R8. [Google Scholar] [CrossRef] [PubMed]
- El-Rahman, S.S.A.; Shehab, G.; Nashaat, H. Epigallocatechin-3-Gallate: The prospective targeting of cancer stem cells and preventing metastasis of chemically-induced mammary cancer in rats. Am. J. Med. Sci. 2017, 354, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yan, L.; Wang, L.; Tai, W.; Wang, W.; Yang, C. Genistein enhances the effect of cisplatin on the inhibition of non-small cell lung cancer A549 cell growth in vitro and in vivo. Oncol. Lett. 2014, 8, 2806–2810. [Google Scholar] [CrossRef]
- Zhu, H.; Cheng, H.; Ren, Y.; Liu, Z.G.; Zhang, Y.F.; De Luo, B. Synergistic inhibitory effects by the combination of gefitinib and genistein on NSCLC with acquired drug-resistance in vitro and in vivo. Mol. Biol. Rep. 2012, 39, 4971–4979. [Google Scholar] [CrossRef]
- Meng, J.; Guo, F.; Xu, H.; Liang, W.; Wang, C.; Yang, X.D. Combination therapy using co-encapsulated resveratrol and paclitaxel in liposomes for drug resistance reversal in breast cancer cells in vivo. Sci. Rep. 2016, 6, 22390. [Google Scholar] [CrossRef]
- Yang, S.; Li, W.; Sun, H.; Wu, B.; Ji, F.; Sun, T.; Chang, H.; Shen, P.; Wang, Y.; Zhou, D. Resveratrol elicits anti-colorectal cancer effect by activating miR-34c-KITLG in vitro and in vivo. BMC Cancer 2015, 15, 969. [Google Scholar] [CrossRef]
- Zhao, W.; Bao, P.; Qi, H.; You, H. Resveratrol down-regulates survivin and induces apoptosis in human multidrug-resistant SPC-A-1/CDDP cells. Oncol. Rep. 2010, 23, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef]
- Lou, S.; Zhao, Z.; Dezort, M.; Lohneis, T.; Zhang, C. Multifunctional Nanosystem for Targeted and Controlled Delivery of Multiple Chemotherapeutic Agents for the Treatment of Drug-Resistant Breast Cancer. ACS Omega 2018, 3, 9210–9219. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Diagaradjane, P.; Anand, P.; Kuzhuvelil, H.B.; Deorukhkar, A.; Gelovani, J.; Guha, S.; Krishnan, S.; Aggarwal, B.B. Curcumin sensitizes human colorectal cancer to capecitabine by modulation of cyclin D1, COX-2, MMP-9, VEGF and CXCR4 expression in an orthotopic mouse model. Int. J. Cancer 2009, 125, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Neerati, P.; Sudhakar, Y.A.; Kanwar, J.R. Curcumin regulates colon cancer by inhibiting P-glycoprotein in in-situ cancerous colon perfusion rat model. J. Cancer Sci. 2013, 5, 313–319. [Google Scholar]
- Howells, L.M.; Sale, S.; Sriramareddy, S.N.; Irving, G.R.; Jones, D.J.; Ottley, C.J.; Pearson, D.G.; Mann, C.D.; Manson, M.M.; Berry, D.P. Curcumin ameliorates oxaliplatin-induced chemoresistance in HCT116 colorectal cancer cells in vitro and in vivo. Int. J. Cancer 2011, 129, 476–486. [Google Scholar] [CrossRef]
- Yan, J.; Wang, Y.; Zhang, X.; Liu, S.; Tian, C.; Wang, H. Targeted nanomedicine for prostate cancer therapy: Docetaxel and curcumin co-encapsulated lipid–polymer hybrid nanoparticles for the enhanced anti-tumor activity in vitro and in vivo. Drug Deliv. 2016, 23, 1757–1762. [Google Scholar] [CrossRef]
- Pramanik, D.; Campbell, N.R.; Das, S.; Gupta, S.; Chenna, V.; Bisht, S.; Sysa-Shah, P.; Bedja, D.; Karikari, C.; Steenbergen, C.; et al. A composite polymer nanoparticle overcomes multidrug resistance and ameliorates doxorubicin-associated cardiomyopathy. Oncotarget 2012, 3, 640–650. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Hill, D.L.; Wang, H.; Zhang, R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007, 67, 1988–1996. [Google Scholar] [CrossRef]
- Cheng, K.-W.; Wong, C.C.; Mattheolabakis, G.; Xie, G.; Huang, L.; Rigas, B. Curcumin enhances the lung cancer chemopreventive efficacy of phospho-sulindac by improving its pharmacokinetics. Int. J. Oncol. 2013, 43, 895–902. [Google Scholar] [CrossRef]
- Cui, T.; Zhang, S.; Sun, H. Co-delivery of doxorubicin and pH-sensitive curcumin prodrug by transferrin-targeted nanoparticles for breast cancer treatment. Oncol. Rep. 2017, 37, 1253–1260. [Google Scholar] [CrossRef]
- Mahammedi, H.; Planchat, E.; Pouget, M.; Durando, X.; Cure, H.; Guy, L.; Van-Praagh, I.; Savareux, L.; Atger, M.; Bayet-Robert, M.; et al. The New Combination Docetaxel, Prednisone and Curcumin in Patients with Castration-Resistant Prostate Cancer: A Pilot Phase II Study. Oncology 2016, 90, 69–78. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. 2010, 9, 8–14. [Google Scholar] [CrossRef]
- D’Archivio, M.; Filesi, C.; Vari, R.; Scazzocchio, B.; Masella, R. Bioavailability of the polyphenols: Status and controversies. Int. J. Mol. Sci. 2010, 11, 1321–1342. [Google Scholar] [CrossRef]
- Tresserra-Rimbau, A.; Lamuela-Raventos, R.M.; Moreno, J.J.; Polyphenols, food and pharma. Current knowledge and directions for future research. Biochem. Pharm. 2018, 156, 186–195. [Google Scholar] [CrossRef]
- Velderrain-Rodriguez, G.R.; Palafox-Carlos, H.; Wall-Medrano, A.; Ayala-Zavala, J.F.; Chen, C.Y.; Robles-Sanchez, M.; Astiazaran-Garcia, H.; Alvarez-Parrilla, E.; Gonzalez-Aguilar, G.A. Phenolic compounds: Their journey after intake. Food Funct. 2014, 5, 189–197. [Google Scholar] [CrossRef]
- Azrad, M.; Vollmer, R.T.; Madden, J.; Dewhirst, M.; Polascik, T.J.; Snyder, D.C.; Ruffin, M.T.; Moul, J.W.; Brenner, D.E.; Demark-Wahnefried, W. Flaxseed-derived enterolactone is inversely associated with tumor cell proliferation in men with localized prostate cancer. J. Med. Food 2013, 16, 357–360. [Google Scholar] [CrossRef]
- Gonzalez-Sarrias, A.; Tome-Carneiro, J.; Bellesia, A.; Tomas-Barberan, F.A.; Espin, J.C. The ellagic acid-derived gut microbiota metabolite, urolithin A, potentiates the anticancer effects of 5-fluorouracil chemotherapy on human colon cancer cells. Food Funct. 2015, 6, 1460–1469. [Google Scholar] [CrossRef]
- Alam, M.N.; Almoyad, M.; Huq, F. Polyphenols in Colorectal Cancer: Current State of Knowledge including Clinical Trials and Molecular Mechanism of Action. Biomed. Res. Int. 2018. [Google Scholar] [CrossRef]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, W.P.; Brown, K. Clinical trials of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef]
- Vinod, B.S.; Maliekal, T.T.; Anto, R.J. Phytochemicals as chemosensitizers: From molecular mechanism to clinical significance. Antioxid. Redox Signal. 2013, 18, 1307–1348. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse effects of cancer chemotherapy: Anything new to improve tolerance and reduce sequelae? Front. Pharm. 2018, 9, 245. [Google Scholar] [CrossRef]
- James, M.I.; Iwuji, C.; Irving, G.; Karmokar, A.; Higgins, J.A.; Griffin-Teal, N.; Thomas, A.; Greaves, P.; Cai, H.; Patel, S.R.; et al. Curcumin inhibits cancer stem cell phenotypes in ex vivo models of colorectal liver metastases, and is clinically safe and tolerable in combination with FOLFOX chemotherapy. Cancer Lett. 2015, 364, 135–141. [Google Scholar] [CrossRef]
- Gorelick, D.A. Pharmacokinetic strategies for treatment of drug overdose and addiction. Future Med. Chem. 2012, 4, 227–243. [Google Scholar] [CrossRef]
- Li, H.; Krstin, S.; Wink, M. Modulation of multidrug resistant in cancer cells by EGCG, tannic acid and curcumin. Phytomedicine 2018, 50, 213–222. [Google Scholar] [CrossRef]
- Hu, F.; Wei, F.; Wang, Y.; Wu, B.; Fang, Y.; Xiong, B. EGCG synergizes the therapeutic effect of cisplatin and oxaliplatin through autophagic pathway in human colorectal cancer cells. J. Pharm. Sci. 2015, 128, 27–34. [Google Scholar] [CrossRef]
- Schonthal, A.H. Adverse effects of concentrated green tea extracts. Mol. Nutr. Food Res. 2011, 55, 874–885. [Google Scholar] [CrossRef]
- Pisters, K.M.; Newman, R.A.; Coldman, B.; Shin, D.M.; Khuri, F.R.; Hong, W.K.; Glisson, B.S.; Lee, J.S. Phase I trial of oral green tea extract in adult patients with solid tumors. J. Clin. Oncol. 2001, 19, 1830–1838. [Google Scholar] [CrossRef]
- Ullmann, U.; Haller, J.; Decourt, J.D.; Girault, J.; Spitzer, V.; Weber, P. Plasma-kinetic characteristics of purified and isolated green tea catechin epigallocatechin gallate (EGCG) after 10 days repeated dosing in healthy volunteers. Int. J. Vitam. Nutr. Res. 2004, 74, 269–278. [Google Scholar] [CrossRef]
- Popat, R.; Plesner, T.; Davies, F.; Cook, G.; Cook, M.; Elliott, P.; Jacobson, E.; Gumbleton, T.; Oakervee, H.; Cavenagh, J. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br. J. Haematol. 2013, 160, 714–717. [Google Scholar] [CrossRef]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Et. Biophys. Acta 2015, 1852, 1178–1185. [Google Scholar] [CrossRef]
- Navarro, V.J.; Bonkovsky, H.L.; Hwang, S.I.; Vega, M.; Barnhart, H.; Serrano, J. Catechins in dietary supplements and hepatotoxicity. Dig. Dis. Sci. 2013, 58, 2682–2690. [Google Scholar] [CrossRef]
- Bonkovsky, H.L. Hepatotoxicity associated with supplements containing Chinese green tea (Camellia sinensis). Ann. Intern. Med. 2006, 144, 68–71. [Google Scholar] [CrossRef]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [CrossRef]
- Granja, A.; Pinheiro, M.; Reis, S. Epigallocatechin gallate nanodelivery systems for cancer therapy. Nutrients 2016, 8, 307. [Google Scholar] [CrossRef]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef]




















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).