Oxidative Stress and Microglial Response in Retinitis Pigmentosa




Abstract
1. Introduction
2. Retinitis Pigmentosa and Oxidative Stress
2.1. Etiology of Retinitis Pigmentosa
2.2. Oxidative Damage in Animal Models of RP
2.2.1. Evidence of Increased Oxidative Damage in the RP Retina
2.2.2. Role of Oxidative Damage in RP
2.2.3. Modification of Anti-Oxidant Genes in RP
2.3. Clinical Evidence of Oxidative Stress in RP
3. Clinical Outcomes of Anti-oxidant Treatments
3.1. Anti-oxidant Supplements for Systemic Diseases
3.2. Anti-oxidant Supplements for Ocular Disorders
3.2.1. Anti-oxidant Supplements for Age-related Macular Degeneration (AMD)
3.2.2. Anti-oxidant Supplements for RP
3.3. Oral NAC Treatments for RP
4. Oxidative DNA Damage and its Repair System
4.1. Mechanisms by Which Oxidative Stress Mediates Retinal Degeneration
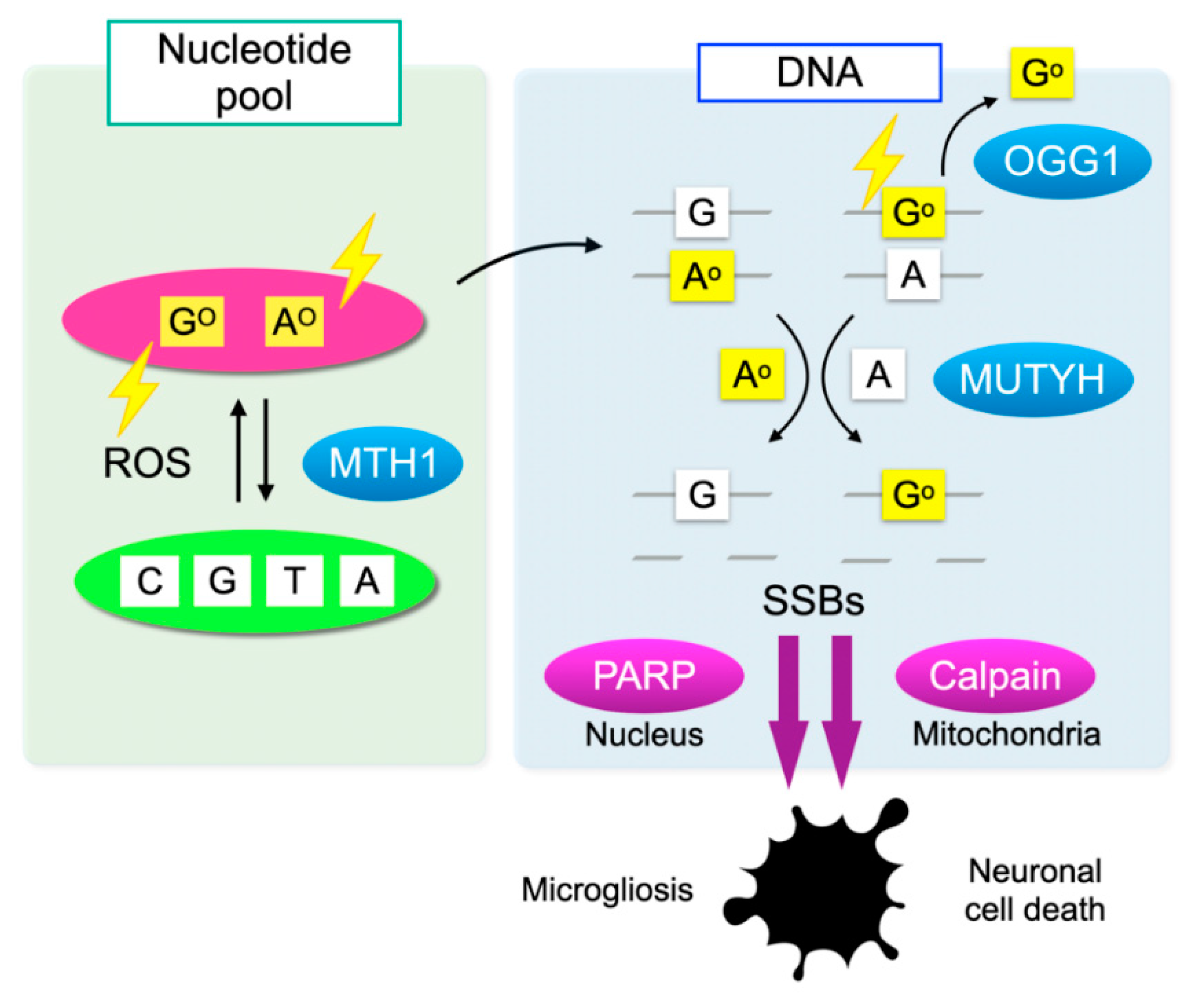
4.2. DNA Oxidation and Mismatched Base Pair Formation
4.3. Counter-Mechanisms against Oxidative DNA Damage
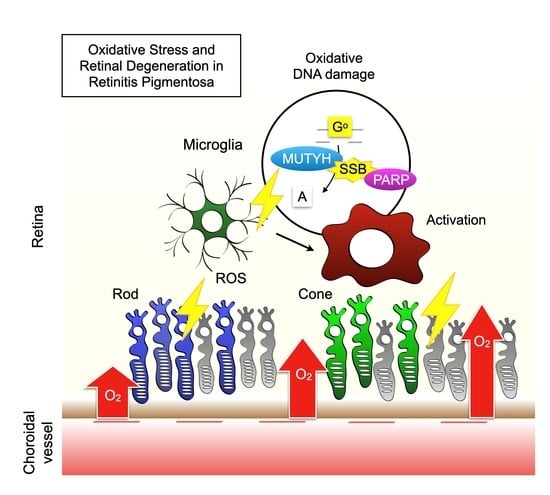
5. DNA Oxidation in Microglia Promotes Inflammation and Degeneration in RP
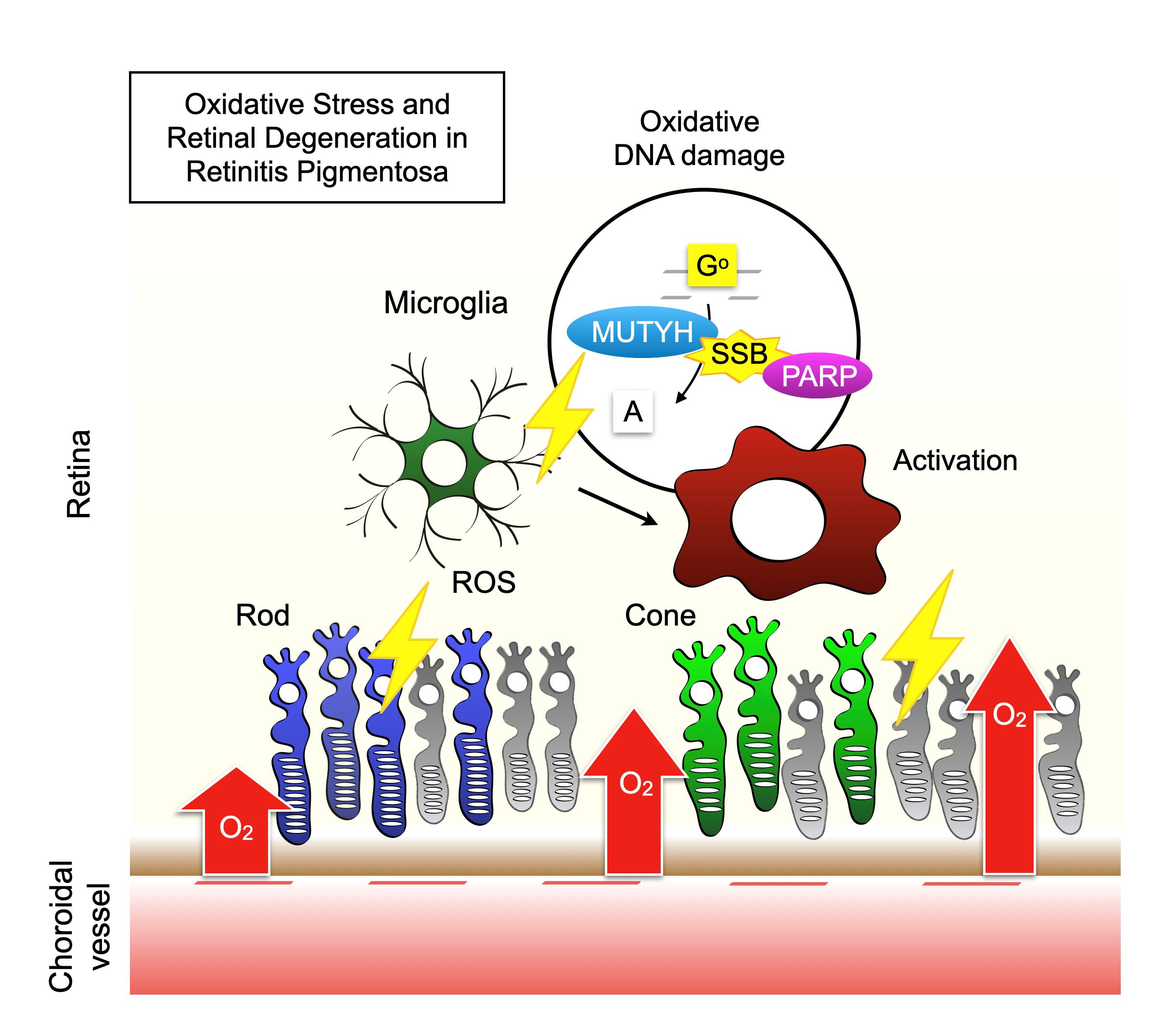
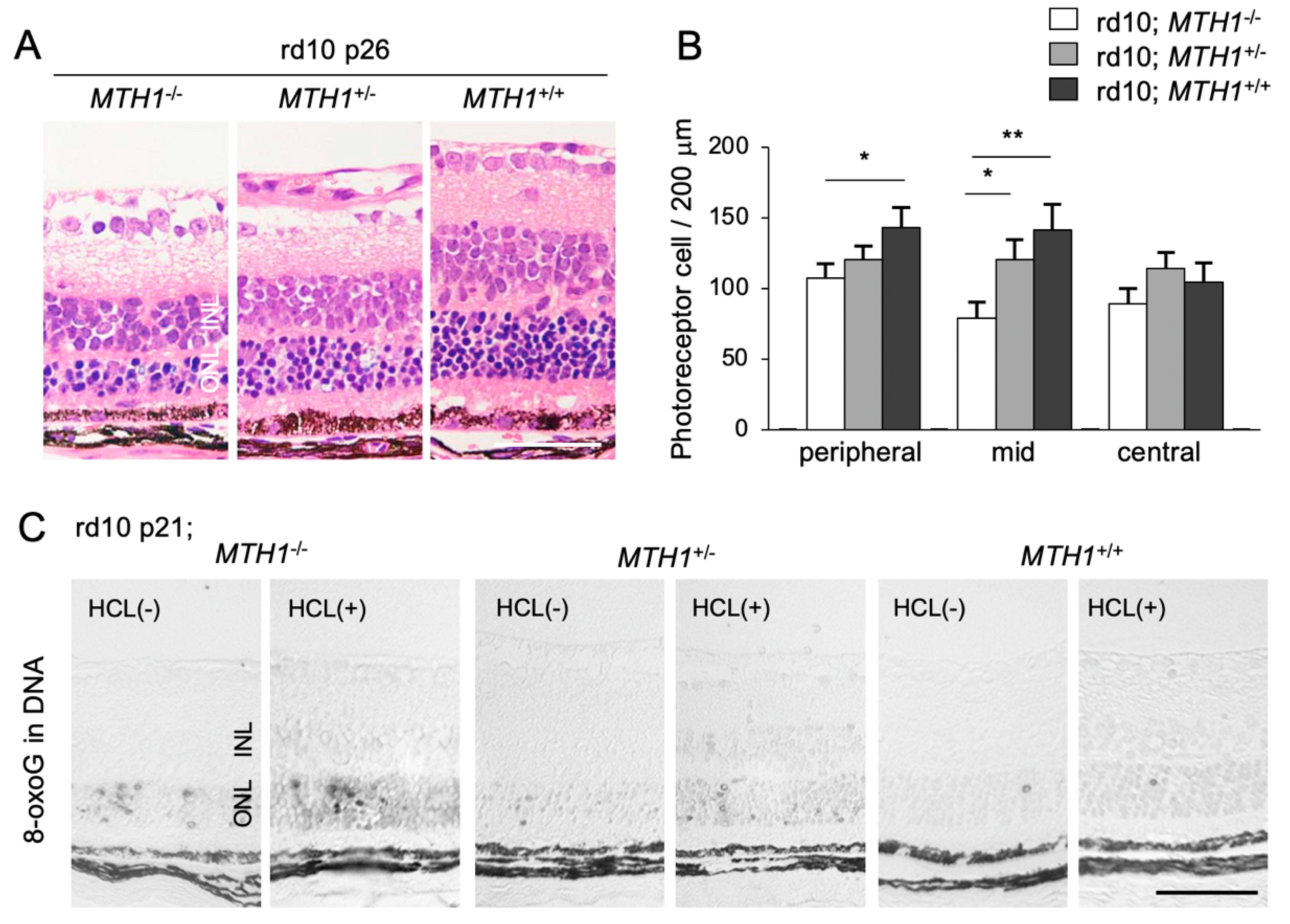
5.1. Accumulation of 8-oxoG in RP
5.2. Role of Oxidative DNA Damage in RP
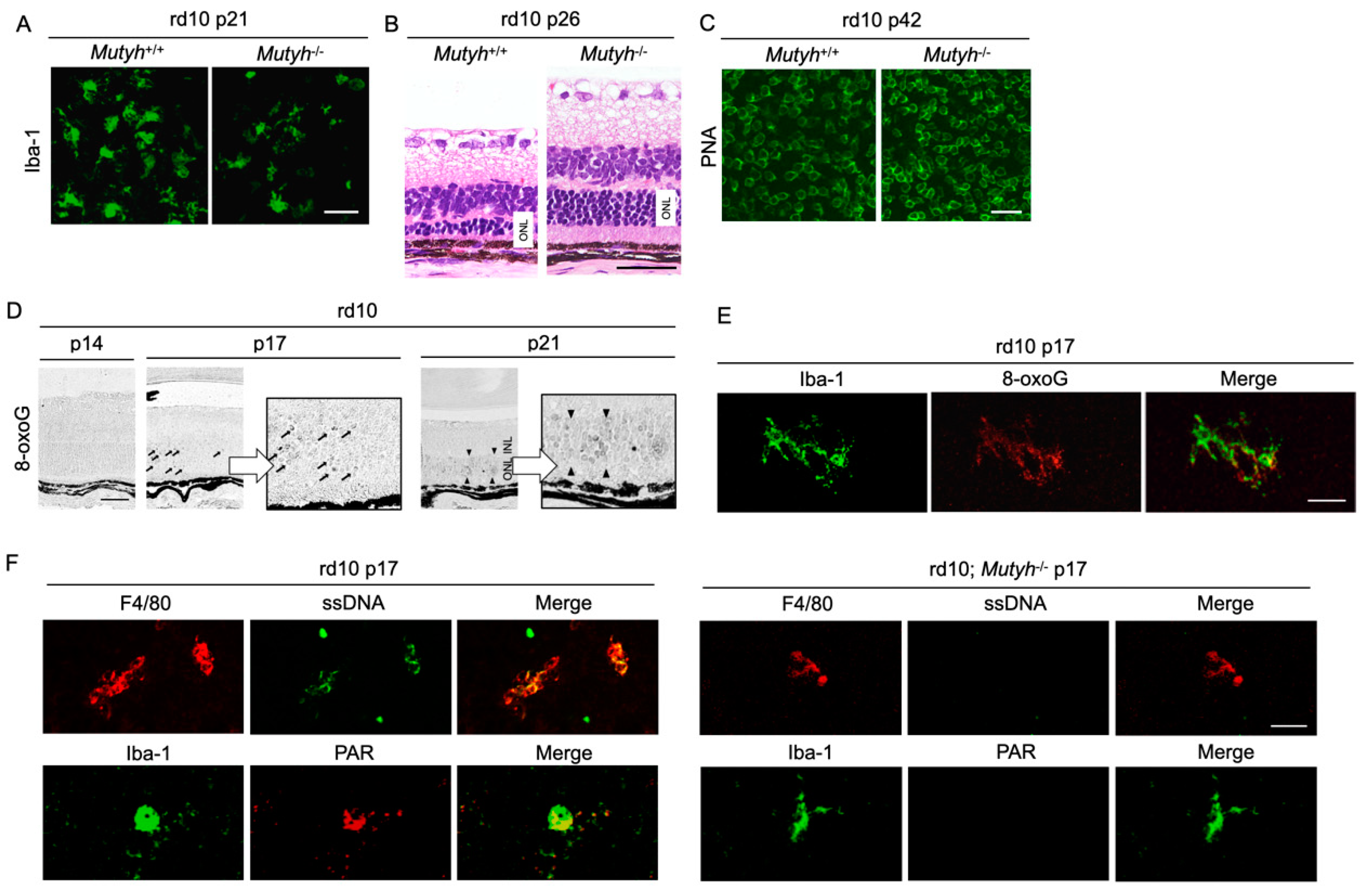
5.3. Oxidative DNA Damage Mediates Microglial Activation through MUTYH-mediated SSB Formation, Thereby Promoting Retinal Degeneration
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Takeya, R.; Sumimoto, H. Molecular mechanism for activation of superoxide-producing NADPH oxidases. Mol. Cells 2003, 16, 271–277. [Google Scholar]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef]
- Morizane, Y.; Morimoto, N.; Fujiwara, A.; Kawasaki, R.; Yamashita, H.; Ogura, Y.; Shiraga, F. Incidence and causes of visual impairment in Japan: The first nation-wide complete enumeration survey of newly certified visually impaired individuals. Jpn. J. Ophthalmol. 2019, 63, 26–33. [Google Scholar] [CrossRef]
- Ikeda, Y.; Nakatake, S.; Funatsu, J.; Fujiwara, K.; Tachibana, T.; Murakami, Y.; Hisatomi, T.; Yoshida, S.; Enaida, H.; Ishibashi, T.; et al. Night-vision aid using see-through display for patients with retinitis pigmentosa. Jpn. J. Ophthalmol. 2019, 63, 181–185. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef]
- Petit, L.; Ma, S.; Cipi, J.; Cheng, S.Y.; Zieger, M.; Hay, N.; Punzo, C. Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis pigmentosa. Cell Rep. 2018, 23, 2629–2642. [Google Scholar] [CrossRef]
- Usui, S.; Oveson, B.C.; Lee, S.Y.; Jo, Y.J.; Yoshida, T.; Miki, A.; Miki, K.; Iwase, T.; Lu, L.; Campochiaro, P.A. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J. Neurochem. 2009, 110, 1028–1037. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Carvalho, L.S.; Rizzi, M.; Powell, K.; Holthaus, S.M.; Azam, S.A.; Duran, Y.; Ribeiro, J.; Luhmann, U.F.; Bainbridge, J.W.; et al. Gene therapy restores vision in rd1 mice after removal of a confounding mutation in Gpr179. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Shen, J.; Yang, X.; Dong, A.; Petters, R.M.; Peng, Y.W.; Wong, F.; Campochiaro, P.A. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J. Cell. Physiol. 2005, 203, 457–464. [Google Scholar] [CrossRef]
- Murakami, Y.; Ikeda, Y.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Oka, S.; De Luca, G.; Yonemitsu, Y.; Bignami, M.; Nakabeppu, Y.; et al. MutT homolog-1 attenuates oxidative DNA damage and delays photoreceptor cell death in inherited retinal degeneration. Am. J. Pathol. 2012, 181, 1378–1386. [Google Scholar] [CrossRef]
- Yamada, H.; Yamada, E.; Hackett, S.F.; Ozaki, H.; Okamoto, N.; Campochiaro, P.A. Hyperoxia causes decreased expression of vascular endothelial growth factor and endothelial cell apoptosis in adult retina. J. Cell. Physiol. 1999, 179, 149–156. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef]
- Lee, S.Y.; Usui, S.; Zafar, A.B.; Oveson, B.C.; Jo, Y.J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell. Physiol. 2011, 226, 1843–1849. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, e5–e12. [Google Scholar] [CrossRef]
- Usui, S.; Oveson, B.C.; Iwase, T.; Lu, L.; Lee, S.Y.; Jo, Y.J.; Wu, Z.; Choi, E.Y.; Samulski, R.J.; Campochiaro, P.A. Overexpression of SOD in retina: Need for increase in H2O2-detoxifying enzyme in same cellular compartment. Free Radic. Biol. Med. 2011, 51, 1347–1354. [Google Scholar] [CrossRef]
- Xiong, W.; MacColl Garfinkel, A.E.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in rpe65 mutation-associated inherited retinal dystrophy: Results of phase 1 and 3 trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Cideciyan, A.V.; Heon, E.; Sheplock, R.; Pearson, A.; WeiYang Yu, C.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2019, 100827. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is there excess oxidative stress and damage in eyes of patients with retinitis pigmentosa? Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef]
- de la Cámara, C.M.F.; Salom, D.; Sequedo, M.D.; Hervas, D.; Marin-Lambies, C.; Aller, E.; Jaijo, T.; Diaz-Llopis, M.; Millan, J.M.; Rodrigo, R. Altered antioxidant-oxidant status in the aqueous humor and peripheral blood of patients with retinitis pigmentosa. PLoS ONE 2013, 8, e74223. [Google Scholar] [CrossRef]
- Ishizu, M.; Murakami, Y.; Fujiwara, K.; Funatsu, J.; Shimokawa, S.; Nakatake, S.; Tachibana, T.; Hisatomi, T.; Koyanagi, Y.; Akiyama, M.; et al. Relationships between serum antioxidant and oxidant statuses and visual function in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4462–4468. [Google Scholar] [CrossRef]
- Sunkara, A.; Raizner, A. Supplemental vitamins and minerals for cardiovascular disease prevention and treatment. Methodist Debakey Cardiovasc. J. 2019, 15, 179–184. [Google Scholar]
- Gaziano, J.M.; Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.; Glynn, R.J.; Buring, J.E. Multivitamins in the prevention of cancer in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2012, 308, 1871–1880. [Google Scholar] [CrossRef]
- Hercberg, S.; Galan, P.; Preziosi, P.; Bertrais, S.; Mennen, L.; Malvy, D.; Roussel, A.M.; Favier, A.; Briancon, S. The SU.VI.MAX Study: A randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch. Intern. Med. 2004, 164, 2335–2342. [Google Scholar] [CrossRef]
- Fortmann, S.P.; Burda, B.U.; Senger, C.A.; Lin, J.S.; Whitlock, E.P. Vitamin and mineral supplements in the primary prevention of cardiovascular disease and cancer: An updated systematic evidence review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2013, 159, 824–834. [Google Scholar] [CrossRef]
- Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar] [CrossRef]
- Tanvetyanon, T.; Bepler, G. Beta-carotene in multivitamins and the possible risk of lung cancer among smokers versus former smokers: A meta-analysis and evaluation of national brands. Cancer 2008, 113, 150–157. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Sperduto, R.D.; Sangiovanni, J.P.; Kurinij, N.; Davis, M.D. Age-Related Eye Disease Study Research, G. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. Ophthalmology 2013, 120, 1604–1611. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Launer, L.J.; Grodstein, F.; Bernstein, P.S.; Age-Related Eye Disease Study 2 (AREDS2) Research Group. Effect of omega-3 fatty acids, lutein/zeaxanthin, or other nutrient supplementation on cognitive function: The AREDS2 Randomized Clinical Trial. JAMA 2015, 314, 791–801. [Google Scholar] [CrossRef]
- Christen, W.G.; Glynn, R.J.; Sesso, H.D.; Kurth, T.; Macfadyen, J.; Bubes, V.; Buring, J.E.; Manson, J.E.; Gaziano, J.M. Vitamins E and C and medical record-confirmed age-related macular degeneration in a randomized trial of male physicians. Ophthalmology 2012, 119, 1642–1649. [Google Scholar] [CrossRef]
- Christen, W.G.; Glynn, R.J.; Manson, J.E.; MacFadyen, J.; Bubes, V.; Schvartz, M.; Buring, J.E.; Sesso, H.D.; Gaziano, J.M. Effects of multivitamin supplement on cataract and age-related macular degeneration in a randomized trial of male physicians. Ophthalmology 2014, 121, 525–534. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Willett, W.C. Omega-3 intake and visual acuity in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 2012, 130, 707–711. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Pearson, N.S.; Fish, G.E.; Spencer, R.; Takacs, A.; Klein, M.; Locke, K.G.; Birch, D.G. Four-year placebo-controlled trial of docosahexaenoic acid in X-linked retinitis pigmentosa (DHAX trial): A randomized clinical trial. JAMA Ophthalmol. 2014, 132, 866–873. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Spencer, R.; Fish, G.E.; Pearson, N.S.; Wang, Y.Z.; Klein, M.; Takacs, A.; Locke, K.G.; Birch, D.G. Docosahexaenoic acid slows visual field progression in X-linked retinitis pigmentosa: Ancillary outcomes of the DHAX Trial. Invest. Ophthalmol. Vis. Sci. 2015, 56, 6646–6653. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Brockhurst, R.J.; Hayes, K.C.; Johnson, E.J.; Anderson, E.J.; Johnson, C.A.; Gaudio, A.R.; et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 2010, 128, 403–411. [Google Scholar]
- Rotenstreich, Y.; Belkin, M.; Sadetzki, S.; Chetrit, A.; Ferman-Attar, G.; Sher, I.; Harari, A.; Shaish, A.; Harats, D. Treatment with 9-cis beta-carotene-rich powder in patients with retinitis pigmentosa: A randomized crossover trial. JAMA Ophthalmol. 2013, 131, 985–992. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Iftikhar, M.; Hafiz, G.; Akhlaq, A.; Tsai, G.; Wehling, D.; Lu, L.; Wall, G.M.; Singh, M.S.; Kong, X. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Investig. 2020, 130, 1527–1541. [Google Scholar] [CrossRef]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Kamiya, H.; Kasai, H. Formation of 2-hydroxydeoxyadenosine triphosphate, an oxidatively damaged nucleotide, and its incorporation by DNA polymerases. Steady-state kinetics of the incorporation. J. Biol. Chem. 1995, 270, 19446–19450. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014, 15, 12543–12557. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Ohta, E.; Abolhassani, N. MTH1 as a nucleotide pool sanitizing enzyme: Friend or foe? Free Radic. Biol. Med. 2017, 107, 151–158. [Google Scholar] [CrossRef]
- Oka, S.; Nakabeppu, Y. DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011, 102, 677–682. [Google Scholar] [CrossRef]
- Sakamoto, K.; Tominaga, Y.; Yamauchi, K.; Nakatsu, Y.; Sakumi, K.; Yoshiyama, K.; Egashira, A.; Kura, S.; Yao, T.; Tsuneyoshi, M.; et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007, 67, 6599–6604. [Google Scholar] [CrossRef]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999, 154, 1423–1429. [Google Scholar] [CrossRef]
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008, 27, 421–432. [Google Scholar] [CrossRef]
- Foti, J.J.; Devadoss, B.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 2012, 336, 315–319. [Google Scholar] [CrossRef]
- Nakatake, S.; Murakami, Y.; Ikeda, Y.; Morioka, N.; Tachibana, T.; Fujiwara, K.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Yoshida, S.; et al. MUTYH promotes oxidative microglial activation and inherited retinal degeneration. JCI Insight 2016, 1, e87781. [Google Scholar] [CrossRef][Green Version]
- Herz, J.; Filiano, A.J.; Smith, A.; Yogev, N.; Kipnis, J. Myeloid cells in the central nervous system. Immunity 2017, 46, 943–956. [Google Scholar] [CrossRef]
- Roh, M.I.; Murakami, Y.; Thanos, A.; Vavvas, D.G.; Miller, J.W. Edaravone, an ROS scavenger, ameliorates photoreceptor cell death after experimental retinal detachment. Invest. Ophthalmol. Vis. Sci. 2011, 52, 3825–3831. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Study Design and Supplements | Years of Follow-Up | n | Outcome | Reference |
|---|---|---|---|---|---|
| PHS-II | 2 × 2 × 2 × 2 RCT, b-carotene, vitamin-E, vitamin-C, multivitamin or placebo | 11.2 | 14,641 | CVD ↔ | [29] |
| Prostate cancer ↓ with multivitamin | |||||
| Other cancer ↔ | |||||
| Mortality ↔ | |||||
| AMD ↔ | |||||
| SU.VI.MAX | 2-arm RCT, Multivitamin or placebo | 7.5 | 13,017 | CVD ↔ | [30] |
| Cancer ↔ (in men ↓) | |||||
| Mortality ↔ (in men ↓) | |||||
| Fortmann et al. | Systematic review | NA | 324,653 | CVD ↔ | [31] |
| Cancer ↔ | |||||
| Mortality ↔ | |||||
| Lung cancer ↑ with b-carotene in current smokers | |||||
| AREDS | 4-arm RCT, (1) antioxidants (vitamin-C, vitamin-E and b-carotene), (2) Zinc, (3) antioxidants and zinc, or (4) placebo. | 6.3 | 3640 | Development of advanced AMD ↓ in subjects with AMD regions | [35] |
| AREDS2 | 4-arm RCT, (1) lutein/zeaxanthin, (2) omega-3 fatty acid, (3) lutein/zeaxanthin plus omega-3 fatty acid or (4) placebo in addition to AREDS formulation (AREDS-F) | 5.0 | 4203 | Development of advanced AMD ↔ with addition of lutein/zeaxanthin and/or omega-3 fatty acid | [36] |
| Secondary randomization, (1) AREDS-F, (2) AREDS-F without b-carotene, (3) AREDS-F with lower zinc, or (4) AREDS-F with no b-carotene and lower zinc | Development of advanced AMD ↓ with AREDS-F plus lutein/zeaxanthin compared with b-carotene containing AREDS-F | ||||
| Study | Study Design and Supplements | Years of Follow-Up | n | Outcome | References |
|---|---|---|---|---|---|
| Berson et al. | 2 × 2 RCT, vitamin A, vitamin E andVitamine A plus vitamin E or placebo | 5.2 | 601 | Cone ERG response ↑ with vitamin A | [39] |
| Visual acuity and field loss ↔ | |||||
| Berson et al. | post hoc analysis of omega-3 intake in 3 clnical trials which had tested vitamin A | 4~6 | 357 | Rate of visual acuity loss ↓ with high omega-3 intake | [40] |
| Cone ERG response ↔ | |||||
| DHAX | 2 arm RCT, DHA or placebo | 4.0 | 78 | Cone ERG response ↔ | [41,42] |
| * all participants received multivitamin | Rate of visual sensitivity loss ↓ with DHA | ||||
| Berson et al. | 2 arm RCT, Lutein or placebo | 4.0 | 225 | Rate of visual sensitivity loss ↔ | [43] |
| * all participants received vitamin A | Cone ERG response | ||||
| Rotenstreich et al. | 2 arm RCT, 9-cis b-carotene or placebo, crossover | 0.7 | 34 | Cone ERG response ↑ with 9-cis b-carotene | [44] |
| Visual acuity and field area ↔ | |||||
| Campochiaro et al. | Phase 1, 3 dose escalation of oral NAC | 0.5 | 30 | Visula acuity ↑ with all dosage | [45] |
| Visual sensitivity ↑ with high dosage | |||||
| Ellipsoid zone width ↔ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, Y.; Nakabeppu, Y.; Sonoda, K.-H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. https://doi.org/10.3390/ijms21197170
Murakami Y, Nakabeppu Y, Sonoda K-H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. International Journal of Molecular Sciences. 2020; 21(19):7170. https://doi.org/10.3390/ijms21197170
Chicago/Turabian StyleMurakami, Yusuke, Yusaku Nakabeppu, and Koh-Hei Sonoda. 2020. "Oxidative Stress and Microglial Response in Retinitis Pigmentosa" International Journal of Molecular Sciences 21, no. 19: 7170. https://doi.org/10.3390/ijms21197170
APA StyleMurakami, Y., Nakabeppu, Y., & Sonoda, K.-H. (2020). Oxidative Stress and Microglial Response in Retinitis Pigmentosa. International Journal of Molecular Sciences, 21(19), 7170. https://doi.org/10.3390/ijms21197170