Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration?


Abstract
1. Introduction
2. Disease Mechanisms of AMD
2.1. Pathophysiology
2.2. Molecular Mechanisms
2.3. RPE In Vitro AMD Studies
3. Prevention and Intervention Strategies of AMD
3.1. Dry AMD
3.2. Wet AMD
3.2.1. Anti-VEGF Drugs
3.2.2. Photodynamic Therapy (PDT)
3.2.3. Stem Cell Therapy
4. Overview of Novel Programmed Cell Death (PCD)
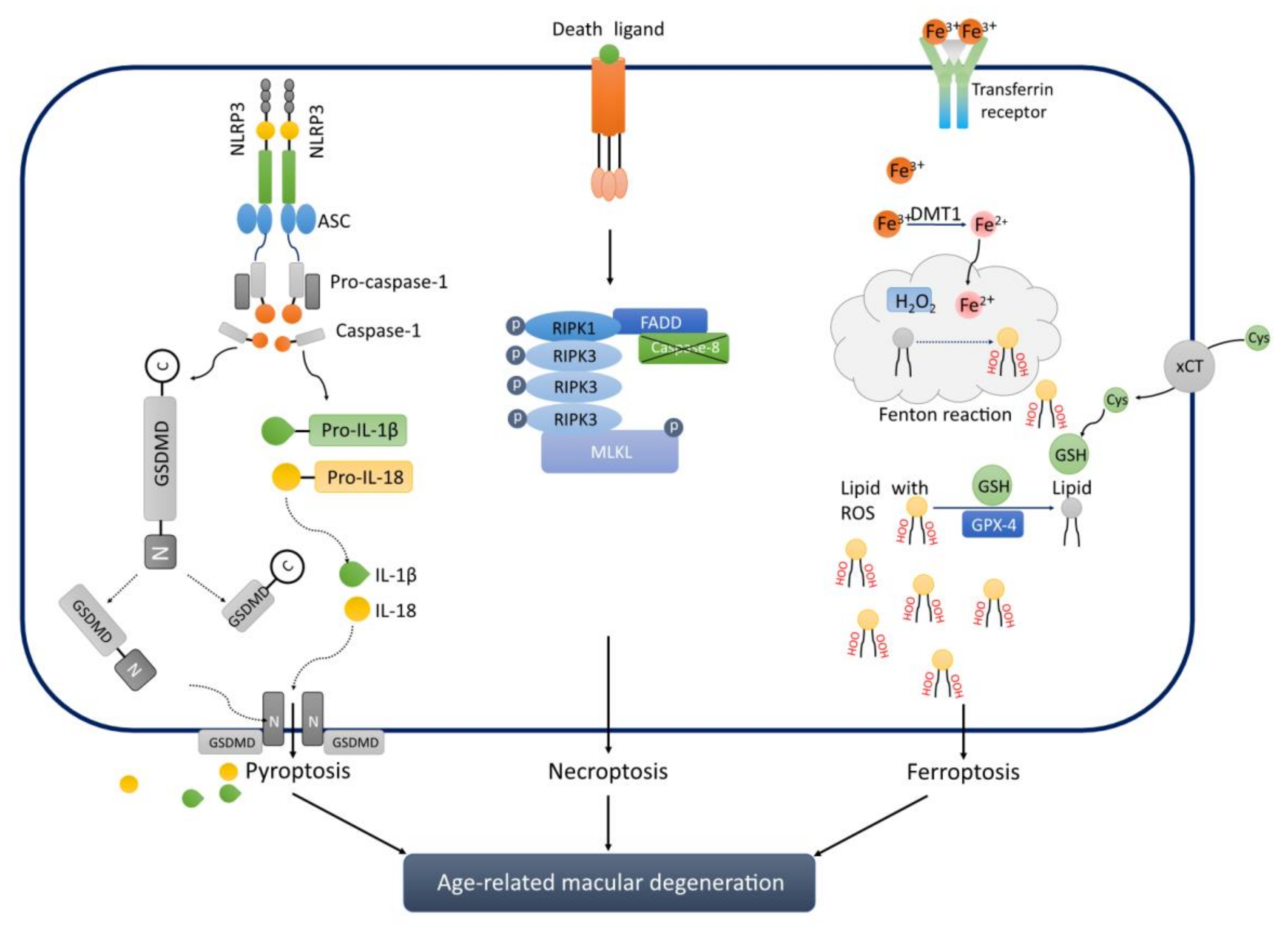
4.1. Pyroptosis
4.1.1. Classical Type of Pyroptosis
4.1.2. Non-Classical TYPE of Pyroptosis
4.2. Necroptosis
4.3. Ferroptosis
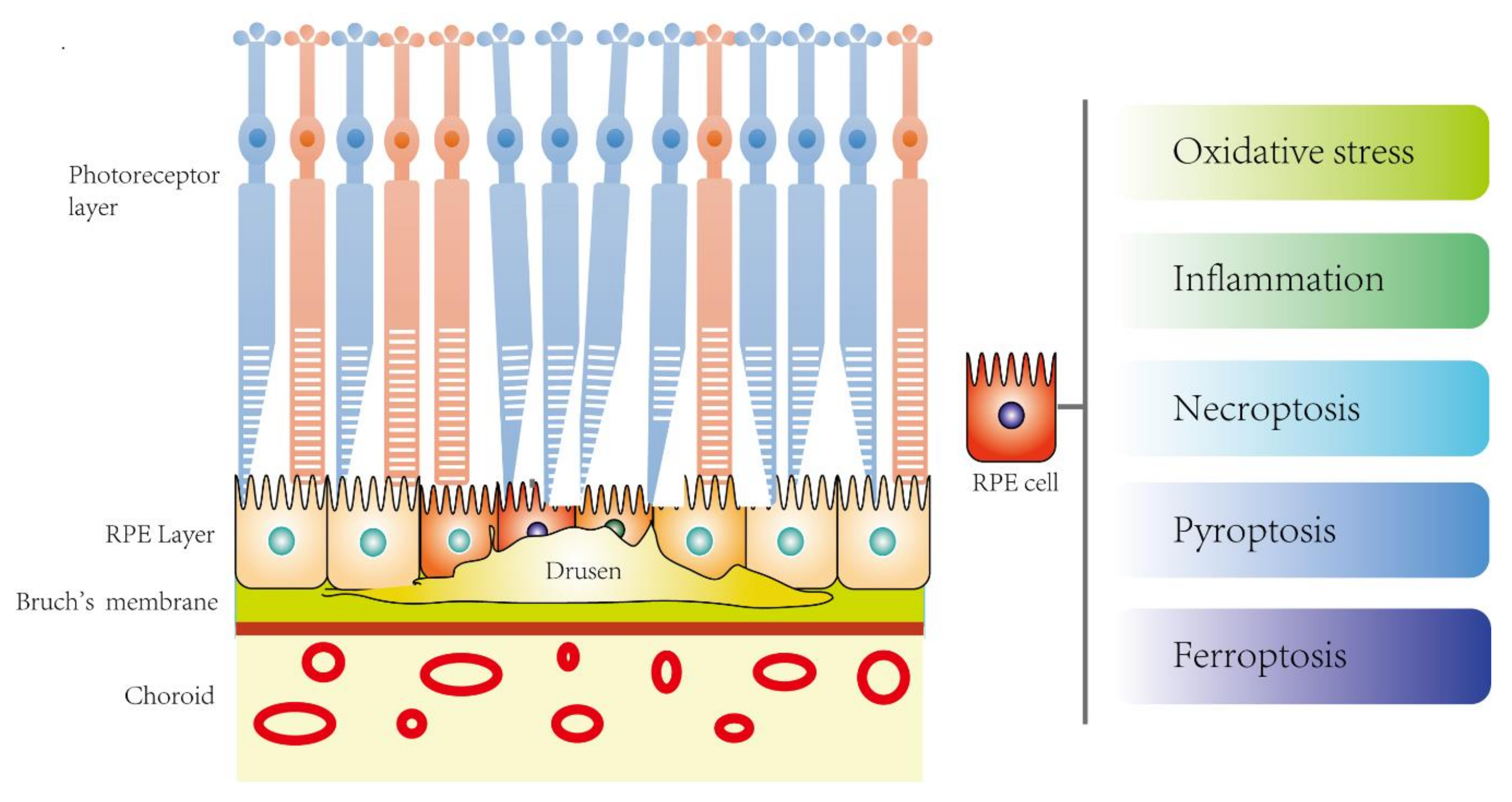
5. Novel PCD is Associated with AMD
5.1. Pyroptosis and AMD
5.2. Necroptosis and AMD
5.3. Ferroptosis and AMD
5.4. Aging and Novel PCD in AMD?
6. Novel PCD: New Future Therapeutic Targets for AMD?
7. Conclusion and Future Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adams, M.K.M.; Simpson, J.A.; Aung, K.Z.; Makeyeva, G.A.; Giles, G.G.; English, D.R.; Hopper, J.; Guymer, R.H.; Baird, P.N.; Robman, L.D. Abdominal Obesity and Age-related Macular Degeneration. Am. J. Epidemiol. 2011, 173, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef]
- Joachim, N.; Mitchell, P.; Burlutsky, G.; Kifley, A.; Wang, J.J. The Incidence and Progression of Age-Related Macular Degeneration over 15 Years The Blue Mountains Eye Study. Ophthalmology 2015, 122, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lan, X.; Wu, J.; Yue, S.; Zhang, H.; Wu, Q.; Zhang, G.; Liu, L. Protocol of global incidence and progression of age-related macular degeneration: A systematic review. Medicine 2019, 98, e14645. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, B.; Frazer-Abel, A.; Leonard, A.; Ratnapriya, R.; Ward, T.; Pietraszkiewicz, A.; O’Quinn, E.; Adams, K.; Swaroop, A.; Wolf, B.J. Association of age-related macular degeneration with complement activation products, smoking, and single nucleotide polymorphisms in South Carolinians of European and African descent. Mol. Vis. 2019, 25, 79–92. [Google Scholar]
- Peeters, A.; Magliano, D.J.; Stevens, J.; Duncan, B.B.; Klein, R.; Wong, T.Y. Changes in Abdominal Obesity and Age-Related Macular Degeneration The Atherosclerosis Risk in Communities Study. Arch. Ophthalmol-Chic 2008, 126, 1554–1560. [Google Scholar] [CrossRef]
- Haymes, S.A.; Lee, J. Effects of task lighting on visual function in age-related macular degeneration. Ophthalmic Physiol. Opt. 2006, 26, 169–179. [Google Scholar] [CrossRef]
- Cougnard-Gregoire, A.; Delyfer, M.N.; Korobelnik, J.F.; Rougier, M.B.; Malet, F.; Le Goff, M.; Dartigues, J.F.; Colin, J.; Barberger-Gateau, P.; Delcourt, C. Long-Term Blood Pressure and Age-Related Macular Degeneration: The ALIENOR Study. Investig. Ophth. Vis. Sci. 2013, 54, 1905–1912. [Google Scholar] [CrossRef]
- Age-Related Macular Degeneration. American Academy of Ophthalmology. Available online: https://www.aao.org/bcscsnippetdetail.aspx?id=9711f063-ed7b-452b-8708-c4dad0d893e8 (accessed on 14 September 2020).
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Sarks, J.P.; Sarks, S.H.; Killingsworth, M.C. Evolution of Soft Drusen in Age-Related Macular Degeneration. Eye 1994, 8, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Luibl, V.; Isas, J.M.; Kayed, R.; Glabe, C.G.; Langen, R.; Chen, J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J. Clin. Investig. 2006, 116, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Bhatia, S.K.; Mazzitello, K.I.; Chrenek, M.A.; Zhang, Q.; Boatright, J.H.; Grossniklaus, H.E.; Jiang, Y.; Nickerson, J.M. RPE Cell and Sheet Properties in Normal and Diseased Eyes. Adv. Exp. Med. Biol. 2016, 854, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Del Priore, L.V.; Ku, Y.H.; Tezel, T.H. Age-related changes in human RPE cell density and apoptosis proportion in situ. Investig. Ophth. Vis. Sci. 2002, 43, 3312–3318. [Google Scholar]
- Kokkinopoulos, I.; Shahabi, G.; Colman, A.; Jeffery, G. Mature Peripheral RPE Cells Have an Intrinsic Capacity to Proliferate; A Potential Regulatory Mechanism for Age-Related Cell Loss. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Chakravarthy, U.; McKay, G.J.; de Jong, P.T.V.M.; Rahu, M.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; Vingerling, J.R.; Vioque, J.; et al. ARMS2 Increases the Risk of Early and Late Age-related Macular Degeneration in the European Eye Study. Ophthalmology 2013, 120, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Decanini, A.; Nordgaard, C.L.; Feng, X.; Ferrington, D.A.; Olsen, T.W. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 2007, 143, 607–615. [Google Scholar] [CrossRef]
- Mullins, R.F.; Skeie, J.M.; Malone, E.A.; Kuehn, M.H. Macular and peripheral distribution of ICAM-1 in the human choriocapillaris and retina. Mol. Vis. 2006, 12, 224–235. [Google Scholar]
- Munch, I.C.; Ek, J.; Kessel, L.; Sander, B.; Almind, G.J.; Brondum-Nielsen, K.; Linneberg, A.; Larsen, M. Small, hard macular drusen and peripheral drusen: Associations with AMD genotypes in the Inter99 Eye Study. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2317–2321. [Google Scholar] [CrossRef]
- Nahavandipour, A.; Krogh Nielsen, M.; Sorensen, T.L.; Subhi, Y. Systemic levels of interleukin-6 in patients with age-related macular degeneration: A systematic review and meta-analysis. Acta Ophthalmol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Masse, A.; Buhannic, L. Understanding age-related macular degeneration. Actual Pharm. 2017, 56, 18–21. [Google Scholar] [CrossRef]
- Bonilha, V.L.; Bell, B.A.; Hu, J.; Milliner, C.; Pauer, G.J.; Hagstrom, S.A.; Radu, R.A.; Hollyfield, J.G. Geographic Atrophy: Confocal Scanning Laser Ophthalmoscopy, Histology, and Inflammation in the Region of Expanding Lesions. Investig. Ophthalmol. Vis. Sci. 2020, 61, 15. [Google Scholar] [CrossRef] [PubMed]
- Bakri, S.J.; Thorne, J.E.; Ho, A.C.; Ehlers, J.P.; Schoenberger, S.D.; Yeh, S.; Kim, S.J. Safety and Efficacy of Anti-Vascular Endothelial Growth Factor Therapies for Neovascular Age-Related Macular Degeneration: A Report by the American Academy of Ophthalmology. Ophthalmology 2019, 126, 55–63. [Google Scholar] [CrossRef]
- Mitchell, P.; Foran, S. Age-Related Eye Disease Study severity scale and simplified severity scale for age-related macular degeneration. Arch. Ophthalmol. 2005, 123, 1598–1599. [Google Scholar] [CrossRef]
- Yehoshua, Z.; Wang, F.H.; Rosenfeld, P.J.; Penha, F.M.; Feuer, W.J.; Gregori, G. Natural History of Drusen Morphology in Age-Related Macular Degeneration Using Spectral Domain Optical Coherence Tomography. Ophthalmology 2011, 118, 2434–2441. [Google Scholar] [CrossRef]
- Celkova, L.; Doyle, S.L.; Campbell, M. NLRP3 Inflammasome and Pathobiology in AMD. J. Clin. Med. 2015, 4, 172–192. [Google Scholar] [CrossRef]
- Dunaief, J.L.; Dentchev, T.; Ying, G.S.; Milam, A.H. The role of apoptosis in age-related macular degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef]
- Feng, L.; Liao, X.; Zhang, Y.; Wang, F. Protective Effects on Age-related Macular Degeneration by Activated Autophagy Induced by Amyloid-beta in Retinal Pigment Epithelial Cells. Discov. Med. 2019, 27, 153–160. [Google Scholar]
- Kim, S.H.; Park, J.W. Morin hydrate attenuates CSE-induced lipid accumulation, ER stress, and oxidative stress in RPE cells: Implications for age-related macular degeneration. Free Radic. Res. 2019, 53, 865–874. [Google Scholar] [CrossRef]
- Dieguez, H.H.; Romeo, H.E.; Alaimo, A.; Gonzalez Fleitas, M.F.; Aranda, M.L.; Rosenstein, R.E.; Dorfman, D. Oxidative stress damage circumscribed to the central temporal retinal pigment epithelium in early experimental non-exudative age-related macular degeneration. Free Radic. Biol. Med. 2019, 131, 72–80. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.J. Macrophages and macular degeneration. J. Ophthalmic. Vis. Res. 2014, 9, 1–2. [Google Scholar] [PubMed]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- Kliffen, M.; Sharma, H.S.; Mooy, C.M.; Kerkvliet, S.; de Jong, P.T. Increased expression of angiogenic growth factors in age-related maculopathy. Br. J. Ophthalmol. 1997, 81, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Tsujinaka, H.; Hirai, H.; Yamashita, M.; Ueda, T.; Ogata, N. Effects of concentration of amyloid beta (Abeta) on viability of cultured retinal pigment epithelial cells. BMC Ophthalmol. 2019, 19, 70. [Google Scholar] [CrossRef]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef]
- Spilsbury, K.; Garrett, K.L.; Shen, W.Y.; Constable, I.J.; Rakoczy, P.E. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am. J. Pathol. 2000, 157, 135–144. [Google Scholar] [CrossRef]
- Kyosseva, S.V. Targeting MAPK Signaling in Age-Related Macular Degeneration. Ophthalmol. Eye Dis. 2016, 8, 23–30. [Google Scholar] [CrossRef]
- Byeon, S.H.; Lee, S.C.; Choi, S.H.; Lee, H.K.; Lee, J.H.; Chu, Y.K.; Kwon, O.W. Vascular Endothelial Growth Factor as an Autocrine Survival Factor for Retinal Pigment Epithelial Cells under Oxidative Stress via the VEGF-R2/PI3K/Akt. Investig. Ophth. Vis. Sci. 2010, 51, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Im, E.; Motiejunaite, R.; Aranda, J.; Park, E.Y.; Federico, L.; Kim, T.I.; Clair, T.; Stracke, M.L.; Smyth, S.; Kazlauskas, A. Phospholipase Cgamma activation drives increased production of autotaxin in endothelial cells and lysophosphatidic acid-dependent regression. Mol. Cell. Biol. 2010, 30, 2401–2410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yazama, F.; Kadonosono, K.; Itoh, N.; Ohno, S. Role of matrix metalloproteinase-7 in angiogenesis associated with age-related macular degeneration. J. Electron. Microsc. 2002, 51, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Lambert, V.; Wielockx, B.; Munaut, C.; Galopin, C.; Jost, M.; Itoh, T.; Werb, Z.; Baker, A.; Libert, C.; Krell, H.W.; et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003, 17, 2290–2292. [Google Scholar] [CrossRef]
- Uno, K.; Bhutto, I.A.; McLeod, D.S.; Merges, C.; Lutty, G.A. Impaired expression of thrombospondin-1 in eyes with age related macular degeneration. Br. J. Ophthalmol. 2006, 90, 48–54. [Google Scholar] [CrossRef]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef]
- Cabral, T.; Mello, L.G.M.; Lima, L.H.; Polido, J.; Regatieri, C.V.; Belfort, R., Jr.; Mahajan, V.B. Retinal and choroidal angiogenesis: A review of new targets. Int. J. Retina Vitreous 2017, 3, 31. [Google Scholar] [CrossRef]
- Hudson, N.; Cahill, M.; Campbell, M. Inner blood-retina barrier involvement in dry age-related macular degeneration (AMD) pathology. Neural. Regen. Res. 2020, 15, 1656–1657. [Google Scholar] [CrossRef]
- Forest, D.L.; Johnson, L.V.; Clegg, D.O. Cellular models and therapies for age-related macular degeneration. Dis. Model. Mech. 2015, 8, 421–427. [Google Scholar] [CrossRef]
- Wang, K.; Yao, Y.; Zhu, X.; Zhang, K.; Zhou, F.; Zhu, L. Amyloid beta induces NLRP3 inflammasome activation in retinal pigment epithelial cells via NADPH oxidase- and mitochondria-dependent ROS production. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, X.; Zhang, K.; Yao, Y.; Zhuang, M.; Tan, C.; Zhou, F.; Zhu, L. Puerarin inhibits amyloid beta-induced NLRP3 inflammasome activation in retinal pigment epithelial cells via suppressing ROS-dependent oxidative and endoplasmic reticulum stresses. Exp. Cell Res. 2017, 357, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; So, K.F.; Lo, A.C.Y.; Lam, W.C. The Effect of Lycium barbarum Polysaccharides on Pyroptosis-Associated Amyloid beta1-40 Oligomers-Induced Adult Retinal Pigment Epithelium 19 Cell Damage. Int. J. Mol. Sci. 2020, 21, 4658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Ng, T.K.; Brelen, M.E.; Wu, D.; Wang, J.X.; Chan, K.P.; Yung, J.S.Y.; Cao, D.; Wang, Y.; Zhang, S.; et al. Continuous exposure to non-lethal doses of sodium iodate induces retinal pigment epithelial cell dysfunction. Sci. Rep. 2016, 6, 37279. [Google Scholar] [CrossRef]
- Lin, Y.C.; Horng, L.Y.; Sung, H.C.; Wu, R.T. Sodium Iodate Disrupted the Mitochondrial-Lysosomal Axis in Cultured Retinal Pigment Epithelial Cells. J. Ocul. Pharmacol. Ther. 2018, 34, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.; Kwon, M.Y.; Woo, J.M.; Chung, S.W. Oxidative Stress-Induced Pentraxin 3 Expression Human Retinal Pigment Epithelial Cells is Involved in the Pathogenesis of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 6028. [Google Scholar] [CrossRef] [PubMed]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Liu, J.; Wang, Z.; Yu, L.L.; Wang, J. Piceatannol Protects Human Retinal Pigment Epithelial Cells against Hydrogen Peroxide Induced Oxidative Stress and Apoptosis through Modulating PI3K/Akt Signaling Pathway. Nutrients 2019, 11, 1515. [Google Scholar] [CrossRef]
- Hu, X.; Wu, X.; Zhao, B.; Wang, Y. Scutellarin protects human retinal pigment epithelial cells against hydrogen peroxide (H2O2)-induced oxidative damage. Cell Biosci. 2019, 9, 12. [Google Scholar] [CrossRef]
- Li, S.; Gaur, U.; Chong, C.M.; Lin, S.; Fang, J.; Zeng, Z.; Wang, H.; Zheng, W. Berberine Protects Human Retinal Pigment Epithelial Cells from Hydrogen Peroxide-Induced Oxidative Damage through Activation of AMPK. Int. J. Mol. Sci. 2018, 19, 1736. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef]
- McCusker, M.M.; Durrani, K.; Payette, M.J.; Suchecki, J. An eye on nutrition: The role of vitamins, essential fatty acids, and antioxidants in age-related macular degeneration, dry eye syndrome, and cataract. Clin. Dermatol. 2016, 34, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Sperduto, R.D.; Sangiovanni, J.P.; Kurinij, N.; Davis, M.D.; Age-Related Eye Disease Study Research Group. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology 2013, 120, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Sperduto, R.D.; Sangiovanni, J.P.; Davis, M.D.; Ferris, F.L.; Age-Related Eye Disease Study Research Group. Ten-year follow-up of age-related macular degeneration in the age-related eye disease study: AREDS report no. 36. JAMA Ophthalmol. 2014, 132, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study 2 Research Group; Chew, E.Y.; Clemons, T.E.; Sangiovanni, J.P.; Danis, R.P.; Ferris, F.L., 3rd; Elman, M.J.; Antoszyk, A.N.; Ruby, A.J.; Orth, D.; et al. Secondary analyses of the effects of lutein/zeaxanthin on age-related macular degeneration progression: AREDS2 report No. 3. JAMA Ophthalmol. 2014, 132, 142–149. [Google Scholar] [CrossRef]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Db Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Merry, G.F.; Munk, M.R.; Dotson, R.S.; Walker, M.G.; Devenyi, R.G. Photobiomodulation reduces drusen volume and improves visual acuity and contrast sensitivity in dry age-related macular degeneration. Acta Ophthalmol. 2017, 95, E270–E277. [Google Scholar] [CrossRef]
- Markowitz, S.N.; Devenyi, R.G.; Munk, M.R.; Croissant, C.L.; Tedford, S.E.; Ruckert, R.; Walker, M.G.; Patino, B.E.; Chen, L.; Nido, M.; et al. A Double-Masked, Randomized, Sham-Controlled, Single-Center Study with Photobiomodulation for the Treatment of Dry Age-Related Macular Degeneration. Retina 2019. [Google Scholar] [CrossRef]
- Turbert, D. Anti-VEGF Treatments. American Academy of Ophthalmology. Available online: https://www.aao.org/eye-health/drugs/anti-vegf-treatments (accessed on 14 September 2020).
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L.; Macular, C.A. Ranibizumab and Bevacizumab for Treatment of Neovascular Age-Related Macular Degeneration. Ophthalmology 2012, 119. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Ying, G.S.; Toth, C.A.; Daniel, E.; Grunwald, J.E.; Martin, D.F.; Maguire, M.G.; Comparison of Age-related Macular Degeneration Treatments Trials Research Group. Macular Morphology and Visual Acuity in Year Five of the Comparison of Age-related Macular Degeneration Treatments Trials. Ophthalmology 2019, 126, 252–260. [Google Scholar] [CrossRef]
- Comparison of Age-related Macular Degeneration Treatments Trials Research Group; Maguire, M.G.; Martin, D.F.; Ying, G.S.; Jaffe, G.J.; Daniel, E.; Grunwald, J.E.; Toth, C.A.; Ferris, F.L., 3rd; Fine, S.L. Five-Year Outcomes with Anti-Vascular Endothelial Growth Factor Treatment of Neovascular Age-Related Macular Degeneration: The Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2016, 123, 1751–1761. [Google Scholar] [CrossRef]
- Daniel, E.; Ying, G.S.; Kim, B.J.; Toth, C.A.; Ferris, F., 3rd; Martin, D.F.; Grunwald, J.E.; Jaffe, G.J.; Dunaief, J.L.; Pan, W.; et al. Five-Year Follow-up of Nonfibrotic Scars in the Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2019, 126, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Alfageme, C.; Nicholson, L.; Hamilton, R.D.; Patel, P.J. Incidence and Long-Term Visual Acuity Outcomes of Retinal Pigment Epithelium Tears after Intravitreal Anti-Vascular Endothelial Growth Factor Treatment of Neovascular Age-Related Macular Degeneration. Retina-J. Ret. Vit. Dis. 2019, 39, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.; Westborg, I.; Lovestam Adrian, M. Twelve per cent of 6142 eyes treated for neovascular age-related macular degeneration (nAMD) presented with low visual outcome within 2 years. Analysis from the Swedish Macula Registry (SMR). Acta Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, A.C.; Reis-Silva, A.; Pinheiro-Costa, J.; Beato, J.; Freitas-da-Costa, P.; Falcao, M.S.; Falcao-Reis, F.; Carneiro, A. Treatment of neovascular age-related macular degeneration with anti-VEGF agents: Retrospective analysis of 5-year outcomes. Clin. Ophthalmol. 2016, 10, 541–546. [Google Scholar] [CrossRef]
- Grunwald, J.E.; Pistilli, M.; Daniel, E.; Ying, G.S.; Pan, W.; Jaffe, G.J.; Toth, C.A.; Hagstrom, S.A.; Maguire, M.G.; Martin, D.F.; et al. Incidence and Growth of Geographic Atrophy during 5 Years of Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2017, 124, 97–104. [Google Scholar] [CrossRef]
- Kaiser, P.K. Verteporfin photodynamic therapy and anti-angiogenic drugs: Potential for combination therapy in exudative age-related macular degeneration. Curr. Med. Res. Opin. 2007, 23, 477–487. [Google Scholar] [CrossRef]
- Van den Bergh, H. Photodynamic therapy of age-related macular degeneration: History and principles. Semin. Ophthalmol. 2001, 16, 181–200. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Gallenga, C.E.; Costagliola, C.; Semeraro, F.; Romano, M.R.; Dell’Omo, R.; Russo, A.; De Nadai, K.; Gemmati, D.; D’Angelo, S.; et al. Impact of methylenetetrahydrofolate reductase C677T polymorphism on the efficacy of photodynamic therapy in patients with neovascular age-related macular degeneration. Sci. Rep. 2019, 9, 2614. [Google Scholar] [CrossRef]
- Cramer, A.O.; MacLaren, R.E. Translating induced pluripotent stem cells from bench to bedside: Application to retinal diseases. Curr. Gene Ther. 2013, 13, 139–151. [Google Scholar] [CrossRef]
- Da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36. [Google Scholar] [CrossRef]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.M.; Mitra, D.; Zhu, D.H.; Thomas, B.B.; et al. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D.; Hubschman, J.P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Sarraf, D.; Ma, J.; Wang, S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Discov. 2016, 2, 16054. [Google Scholar] [CrossRef]
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Shi, J.J.; Gao, W.Q.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yuan, Y.H.; Chen, N.H.; Wang, H.B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K.; Stockwell, B.R. SnapShot: Ferroptosis. Cell 2020, 181, 1188. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Wooff, Y.; Fernando, N.; Wong, J.H.C.; Dietrich, C.; Aggio-Bruce, R.; Chu-Tan, J.A.; Robertson, A.A.B.; Doyle, S.L.; Man, S.M.; Natoli, R. Caspase-1-dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration. Sci. Rep. 2020, 10, 2263. [Google Scholar] [CrossRef]
- Gao, J.; Cui, J.Z.; To, E.; Cao, S.; Matsubara, J.A. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J. Neuroinflammation 2018, 15, 15. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Rozing, M.P.; Durhuus, J.A.; Krogh Nielsen, M.; Subhi, Y.; Kirkwood, T.B.; Westendorp, R.G.; Sorensen, T.L. Age-related macular degeneration: A two-level model hypothesis. Prog. Retin. Eye Res. 2020, 76, 100825. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.E.; Aronson, J.K. Remdesivir in covid-19. BMJ 2020, 369, m1610. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E.; et al. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis. 2019, 10, 367–382. [Google Scholar] [CrossRef]
- Tower, J. Programmed cell death in aging. Ageing Res. Rev. 2015, 23, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, W.T.; Hu, L.C.; Li, J.X.; Fang, Y.; Wang, X.; Xu, X.Z.; Wang, Z.; Huang, K.; Han, J.H. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Sun, H.J.; Jin, X.M.; Xu, J.; Xiao, Q. Baicalin Alleviates Age-Related Macular Degeneration via miR-223/NLRP3-Regulated Pyroptosis. Pharmacology 2020, 105, 28–38. [Google Scholar] [CrossRef]
- Fang, P.; Yu, M.; Min, W.; Han, S.; Shi, M.; Zhang, Z.; Bo, P. Beneficial effect of baicalin on insulin sensitivity in adipocytes of diet-induced obese mice. Diabetes Res. Clin. Pract. 2018, 139, 262–271. [Google Scholar] [CrossRef]
- Huang, P.; Liu, W.; Chen, J.; Hu, Y.; Wang, Y.; Sun, J.; Feng, J. TRIM31 inhibits NLRP3 inflammasome and pyroptosis of retinal pigment epithelial cells through ubiquitination of NLRP3. Cell Biol. Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Yu, S.; Ji, W.; Li, H.; Gao, Y. The Contribution of Necroptosis in Neurodegenerative Diseases. Neurochem. Res. 2017, 42, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, D.; Sun, H.Y.; Wang, W.W.; Wu, H.; Kong, W.; Kong, W.J. Relieving ferroptosis may partially reverse neurodegeneration of the auditory cortex. FEBS J. 2020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type | Morphology | Activated/Increased Molecules | Inactivated/Decreased Molecules | Type of Cell Membrane Pores | References |
|---|---|---|---|---|---|
| Pyroptosis | Cell swelling Membrane blebbing Membrane pore Membrane rupture Pyroptosis bodies | NLRP3, ASC, Pro-caspase-1, and Gasdermin D | N/A | Gasdermin D-N-dependent | [90,91] |
| Necroptosis | Cell swelling Membrane pore Membrane rupture Necrotizing bodies Nucleus chromatin condensation | RIPK1, RIPK3 and MLKL | Caspase-8 | MLKL-dependent | [92] |
| Ferroptosis | Membrane vacuolated Membrane rupture Membrane density increase Cytoplasm rounding-up | Iron accumulation Lipid reactive oxygen species | GPx-4, GSH, xCT | Lipid reactive oxygen species-dependent | [93] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; So, K.-F.; Lam, W.C.; Lo, A.C.Y. Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration? Int. J. Mol. Sci. 2020, 21, 7279. https://doi.org/10.3390/ijms21197279
Yang M, So K-F, Lam WC, Lo ACY. Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration? International Journal of Molecular Sciences. 2020; 21(19):7279. https://doi.org/10.3390/ijms21197279
Chicago/Turabian StyleYang, Ming, Kwok-Fai So, Wai Ching Lam, and Amy Cheuk Yin Lo. 2020. "Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration?" International Journal of Molecular Sciences 21, no. 19: 7279. https://doi.org/10.3390/ijms21197279
APA StyleYang, M., So, K.-F., Lam, W. C., & Lo, A. C. Y. (2020). Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration? International Journal of Molecular Sciences, 21(19), 7279. https://doi.org/10.3390/ijms21197279